Glycogen storage disease
Glycogen storage diseases are a diverse group of rare inherited metabolic disorders (glycogen storage disease type 0 to 15) involving inherited defect in one of the enzymes responsible for forming glycogen, or for releasing glucose from glycogen as it is needed by the body during activity and/or between meals (see Table 1). All glycogen storage diseases are due to a failure to use or store glycogen 1. In the past, glycogen storage diseases were also named by the discovering physician. Glycogen storage disease can affect the liver, the muscles or both. Diagnosis of the glycogen storage disease variant is made on the basis of an individual’s symptoms, the results of a physical examination and of biochemical tests.
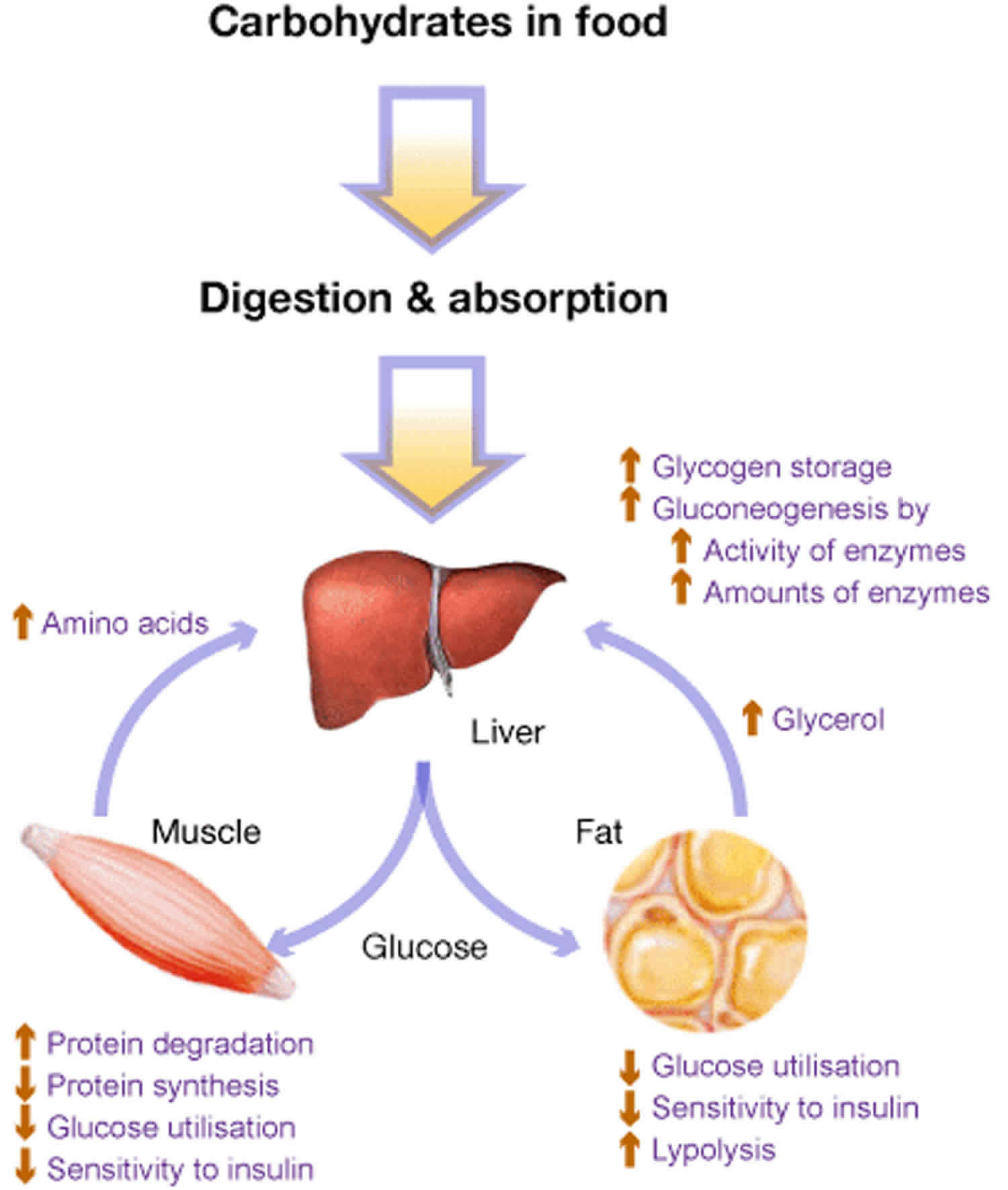
To be a fit and healthy individual must feed their body regularly with a variety of foods and water. This allows growth, provides energy and repairs tissues. The foods that you eat are broken down into smaller packages and either used for growth and repair, stored for when they’re needed for periods between meals or they’re disposed of as waste. Although this explanation describes the basic processes in the body, it is of course much more complex.
Glycogen is a branched polymer with its monomeric units being glucose. After a meal, the level of glucose in plasma increases and stimulates the storage of excess glucose in cytoplasmic glycogen spherical. The liver contains the highest percent glycogen by weight (about 10%) whereas muscle can store about 2% by weight. Nevertheless, since the total muscle mass is greater than liver mass, the total mass of glycogen in muscle is about twice that of the liver. When needed, the glycogen polymer can be broken down into glucose monomers and utilized for energy production. Many of the enzymes and transporters for these processes are key to the cause of glycogen storage diseases.
An increasing number of glycogen storage diseases are being identified, but some are very rare. Glycogen storage diseases affect roughly one child in 20,000 2. The underlying problem in all glycogen storage disease’s is the use or storage of glycogen. Glycogen is a complex material that is composed of glucose molecules that are linked together (like a tree with many branches). Most of the cells in your body rely on glucose as their main energy source. Your body controls its levels of glucose in the blood very carefully using different hormones. Immediately after a meal, the level of glucose rises and there becomes more sugar in the blood than the body needs at that time. The extra glucose is then moved to the liver and muscles and stored as glycogen, serving as a store for when the body’s sugar level drops below the normal level. You have specific enzymes to help us form or release this energy as it’s needed. Enzymes are protein molecules that speed up chemical reactions in the body.
The moving of glucose to glycogen and back again takes many complicated steps and the body requires enzymes to do this. These enzymes are responsible for creating the glycogen from glucose, moving the glycogen to and from the storage areas within the cells and extracting glucose from the glycogen when needed. A different enzyme is required at each step. People affected with glycogen storage disease have a problem in one of these many enzymes. The enzyme that’s not working fails to complete its step and the process comes to a stop.
In general, inborn errors of metabolisms result from the lack or insufficient level of specific enzymes that are needed to: (1) convert fat or carbohydrates to energy; (2) breakdown amino acids or other metabolites, allowing them to accumulate and become toxic. glycogen storage diseases, depending on the specific type, can result from a failure to convert glycogen into energy and/or a toxic glycogen accumulation.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Table 1. Glycogen storage disease types
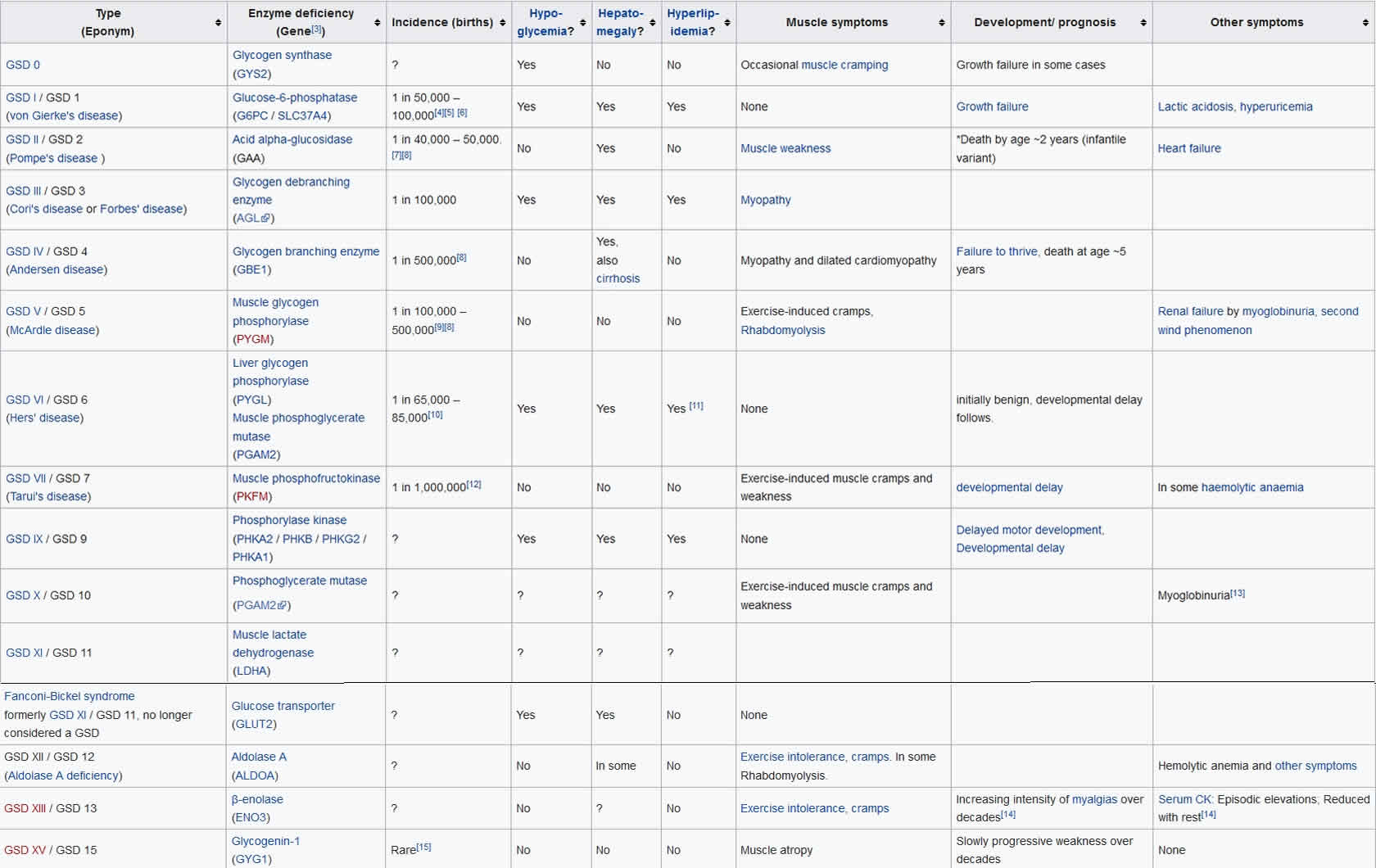
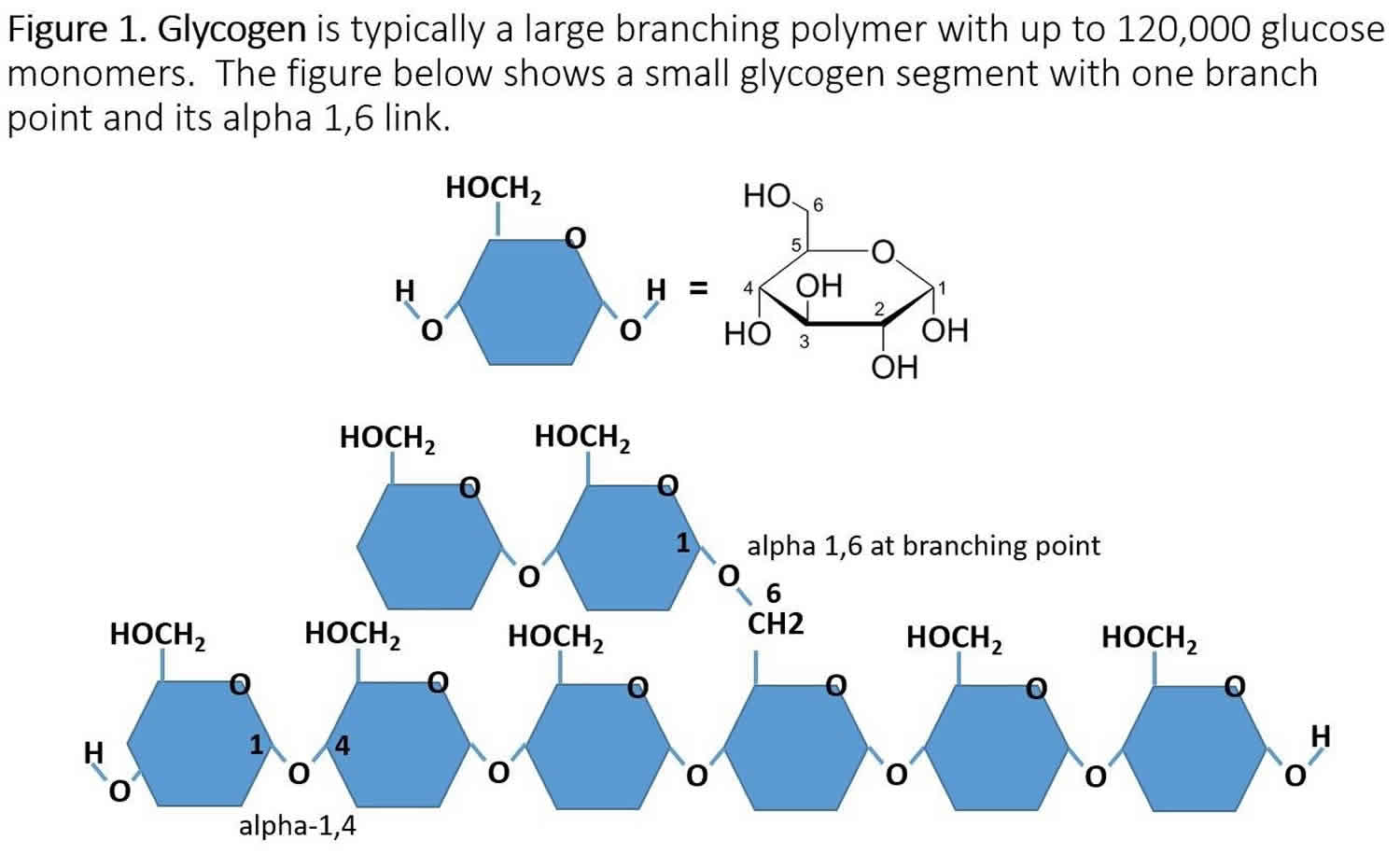
Figure 2. Glycogen metabolism
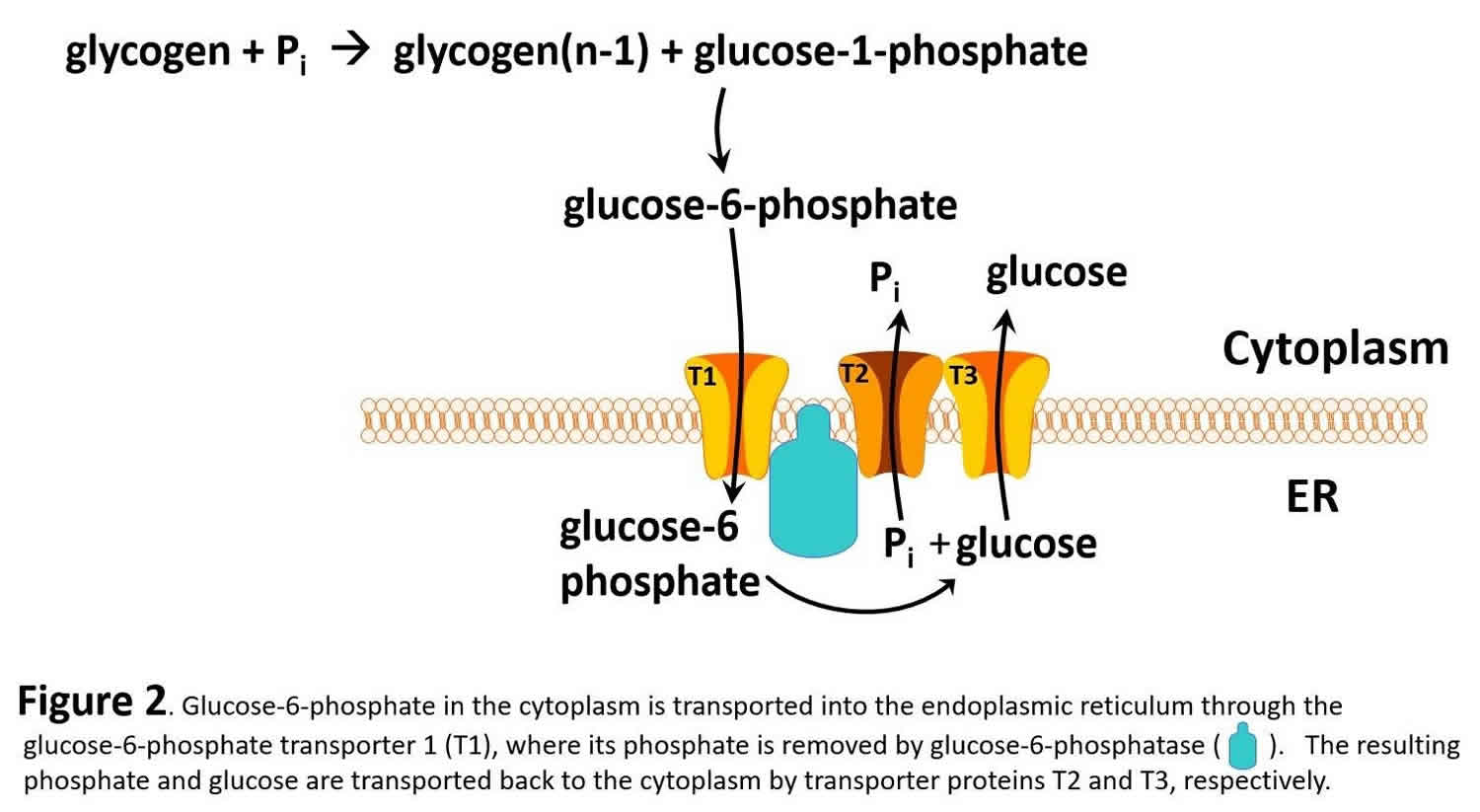
Figure 3. Glycogenolysis
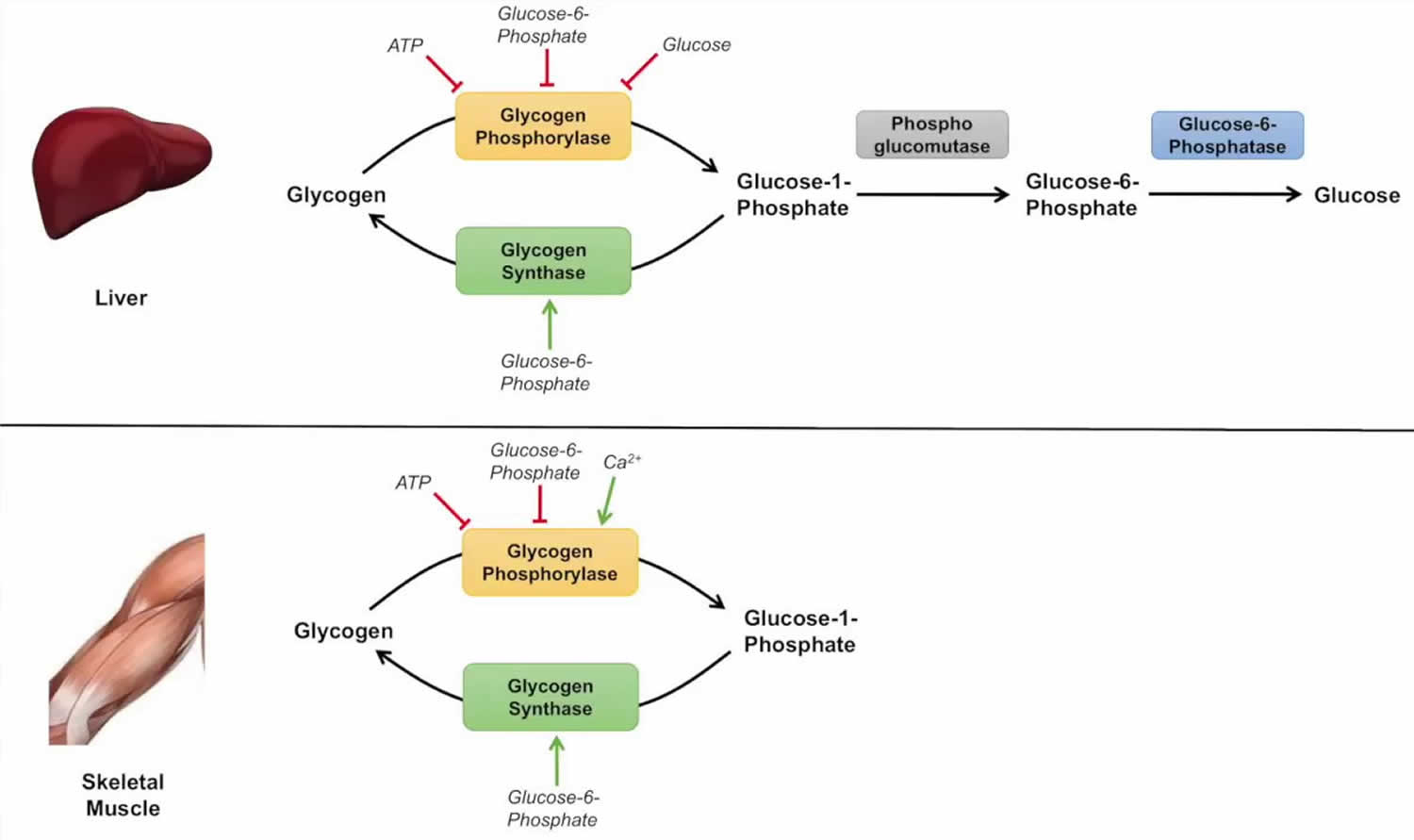
Figure 4. Glycogenolysis pathway
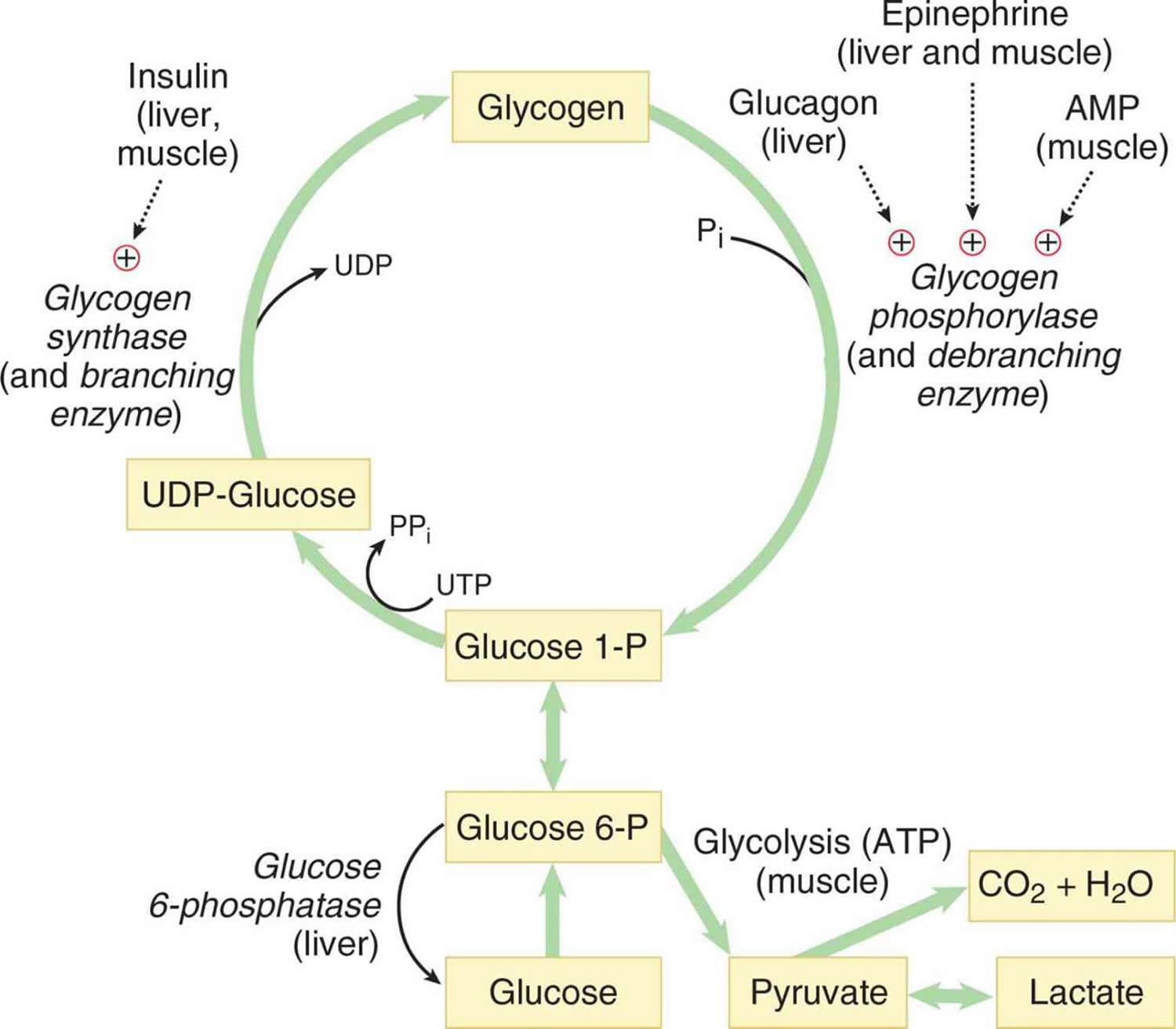
Abbreviations: UDP-Glucose = uracil diphosphate glucose
Glycogen storage disease causes
The cause of glycogen storage disease is best understood by following the metabolic events leading to the synthesis (glycogenesis) and degradation of glycogen (glycogenolysis) 4. Genetic defects in the enzymes and transporters involved in either glycogenesis or glycogenolysis are actual or potential causes of all glycogen storage diseases 5. Excess dietary glucose is stored in glycogen and glycogen synthesis is, in part, accomplished by glycogen synthase. As indicated in Table 1, there are two distinct forms of glycogen synthase, one in the liver encoded by the GYS2 gene and one in skeletal muscle encoded by the GYS1 gene. Both forms of glycogen synthase work by linking (alpha-1,4 links) a glucose monomer to the growing glycogen polymer. As indicated in Figure 1, glycogen has two different types of linkages, alpha-1,4 links, and alpha-1,6 links. About 95% of linkages in glycogen are alpha-1,4 links. The absence or malfunction of liver glycogen synthase due to mutations in the GYS2 gene will prevent glycogen from being synthesized in the liver, and this is the cause of glycogen storage disease type 0a (Table 1). Similarly, the absence or malfunction of muscle glycogen synthase due to mutations in the GYS1 gene will prevent glycogen from being synthesized in muscles, and this is the cause of glycogen storage disease type 0b (Table 1).
While glycogen synthase can catalyze the alpha-1,4 glucose linkages in glycogen, a different enzyme, glycogen branching enzyme (GBE1 gene symbol), is needed to produce the branching alpha-1,6 linkages (Figure 1). Mutations in the glycogen branching enzyme can result in the production of glycogen with an abnormal structure, and this is the cause of glycogen storage disease type 4 (Table 1). The abnormal glycogen structures are called polyglucosan bodies: they can accumulate in all cells, but most markedly in liver and muscle cells. Polyglucosan bodies do not effectively undergo glycogenolysis, and in muscle tissue, this can cause weakness and myopathy. In the liver, the accumulation of polyglucosan bodies causes hepatomegaly.
While glycogen storage disease 0a and glycogen storage disease 0b are due to insufficient storage of glycogen, most glycogen storage diseases are due to an inability to remove glucose from glycogen (glycogenolysis) resulting in excess glycogen tissue storage. The first step in glycogenolysis is the release of glucose-1-phosphate (G-1-P) from glycogen by the action of glycogen phosphorylase.
- Glycogen + P -> glycogen(n-1) + glucose-1-phosphate (G-1-P)
Glycogen storage disease type 5 is caused by mutations in the glycogen phosphorylase gene specific for muscle (PYGM). Mutations in the glycogen phosphorylase gene specific for liver (PYGL) cause glycogen storage disease type 6.
The glucose-1-phosphate (G-1-P) released by glycogen phosphorylase is converted to glucose-6-phosphate (G-6-P) by the action of phosphoglucomutase.
- Glucose-1-phosphate (G-1-P) -> glucose-6-phosphate (G-6-P)
Glucose-6-phosphate in the liver is, in turn, converted to glucose by glucose-6-phosphatase (gene name G6PC) and the resulting glucose is released into the blood as an energy source for other tissues/organs, such as the brain (see Figure 2).
- Glucose-6-phosphate (G-6-P) -> Glucose + Phosphate (liver not muscle) -> into blood
It should be noted that muscle lacks glucose-6-phosphatase and therefore does not release glucose into the blood. glycogen storage diseases type 1 is the result of genetic disorders in the metabolism of glucose-6-phosphatase 6. Glycogen storage disease type 1a also called von Gierke disease, is caused by mutations in the G6PC gene (Table 1). Glucose-6-phosphate is synthesized in the cytoplasm of hepatocytes and must be transported into the lumen of the endoplasmic reticulum where it is acted upon by glucose-6-phosphatase yielding glucose which is transported back to the cytoplasm and then through the hepatic GLUT2 transporter into the blood. Glucose-6-phosphate translocase1 (G6PT1) is the transporter protein that provides a G-6-P channel between the cytoplasm and the endoplasmic reticulum. The G6PT protein is made of three subunits termed G6PT1, G6PT2, and G6PT3 (see Figure 2). Mutations in the SLC37A4 gene, which encodes the G6PT1 protein, are responsible for glycogen storage disease type Ib (Figure 1). Fanconi-Bickel disease is a rare glycogen storage disease caused by a GLUT2 deficiency (gene name SLC2A2). GLUT2 deficiency results in a failure to export glucose, an increased intracellular glucose level and reduced degradation of glycogen: eventually there is increased glycogen storage and hepatomegaly.
As mentioned above, glycogen is a branched polymer. While glycogen phosphorylase works well at removing glucose from alpha-(1,4)-linkages, it does not work at branch points. Branch points are alpha-1,6 linkages: a glycogen debranching enzyme is required, which in mammals is called “ammylo-alpha-1,6-glucosidase, 4-alpha-Glucanotransferase” with the gene name AGL. glycogen storage disease type 3 is caused by mutations in the AGL gene (Figure 1), resulting in either a nonfunctional glycogen debranching enzyme (glycogen storage disease type 3a or type 3b) or a glycogen debranching enzyme with reduced function (glycogen storage disease type 3c and 3d) 7.
Glycogen storage disease 2 is unique among glycogen storage disease since it is also classified as lysosomal storage disease 8. Lysosomes are subcellular organelles that recycle cellular macromolecules. All lysosomal storage diseases are caused by a missing or nonfunctional lysosomal enzyme. In the case of glycogen storage disease 2, this enzyme is lysosomal acid alpha-glucosidase (gene name GAA), which breaks down glycogen into glucose for use as a cellular energy source. Mutation in the GAA gene results in the toxic accumulation of glycogen in lysosomes.
Glycogen storage disease symptoms
Glycogen storage diseases are a diverse set of rare inborn errors of carbohydrate metabolism that can have a very variable phenotypic presentation even within same glycogen storage disease type. Obtaining a family pedigree is useful in establishing the mode of inheritance. Most glycogen storage diseases show an autosomal recessive inheritance, but a few, e.g., a subtype of glycogen storage disease-9) show an X-linked inheritance.
Very general symptoms/signs would include:
- Failure to grow
- Heat intolerance
- Exercise intolerance
- Hypoglycemia
- Hepatomegaly
- Low muscle tone
- Acidosis
- Hyperlipidemia.
In glycogen storage disease type 1, glycogenolysis in the liver results in increased lactic acid production (lactic acidosis) due to the intracellular accumulation of glucose-6-phosphate which stimulates the glycolytic pathway.
Glycogen storage disease diagnosis
The evaluation would include (1) a biopsy of the affected tissue to measure glycogen content and appropriate enzymatic assays, (2) blood and urine tests, and (3) an MRI/CT scan. The blood tests would include serum glucose, anion gap, serum lactate, pH, serum uric acid, a lipid panel, serum gamma-glutamyltransferase, serum alkaline phosphatase, calcium, phosphorus, urea, and creatinine levels, complete blood count (CBC) and differential. Urine analysis would include testing for aminoaciduria, proteinuria, and microalbuminuria in older patients as well as excreted uric acid and calcium. Liver and kidney MRI (or CT) are routinely done for adults and ultrasonography for patients less than 16 years of age.
Glycogen storage disease treatment
At present, there is no cure for any glycogen storage disease, and most treatments attempt to alleviate signs/symptoms 5. Key overall goals are to treat or avoid hypoglycemia, hyperlactatemia, hyperuricemia, and hyperlipidemia. Hypoglycemia is avoided by consuming starch and an optimal, physically modified form, Glycosade, is now commercially available. Hyperuricemia is treated with allopurinol and hyperlipidemia with statins. glycogen storage disease type 2 can now be treated with enzyme replacement therapy (ERT), using recombinant alglucosidase alfa (Lumizyme) which degrades lysosomal glycogen 9. There is ongoing research to use enzyme replacement therapy with other forms of glycogen storage disease. Liver transplantation should be considered for patients with glycogen storage disease type 4 classical and progressive hepatic forms and for glycogen storage diseases that have progressed to hepatic malignancy or failure.
Glycogen storage disease type 0
Glycogen storage disease type 0 also known as GSD 0, is a condition caused by the body’s inability to form a complex sugar called glycogen, which is a major source of stored energy in the body.
Glycogen storage disease type 0 has two types:
- Muscle glycogen storage disease type 0, glycogen formation in the muscles is impaired,
- Liver glycogen storage disease type 0, glycogen formation in the liver is impaired.
The signs and symptoms of muscle glycogen storage disease type 0 typically begin in early childhood. Affected individuals often experience muscle pain and weakness or episodes of fainting (syncope) following moderate physical activity, such as walking up stairs. The loss of consciousness that occurs with fainting typically lasts up to several hours. Some individuals with muscle glycogen storage disease type 0 have a disruption of the heart’s normal rhythm (arrhythmia) known as long QT syndrome. In all affected individuals, muscle glycogen storage disease type 0 impairs the heart’s ability to effectively pump blood and increases the risk of cardiac arrest and sudden death, particularly after physical activity. Sudden death from cardiac arrest can occur in childhood or adolescence in people with muscle glycogen storage disease type 0.
Individuals with liver glycogen storage disease type 0 usually show signs and symptoms of the disorder in infancy. People with this disorder develop low blood sugar (hypoglycemia) after going long periods of time without food (fasting). Signs of hypoglycemia become apparent when affected infants begin sleeping through the night and stop late-night feedings; these infants exhibit extreme tiredness (lethargy), pale skin (pallor), and nausea. During episodes of fasting, ketone levels in the blood may increase (ketosis). Ketones are molecules produced during the breakdown of fats, which occurs when stored sugars (such as glycogen) are unavailable. These short-term signs and symptoms of liver glycogen storage disease 0 often improve when food is eaten and sugar levels in the body return to normal. The features of liver glycogen storage disease type 0 vary; they can be mild and go unnoticed for years, or they can include developmental delay and growth failure.
Glycogen storage disease type 0 affects both males and females and cases have been seen all around the world. The prevalence of glycogen storage disease type 0 is unknown; fewer than 10 people with the muscle type and fewer than 30 people with the liver type have been described in the scientific literature 10. Because some people with muscle glycogen storage disease type 0 die from sudden cardiac arrest early in life before a diagnosis is made and many with liver glycogen storage disease type 0 have mild signs and symptoms, it is thought that glycogen storage disease type 0 may be underdiagnosed.
Glycogen storage disease type 0 causes
Mutations in the GYS1 gene cause muscle glycogen storage disease type 0, and mutations in the GYS2 gene cause liver glycogen storage disease type 0. These genes provide instructions for making different versions of an enzyme called glycogen synthase. Both versions of glycogen synthase have the same function, to form glycogen molecules by linking together molecules of the simple sugar glucose, although they perform this function in different regions of the body.
The GYS1 gene provides instructions for making muscle glycogen synthase; this form of the enzyme is produced in most cells, but it is especially abundant in heart (cardiac) muscle and the muscles used for movement (skeletal muscles). During cardiac muscle contractions or rapid or sustained movement of skeletal muscle, glycogen stored in muscle cells is broken down to supply the cells with energy.
The GYS2 gene provides instructions for making liver glycogen synthase, which is produced solely in liver cells. Glycogen that is stored in the liver can be broken down rapidly when glucose is needed to maintain normal blood sugar levels between meals.
Mutations in the GYS1 or GYS2 gene lead to a lack of functional glycogen synthase, which prevents the production of glycogen from glucose. Mutations that cause glycogen storage disease type 0 result in a complete absence of glycogen in either liver or muscle cells. As a result, these cells do not have glycogen as a source of stored energy to draw upon following physical activity or fasting. This shortage of glycogen leads to the signs and symptoms of glycogen storage disease type 0.
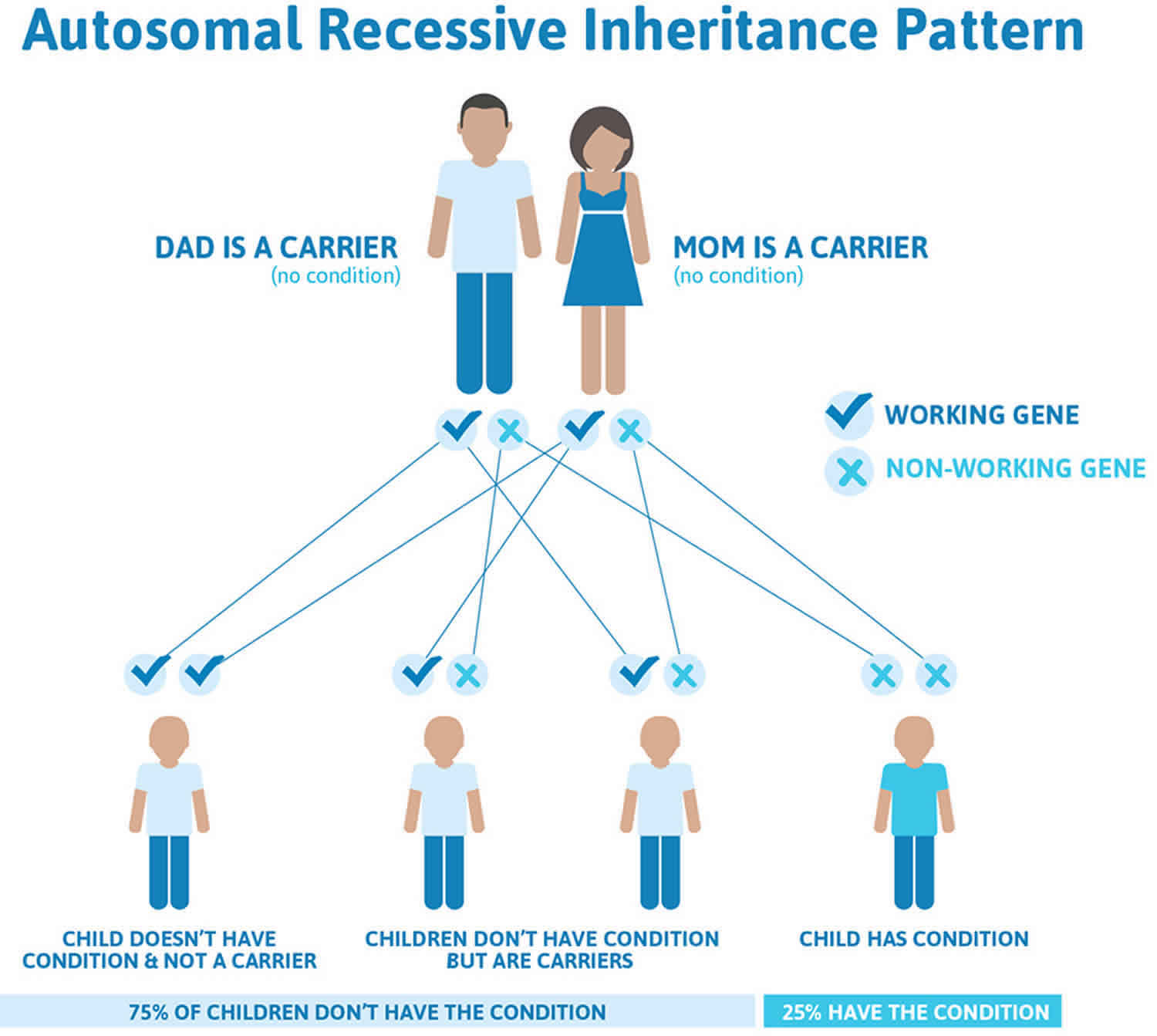
Glycogen storage disease type 0 inheritance pattern
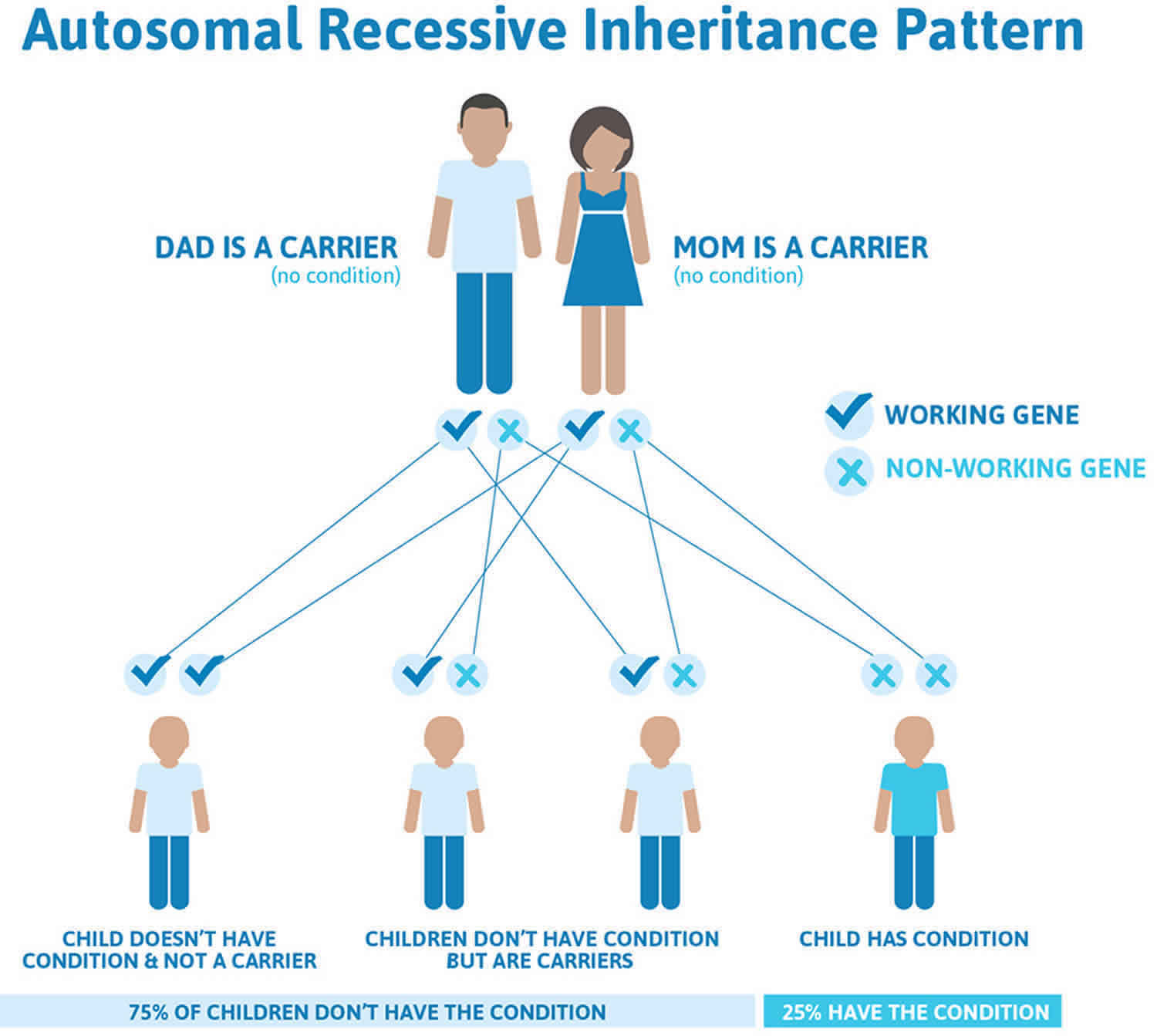
Glycogen storage disease type 0 is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Figure 5. Glycogen storage disease type 0 autosomal recessive pattern
Glycogen storage disease type 0 diagnosis
Children with glycogen storage disease type 0 may become tired more quickly than their peers. There may be muscle cramps from accumulated lactic acid. Children may have a mild growth delay, but in general will develop normally, with normal appearance and liver size.
A history of needing frequent meals or snacks, tiring quickly and hypoglycemia may suggest glycogen storage disease type 0. Detailed blood and urine tests may show patterns that are unique to glycogen storage disease type 0 and a liver biopsy will show very little glycogen. DNA testing is now available, the condition being caused by a change in the glycogen synthase-2 (GYS2) gene.
Glycogen storage disease type 0 treatment
Glycogen storage disease type 0 treatment aims to prevent hypoglycemia by taking snacks every 3-4 hours. Uncooked cornstarch can act as a ‘slow release’ form of glucose and may prevent hypoglycemia overnight. A diet higher than normal in protein may help with the cramping, tiredness and fatigue.
Glycogen storage disease type 1
Glycogen storage disease type 1 also known as von Gierke disease, glucose-6-phosphatase deficiency or GSD1, is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body’s cells. The accumulation of glycogen in certain organs and tissues, especially the liver, kidneys, and small intestines, impairs their ability to function normally.
Signs and symptoms of glycogen storage disease type 1 (von Gierke disease) typically appear around the age of 3 or 4 months, when babies start to sleep through the night and do not eat as frequently as newborns. Affected infants may have low blood sugar (hypoglycemia), which can lead to seizures. They can also have a buildup of lactic acid in the body (lactic acidosis), high blood levels of a waste product called uric acid (hyperuricemia), and excess amounts of fats in the blood (hyperlipidemia). As they get older, children with glycogen storage disease type 1 have thin arms and legs and short stature. An enlarged liver may give the appearance of a protruding abdomen. The kidneys may also be enlarged. Affected individuals may also have diarrhea and deposits of cholesterol in the skin (xanthomas).
People with glycogen storage disease type 1 may experience delayed puberty. Beginning in young to mid-adulthood, affected individuals may have thinning of the bones (osteoporosis), a form of arthritis resulting from uric acid crystals in the joints (gout), kidney disease, and high blood pressure in the blood vessels that supply the lungs (pulmonary hypertension). Females with this condition may also have abnormal development of the ovaries (polycystic ovaries). In affected teens and adults, tumors called adenomas may form in the liver. Adenomas are usually noncancerous (benign), but occasionally these tumors can become cancerous (malignant).
Researchers have described two types of glycogen storage disease type 1, which differ in their signs and symptoms and genetic cause. These types are known as glycogen storage disease type 1a (GSD1a) and glycogen storage disease type 1b (GSD1b). Two other forms of glycogen storage disease type 1 have been described, and they were originally named types 1c and 1d. However, these types are now known to be variations of glycogen storage disease type 1b; for this reason, glycogen storage disease type 1b is sometimes called glycogen storage disease type 1 non-a.
Many people with glycogen storage disease type 1b have a shortage of white blood cells (neutropenia), which can make them prone to recurrent bacterial infections. Neutropenia is usually apparent by age 1. Many affected individuals also have inflammation of the intestinal walls (inflammatory bowel disease). People with glycogen storage disease type 1b may have oral problems including cavities, inflammation of the gums (gingivitis), chronic gum (periodontal) disease, abnormal tooth development, and open sores (ulcers) in the mouth. The neutropenia and oral problems are specific to people with glycogen storage disease type 1b and are typically not seen in people with glycogen storage disease type 1a.
The overall incidence of glycogen storage disease type 1 is 1 in 100,000 individuals 11. Glycogen storage disease type 1a is more common than glycogen storage disease type 1b, accounting for 80 percent of all glycogen storage disease type 1 cases.
The management of glycogen storage disease type 1 is lifelong. Diet is the cornerstone of treatment and many types of foods are restricted, severely limiting dietary options. Glycogen storage disease type 1 children should minimize foods containing sucrose (table sugar,) fructose (sugar found in fruits,) and lactose and galactose (sugars found in milk products) because these sugars end up as glycogen trapped in the liver. They also must be fed every one to four hours in order to maintain blood glucose at an appropriate level. Because this can often be very difficult for the child to tolerate and because missing a meal or a feeding time can have catastrophic effects, most have a gastric or nasogastric tube placed. In infancy, the tube is critical for frequent feeds during the day and for using a continuous feeding pump at night. This alternative route of ingestion will also help during times of normal childhood illnesses when hypoglycemia and acidosis can occur more often. A consequence of having to eat so frequently is that glycogen storage disease type 1 children often have problems ingesting food by mouth. They must undergo intensive therapy to relearn sucking, swallowing and even speech patterns.
Glycogen storage disease type 1a
Glycogen storage disease type 1a (von Gierke disease or glucose‐6‐phosphatase deficiency) is an error in the enzyme called glucose-6-phosphatase that is mainly found in the liver but also in the kidney and small intestine is the cause of glycogen storage disease type 1.
The last step of moving stored glycogen is the change of glucose-6-phosphate into glucose. In glycogen storage disease type 1a this step does not happen and so glucose (sugar) cannot be delivered from the liver into the bloodstream.
Glycogen storage disease type 1b
Glycogen storage disease type 1b (von Gierke disease or glucose‐6‐phosphatase translocase deficiency) is caused by the absence or deficiency of an enzyme called glucose-6‐phosphatase translocase.
For the last step of moving stored glycogen into glucose, glucose-6‐phosphate must be moved to a certain area of the cell, but because the enzyme that is required for the move is missing or not working properly, the glucose-6-phosphatase does not arrive in the right place and the last step does not happen and so glucose (sugar) cannot be delivered from the liver into the bloodstream.
Glycogen storage disease type 1 causes
Mutations in two genes, G6PC and SLC37A4, cause glycogen storage disease type 1. G6PC gene mutations cause glycogen storage disease type 1a, and SLC37A4 gene mutations cause glycogen storage disease type 1b.
The proteins produced from the G6PC and SLC37A4 genes work together to break down a type of sugar molecule called glucose 6-phosphate. The breakdown of this molecule produces the simple sugar glucose, which is the primary energy source for most cells in the body.
Mutations in the G6PC and SLC37A4 genes prevent the effective breakdown of glucose 6-phosphate. Glucose 6-phosphate that is not broken down to glucose is converted to glycogen and fat so it can be stored within cells. Too much glycogen and fat stored within a cell can be toxic. This buildup damages organs and tissues throughout the body, particularly the liver and kidneys, leading to the signs and symptoms of glycogen storage disease type 1.
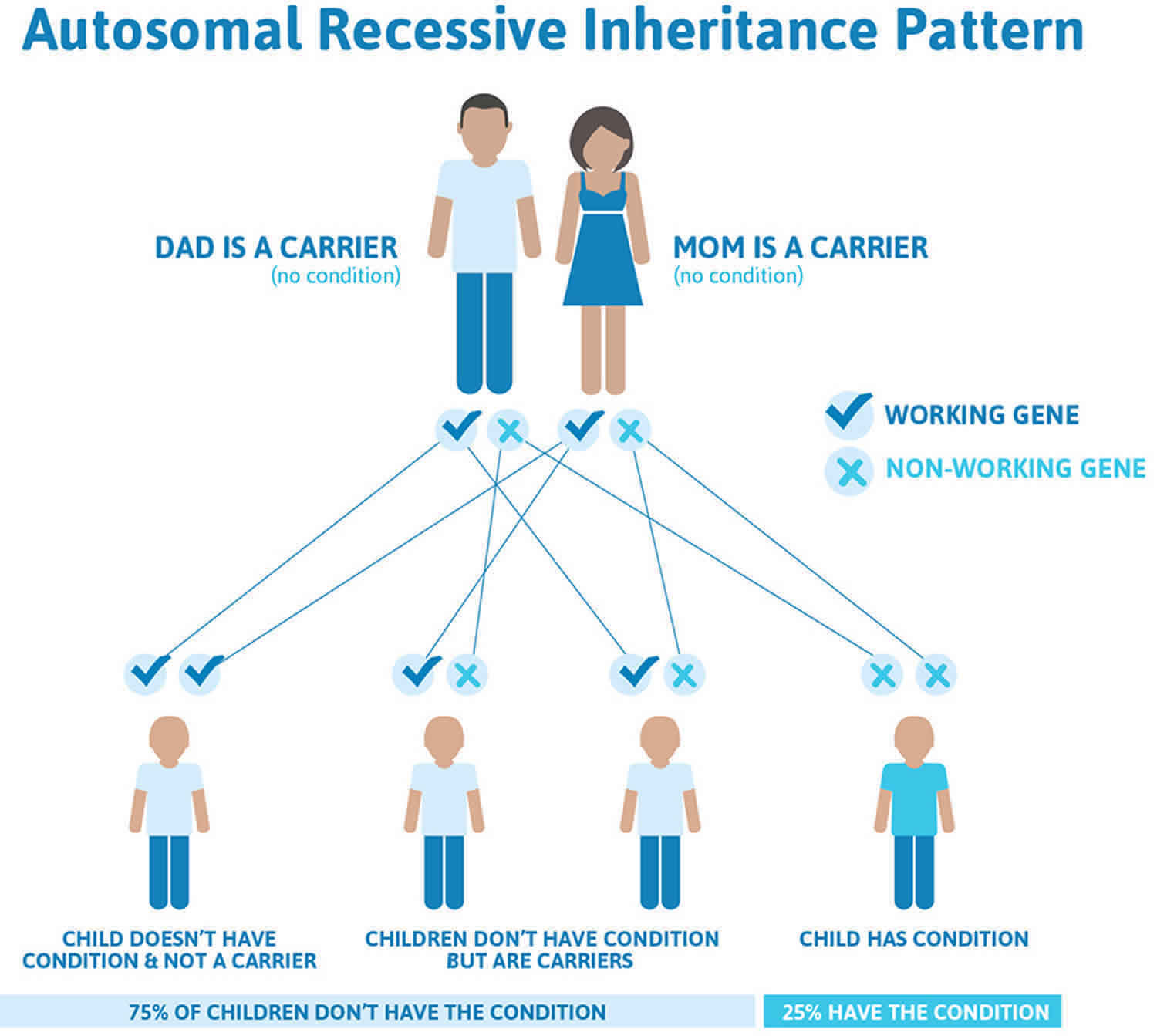
Glycogen storage disease type 1 inheritance pattern
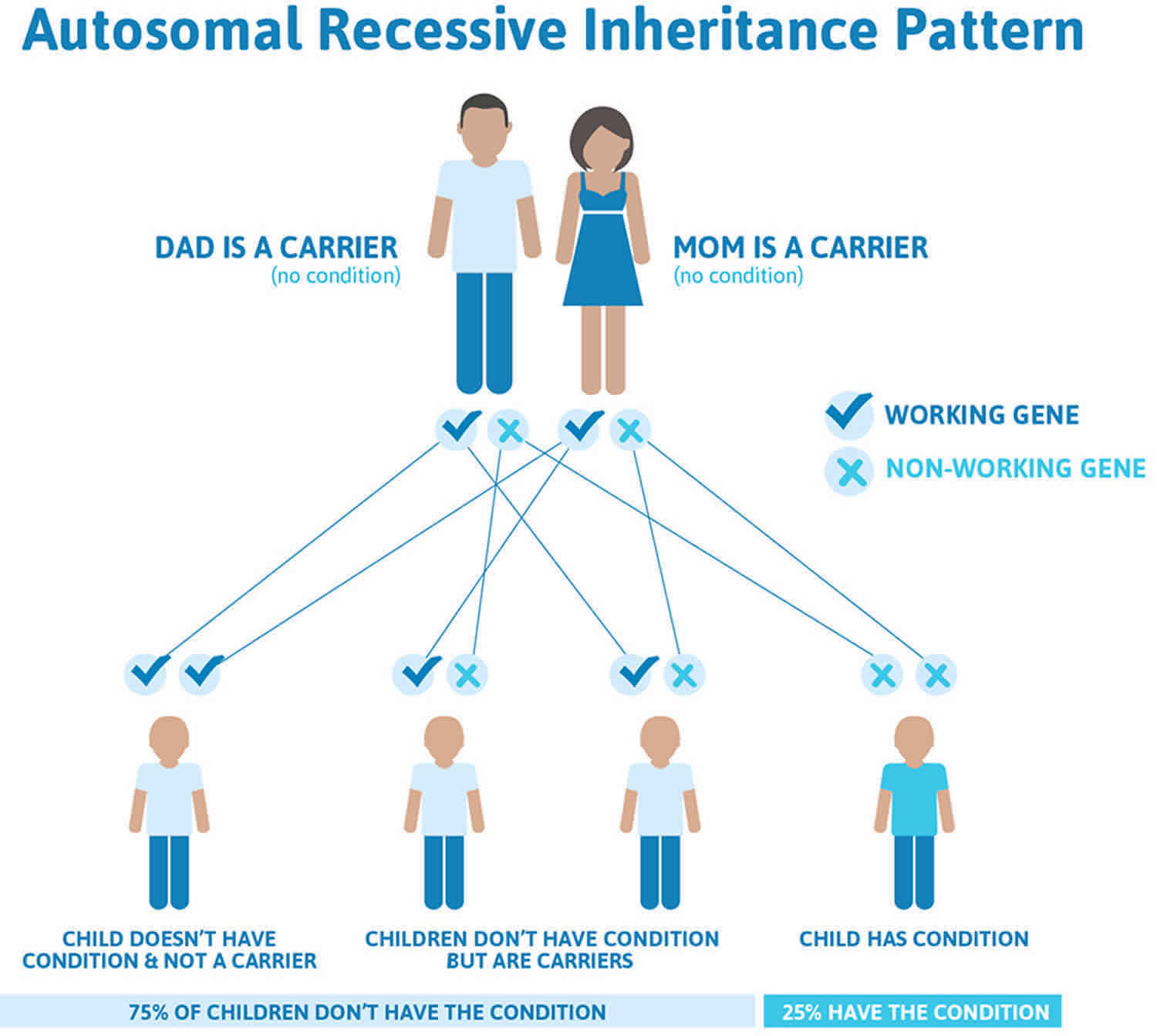
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Figure 6. Glycogen storage disease type 1 autosomal recessive pattern
Glycogen storage disease type 1 symptoms
The primary symptom of glycogen storage disease type 1 in infancy is a low blood sugar level (hypoglycemia). Symptoms of glycogen storage disease type 1 usually begin at three to four months of age and include enlargement of the liver (hepatomegaly), kidney (nephromegaly), elevated levels of lactate, uric acid and lipids (both total lipids and triglycerides), and seizures caused by repeated episodes of hypoglycemia. Continued low blood sugar can lead to delayed growth and development and muscle weakness. Affected children typically have doll-like faces with fat cheeks, relatively thin extremities, short stature, and protuberant abdomen.
High lipid levels can lead to the formation of fatty skin growths called xanthomas. Other conditions that can be associated with untreated glycogen storage disease type 1 include osteoporosis, delayed puberty, gout (arthritis caused by accumulation of uric acid), kidney disease, pulmonary hypertension (high blood pressure in the arteries that supply the lungs), hepatic adenoma (benign liver tumors), polycystic ovaries in females, an inflammation of the pancreas (pancreatitis), diarrhea and changes in brain function.
Impaired platelet function can lead to a bleeding tendency with frequent nose bleeds (epistaxis). In general glycogen storage disease type Ib patients have similar clinical manifestations as type Ia patients, but in addition glycogen storage disease type 1b is associated with impaired neutrophil and monocyte function as well as chronic neutropenia after the first few years of life, all of which result in recurrent bacterial infections and oral and intestinal mucosal ulcers.
Early diagnosis and effective treatment can result in normal growth and puberty and many affected individuals live into adulthood and enjoy normal life activities. Many female patients have had successful pregnancies.
Glycogen storage disease type 1 diagnosis
Glycogen storage disease type 1 is diagnosed by laboratory tests that indicate abnormal levels of glucose, lactate, uric acid, triglycerides and cholesterol. Molecular genetic testing for the G6PC and SLC37A4 genes is available to confirm a diagnosis. Molecular genetic testing can also be used for carrier testing and prenatal diagnosis. Liver biopsy can also be used to prove specific enzyme deficiency for glycogen storage disease type 1a.
Glycogen storage disease type 1 treatment
Glycogen storage disease type 1 is treated with a special diet in order to maintain normal glucose levels, prevent hypoglycemia and maximize growth and development. Frequent small servings of carbohydrates during the day must be maintained throughout life. Calcium, vitamin D and iron supplements may be recommended. Feeding of uncooked cornstarch is used to improve blood levels of glucose. Allopurinol, a drug capable of reducing the level of uric acid in the blood, may be useful to control the symptoms of gout-like arthritis during the adolescent years. Medications may be prescribed to lower lipid levels and prevent and/or treat kidney disease. Human granulocyte colony stimulating factor (GCSF) may be used to treat recurrent infections in glycogen storage disease type Ib patients. Liver tumors (adenomas) can be treated with surgery or a procedure in which current is used to heat and eliminate the tumor (radiofrequency ablation). Kidney and/or liver transplantation are sometimes considered if other therapies are unsuccessful or where liver adenomas keep growing.
Individuals with glycogen storage disease type 1 should be monitored at least annually with kidney and liver ultrasound and routine blood work specifically used for monitoring glycogen storage disease patients.
Genetic counseling is recommended for affected individuals and their families.
Glycogen storage disease type 2
Glycogen storage disease type 2 also called Pompe disease, is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body’s cells. The accumulation of glycogen in certain organs and tissues, especially muscles, impairs their ability to function normally.
Researchers have described three types of Pompe disease, which differ in severity and the age at which they appear. These types are known as classic infantile-onset, non-classic infantile-onset, and late-onset.
The classic form of infantile-onset Pompe disease begins within a few months of birth. Infants with this disorder typically experience muscle weakness (myopathy), poor muscle tone (hypotonia), an enlarged liver (hepatomegaly), and heart defects. Affected infants may also fail to gain weight and grow at the expected rate (failure to thrive) and have breathing problems. If untreated, this form of Pompe disease leads to death from heart failure in the first year of life.
The non-classic form of infantile-onset Pompe disease usually appears by age 1. It is characterized by delayed motor skills (such as rolling over and sitting) and progressive muscle weakness. The heart may be abnormally large (cardiomegaly), but affected individuals usually do not experience heart failure. The muscle weakness in this disorder leads to serious breathing problems, and most children with non-classic infantile-onset Pompe disease live only into early childhood.
The late-onset type of Pompe disease may not become apparent until later in childhood, adolescence, or adulthood. Late-onset Pompe disease is usually milder than the infantile-onset forms of this disorder and is less likely to involve the heart. Most individuals with late-onset Pompe disease experience progressive muscle weakness, especially in the legs and the trunk, including the muscles that control breathing. As the disorder progresses, breathing problems can lead to respiratory failure.
Glycogen storage disease type 2 or Pompe disease affects about 1 in 40,000 people in the United States 12. The incidence of this disorder varies among different ethnic groups.
Glycogen storage disease type 2 causes
Mutations in the GAA gene cause glycogen storage disease type 2 or Pompe disease. The GAA gene provides instructions for producing an enzyme called acid alpha-glucosidase (also known as acid maltase). This enzyme is active in lysosomes, which are structures that serve as recycling centers within cells. The enzyme normally breaks down glycogen into a simpler sugar called glucose, which is the main energy source for most cells.
Mutations in the GAA gene prevent acid alpha-glucosidase from breaking down glycogen effectively, which allows this sugar to build up to toxic levels in lysosomes. This buildup damages organs and tissues throughout the body, particularly the muscles, leading to the progressive signs and symptoms of glycogen storage disease type 2 or Pompe disease.
Glycogen storage disease type 2 inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Figure 7. Glycogen storage disease type 2 autosomal recessive pattern
Glycogen storage disease type 2 signs and symptoms
Patients with the classic infantile form of Pompe disease are the most severely affected. Although hardly any symptoms may be apparent at birth, the disease usually presents within the first three months of life with rapidly progressive muscle weakness (floppy infants), diminished muscle tone (hypotonia), respiratory insufficiency, and a type of heart disease known as hypertrophic cardiomyopathy, a condition characterized by abnormal thickening of the walls of the heart (mainly the left chamber and the wall between the left and right chamber) resulting in diminished cardiac function. These problems together culminate in cardio-respiratory failure within the first 2 years of life.
Many infants have a large, protruding tongue and a moderate enlargement of the liver. The legs often rest in a frog position and feel firm on palpation (pseudo-hypertrophy).
Feeding and swallowing problems as well as respiratory difficulties, which are often combined with respiratory tract infections, are common. Major developmental milestones such as rolling over, sitting up, and standing are delayed or not achieved. Mental development is usually normal. Virtually all infants experience hearing loss. The classic infantile form of Pompe disease is characterized by a total lack of acid alpha-glucosidase (GAA) activity and by a rapid buildup of glycogen in skeletal muscle and heart.
Childhood Pompe disease typically presents during childhood and adult Pompe disease during adulthood. Both these forms of Pompe disease are often grouped together as late-onset Pompe disease (abbreviated as LOPD) despite the fact that the time of presentation can vary from the first year to the eighth decade. Patients who develop symptoms early in life tend to be the more severely affected and to have a faster rate of disease progression than those who develop symptoms later in life. Both children and adults usually have more GAA activity present than those who show symptoms as infants, and the glycogen buildup is not usually as rapid. However, symptoms do progress, can greatly affect the quality of life, and diminish the lifespan of affected individuals.
Childhood and adult Pompe disease are associated with progressive weakness of mainly the proximal muscles (limb girdle, upper arms and upper legs), and varying degrees of respiratory weakness due to dysfunction of the diaphragm and intercostal muscles (muscle between ribs). The lower limbs are more affected than the upper limbs. The extent of muscle involvement is highly variable. The muscles adjacent to the spinal column (para-spinal muscles) and neck are usually also involved. Weakness of the para-spinal muscles around puberty can cause abnormal curvature of the spine (scoliosis). As a result of the combination of these serious symptoms, affected individuals may become wheelchair and/or ventilator dependent.
Other symptoms can include chewing and swallowing difficulties and drooping of the upper eyelids (ptosis). Additionally, blood vessel abnormalities due to smooth muscle weakness and problems of the urinary and digestive systems have been reported.
Glycogen storage disease type 2 diagnosis
A diagnosis of glycogen storage disease type 2 or Pompe disease is based upon a thorough clinical evaluation, a detailed patient and family history, and a variety of tests. Prenatal diagnosis is possible when a pregnancy is believed to be at risk for Pompe disease.
Clinical testing and work up
In individuals suspected of having glycogen storage disease type 2 or Pompe disease, blood can be drawn and the function/activity of the GAA enzyme can be measured in white blood cells (leukocytes), but only if the proper assay conditions are being used and acarbose is added to the reaction mixture to inhibit the activity of glucoamylase. The isolation of lymphocytes to prevent the interference of glucoamylase is not advised, as the successful isolation of lymphocytes is not only time consuming, but also error prone when the blood sample is not sufficiently fresh.
Alternatively, the GAA enzyme activity/functional assay can also be performed on a dried blood spot, but the assay is not any quicker or more sensitive than the leukocyte assay and also requires the use of acarbose to inhibit the glucoamylase activity. The advantage of the bloodspot test is that it allows convenient shipment of samples if a certified diagnostic laboratory test is not locally available. Additionally, dried blood spot testing allows for mass screening. The blood spot test is without any argument the most convenient methodology for the screening of large populations of newborns and, for instance, large numbers of patients with undiagnosed limb-girdle muscular dystrophies and CK-emias.
When a diagnosis of glycogen storage disease type 2 or Pompe disease is based on a leukocyte or blood spot assay, it must be confirmed through molecular genetic testing (DNA analysis) or by another enzyme assay, preferably using cultured skin fibroblasts obtained by a skin biopsy. More invasive muscle biopsies are not needed and not optimal for obtaining material for GAA enzyme activity/function assays. The advantage of DNA analysis over a GAA enzyme activity assay is that the DNA test discriminates unequivocally between carriers and affected children/adults, whereas the enzyme assay does not in all cases.
The taking of a skin biopsy and the growing of a skin fibroblasts culture may not be feasible in every diagnostic setting, but should always be considered as there are important advantages to this procedure. The GAA enzyme activity test on this material is superior over others as it is the most sensitive test and discriminates best between classic-infantile, childhood and adult Pompe disease. Cultured fibroblasts can be stored forever and used as eternal source of GAA enzyme, DNA and mRNA for all kinds of present day and future sophisticated analyses.
A variety of additional tests may be performed to detect or assess symptoms potentially associated with Pompe disease such as sleep studies, breathing tests to measure lung capacity, and electromyography, a test to measure muscle function. Muscle MRI’s are also used to measure the degree of damage that has occurred to the muscles.
Specific tests may also performed to assess the heart including chest x-rays, electrocardiogram, and echocardiogram. Chest x-rays allows physicians to assess the size of the heart, which can be enlarged in some infants with Pompe disease. An electrocardiogram measures the electrical activity of the heart and can detect abnormal heart rhythms. An echocardiogram uses reflected sound waves to create a picture of the heart and can reveal abnormal thickening of the heart muscle tissue.
If the specific GAA gene mutations in both parents are known, prenatal diagnosis is possible through chorionic villi sampling or amniocentesis. Pre-implantation genetic diagnosis (testing an embryo to determine whether it has the same genetic abnormalities as the parents) may also be an option. Pre-implantation genetic diagnosis can be performed on embryos created through in vitro fertilization. Families interested in pre-implantation genetic diagnosis should seek the counsel of a certified genetics professional.
Glycogen storage disease type 2 treatment
The treatment of glycogen storage disease type 2 or Pompe disease is disease-specific, symptomatic, and supportive. Treatment requires the coordinated efforts of a team of specialists with expertise in treating neuromuscular disorders. Pediatricians or internists, neurologists, orthopedists, cardiologists, dieticians, and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment. Genetic counseling is of utmost importance for affected individuals and their families.
Enzyme replacement therapy
Enzyme replacement therapy is an approved treatment for all patients with glycogen storage disease type 2 or Pompe disease. It involves the intravenous administration of recombinant human acid α-glucosidase. This treatment, manufactured by Genzyme, a Sanofi Corporation, is Lumizyme (marketed as Myozyme outside the United States), and was first approved by the U.S. Food and Drug Administration (FDA) in 2006. It has been approved for all patients with glycogen storage disease type 2 or Pompe disease.
Supportive therapies
Additional treatment of glycogen storage disease type 2 or Pompe disease is symptomatic and supportive. Respiratory support may be required, as most patients have some degree of respiratory compromise and/or respiratory failure. Physical therapy may be helpful to strengthen respiratory muscles. Some patients may need respiratory assistance through mechanical ventilation (i.e. bipap or volume ventilators) during the night and/or periods of the day. In addition, it may be necessary for additional support during respiratory tract infections. Mechanical ventilation support can be through noninvasive or invasive techniques. The decision about the duration of respiratory support is best made by the family in careful consultation with the patient’s physicians and other members of the healthcare team based upon the specifics of the patient.
Physiotherapy is recommended to improve strength and physical ability. Occupational therapy, including the use of canes or walkers, may be necessary. Eventually, some individuals may require the use of a wheelchair. Speech therapy can be beneficial to improve articulation and speech for some patients.
Orthopedic devices including braces may be recommended for some patients. Surgery may be required for certain orthopedic symptoms such as contractures or spinal deformity.
Since glycogen storage disease type 2 or Pompe disease can weaken muscles used for chewing and swallowing, adequate measures may be required to ensure proper nutrition and weight gain. Some patients may need specialized, high-calorie diets and may need to learn techniques to change the size and texture of food to lower the risk of aspiration. Some infants may require the insertion of a feeding tube that is run through the nose, down the esophagus and into the stomach (nasogastric tube). In some children, a feeding tube may need to be inserted directly into the stomach through a small surgical opening in the abdominal wall. Some individuals with late onset glycogen storage disease type 2 or Pompe disease may require a soft diet, but few require feeding tubes.
Investigational therapies
Gene therapy remains an exciting option and progress continues. Gene therapy in glycogen storage disease type 2 or Pompe disease is directed toward restoring the acid a-glucosidase production and activity in crucial tissues like the diaphragm in order to improve respiratory capacity. Other gene therapy efforts seek to restore the body’s ability to produce acid a-glucosidase by transducing the functional GAA gene in liver cells in vivo, or in bone marrow stem cells ex vivo followed by stem cell transplantation. At this time multiple groups are working to advance this therapy to the clinic.
Several modifications to the recombinant human acid a-glucosidase that is presently used for enzyme replacement therapy are currently being explored. For example, carbohydrate side chains are being modified to improve uptake by muscle cells. Small molecule therapies to enhance the function of patient’s residual, endogenous acid a-glucosidase, as well as to stabilize the intravenously administered form of rhGAA are also currently in development.
Glycogen storage disease type 3
Glycogen storage disease type 3 also known as Cori disease or Forbes disease, is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body’s cells. The accumulated glycogen is structurally abnormal and impairs the function of certain organs and tissues, especially the liver and muscles.
Glycogen storage disease type 3 is divided into types 3a, 3b, 3c, and 3d, which are distinguished by their pattern of signs and symptoms. glycogen storage disease types 3a and 3c mainly affect the liver and muscles, and glycogen storage disease types 3b and 3d typically affect only the liver. It is very difficult to distinguish between the types of glycogen storage disease type 3 that affect the same tissues. Glycogen storage disease types 3a and 3b are the most common forms of this condition.
Beginning in infancy, individuals with any type of glycogen storage disease type 3 may have low blood sugar (hypoglycemia), excess amounts of fats in the blood (hyperlipidemia), and elevated blood levels of liver enzymes. As they get older, children with this condition typically develop an enlarged liver (hepatomegaly). Liver size usually returns to normal during adolescence, but some affected individuals develop chronic liver disease (cirrhosis) and liver failure later in life. People with glycogen storage disease3 often have slow growth because of their liver problems, which can lead to short stature. In a small percentage of people with glycogen storage disease type 3, noncancerous (benign) tumors called adenomas may form in the liver.
Individuals with glycogen storage disease type 3a may develop muscle weakness (myopathy) later in life. These muscle problems can affect both heart (cardiac) muscle and the muscles that are used for movement (skeletal muscles). Muscle involvement varies greatly among affected individuals. The first signs and symptoms are typically poor muscle tone (hypotonia) and mild myopathy in early childhood. The myopathy may become severe by early to mid-adulthood. Some people with glycogen storage disease type 3a have a weakened heart muscle (cardiomyopathy), but affected individuals usually do not experience heart failure. Other people affected with glycogen storage disease3a have no cardiac muscle problems.
The incidence of glycogen storage disease type 3 in the United States is 1 in 100,000 individuals 13. Glycogen storage disease type 3 is seen more frequently in people of North African Jewish ancestry; in this population, 1 in 5,400 individuals are estimated to be affected.
Glycogen storage disease type 3a is the most common form of glycogen storage disease type 3, accounting for about 85 percent of all cases. glycogen storage disease type 3b accounts for about 15 percent of cases. Glycogen storage disease types 3c and 3d are very rare, and their signs and symptoms are poorly defined. Only a small number of affected individuals have been suspected to have glycogen storage disease types 3c and 3d.
Glycogen storage disease type 3 causes
Mutations in the AGL gene cause glycogen storage disease type 3. The AGL gene provides instructions for making the glycogen debranching enzyme. This enzyme is involved in the breakdown of glycogen, which is a major source of stored energy in the body. Between meals the body breaks down stores of energy, such as glycogen, to use for fuel.
Most AGL gene mutations lead to the production of a nonfunctional glycogen debranching enzyme. These mutations typically cause glycogen storage disease types 3a and 3b. The mutations that cause glycogen storage disease types 3c and 3d are thought to lead to the production of an enzyme with reduced function. All AGL gene mutations lead to storage of abnormal, partially broken down glycogen molecules within cells. A buildup of abnormal glycogen damages organs and tissues throughout the body, particularly the liver and muscles, leading to the signs and symptoms of glycogen storage disease type 3.
Glycogen storage disease type 3 inheritance pattern
Glycogen storage disease type 3 is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Figure 8. Glycogen storage disease type 3 autosomal recessive inheritance pattern
Glycogen storage disease type 3 symptoms
The median age at the first clinical presentations is in the first year of life. Most common presenting symptoms are enlarged liver (hepatomegaly) (98%), low blood sugar (hypoglycemia) (53%), failure to thrive (49%) and recurrent illness and/or infections (17%). Symptoms and signs of glycogen storage disease type 3, at least during the first 4 to 6 years of life, may be indistinguishable from glycogen storage disease type 1. The amount of glycogen in the liver and muscles is abnormally high, the liver is enlarged, and the abdomen protrudes. The muscles tend to be flaccid or weak.
A typical child with glycogen storage disease type 3 has short stature, low blood sugar after fasting that does not respond to the hormone glucagon, and an elevated level of fatty substances in the blood, known as hyperlipidemia. Hypoglycemia is usually associated with increased ketone bodies, and ketonemia can precede hypoglycemia, reflecting activation of burning fat stores. Patients with glycogen storage disease type 3 may also have difficulty fighting infections, and may experience unusually frequent nosebleeds. Enlarged heart muscle (cardiac hypertrophy) is common in individuals with glycogen storage disease type 3a and can already appear in early childhood. However, in most children, heart function remains within normal limits. Children with glycogen storage disease type 3 often grow slowly during childhood and puberty may be delayed, but their adult height is usually normal. Most signs and symptoms improve significantly with adequate dietary management.
In adulthood, the liver manifestations of the disease usually subside, but progression to liver scarring (cirrhosis) and malignancy (carcinoma) may occur. Despite dietary management, muscle disease can get worse. As the cohort of adult glycogen storage disease type 3 patients is still relatively young and small, the course of the disease over time is incompletely described.
Some affected individuals may have virtually no symptoms (asymptomatic) other than a protruding abdomen and an enlarged liver in childhood. These patients tend to lose these few symptoms during adolescence when their liver decreases progressively in size.
Glycogen storage disease type 3 diagnosis
An enlarged liver and low blood sugar with high levels of ketones, transaminases, lipids and creatine kinase is indicative of glycogen storage disease type 3. Uric acid and fasting lactic acid levels are usually normal. In glycogen storage disease type 3b creatine kinase can be normal. Molecular genetic testing for mutations in the AGL gene can be used to confirm the diagnosis. Nowadays, liver and muscle biopsies are uncommon. In many countries besides the United States, studies in blood cells and skin fibroblasts are clinically available to confirm glycogen debranching enzyme deficiency.
Glycogen storage disease type 3 treatment
Dietary management is the cornerstone treatment of glycogen storage disease type 3 14.
- Infants and children with glycogen storage disease type 3 are treated with a high-protein diet every 3-4 hours. The recommended daily amount of protein is ± 3-4 grams per kg bodyweight per day and should be well divided during the day. Cornstarch may already be introduced in the first year of life. This is a dietary complex starch like glycogen and the dose/frequency of supplementation is titrated to maintain normoglycemia. Although fructose and galactose can be metabolized, the (extend of) restrictions of so-called simple/fast carbohydrates is a matter of debate. These simple sugars include glucose, galactose (dairy sugar), lactose (galactose + glucose), fructose (fruit sugar), sucrose (fructose + glucose) and maltodextrin. The latter is frequently used as a food additive and typically a mixture of 3-17 glucose units. Special formulas are not required. Fasting should be avoided and for the overnight fast, (a combination of) a bedtime snack, frequent feeds, cornstarch, and/or continuous nocturnal gastric drip feeding may be needed.
- Adolescents and adults have lower basic carbohydrate requirements. The recommended daily amount of protein is ± 25 % of the total caloric intake. A bedtime snack or an overnight high protein formula may be prescribed for patients with myopathy.
- Good dietary control includes at home monitoring of blood glucose and ketones. Based on clinical observations, it is believed that the diet can prevent or resolve heart and/or muscle disease.
- The role of and indications for ketogenic diets (and variations, including Atkins diet) and medium chain triglycerides (MCT) oil are debatable and deserve further, systematic research.
Liver transplantation is indicated only for patients with severe hepatic cirrhosis, liver dysfunction and /or liver cancer (hepatocellular carcinoma).
Glycogen storage disease type 4
Glycogen storage disease type 4 also called Andersen disease, is an inherited disorder caused by the buildup of a complex sugar called glycogen in the body’s cells. The accumulated glycogen is structurally abnormal and impairs the function of certain organs and tissues, especially the liver and muscles. There are five types of glycogen storage disease type 4, which are distinguished by their severity, signs, and symptoms.
The fatal perinatal neuromuscular type is the most severe form of glycogen storage disease type 4, with signs developing before birth. Excess fluid may build up around the fetus (polyhydramnios) and in the fetus’ body. Affected fetuses have a condition called fetal akinesia deformation sequence, which causes a decrease in fetal movement and can lead to joint stiffness (arthrogryposis) after birth. Infants with the fatal perinatal neuromuscular type of glycogen storage disease type 4 have very low muscle tone (severe hypotonia) and muscle wasting (atrophy). These infants usually do not survive past the newborn period due to weakened heart and breathing muscles.
The congenital muscular type of glycogen storage disease type 4 is usually not evident before birth but develops in early infancy. Affected infants have severe hypotonia, which affects the muscles needed for breathing. These babies often have dilated cardiomyopathy, which enlarges and weakens the heart (cardiac) muscle, preventing the heart from pumping blood efficiently. Infants with the congenital muscular type of glycogen storage disease type 4 typically survive only a few months.
The progressive hepatic type is the most common form of glycogen storage disease type 4. Within the first months of life, affected infants have difficulty gaining weight and growing at the expected rate (failure to thrive) and develop an enlarged liver (hepatomegaly). Children with this type develop a form of liver disease called cirrhosis that often is irreversible. High blood pressure in the vein that supplies blood to the liver (portal hypertension) and an abnormal buildup of fluid in the abdominal cavity (ascites) can also occur. By age 1 or 2, affected children develop hypotonia. Children with the progressive hepatic type of glycogen storage disease type 4 often die of liver failure in early childhood.
The non-progressive hepatic type of glycogen storage disease type 4 has many of the same features as the progressive hepatic type, but the liver disease is not as severe. In the non-progressive hepatic type, hepatomegaly and liver disease are usually evident in early childhood, but affected individuals typically do not develop cirrhosis. People with this type of the disorder can also have hypotonia and muscle weakness (myopathy). Most individuals with this type survive into adulthood, although life expectancy varies depending on the severity of the signs and symptoms.
The childhood neuromuscular type of glycogen storage disease type 4 develops in late childhood and is characterized by myopathy and dilated cardiomyopathy. The severity of this type of glycogen storage disease type 4 varies greatly; some people have only mild muscle weakness while others have severe cardiomyopathy and die in early adulthood.
Glycogen storage disease type 4 is estimated to occur in 1 in 600,000 to 800,000 individuals worldwide 15. Glycogen storage disease type 4 accounts for roughly 3 percent of all cases of glycogen storage disease.
Glycogen storage disease type 4 causes
Mutations in the GBE1 gene cause glycogen storage disease type 4. The GBE1 gene provides instructions for making the glycogen branching enzyme. This enzyme is involved in the production of glycogen, which is a major source of stored energy in the body. GBE1 gene mutations that cause glycogen storage disease type 4 lead to a shortage (deficiency) of the glycogen branching enzyme. As a result, glycogen is not formed properly. Abnormal glycogen molecules called polyglucosan bodies accumulate in cells, leading to damage and cell death. Polyglucosan bodies accumulate in cells throughout the body, but liver cells and muscle cells are most severely affected in glycogen storage disease type 4. Glycogen accumulation in the liver leads to hepatomegaly and interferes with liver functioning. The inability of muscle cells to break down glycogen for energy leads to muscle weakness and wasting.
Generally, the severity of the disorder is linked to the amount of functional glycogen branching enzyme that is produced. Individuals with the fatal perinatal neuromuscular type tend to produce less than 5 percent of usable enzyme, while those with the childhood neuromuscular type may have around 20 percent of enzyme function. The other types of glycogen storage disease type 4 are usually associated with between 5 and 20 percent of working enzyme. These estimates, however, vary among the different types.
Glycogen storage disease type 4 inheritance pattern
Glycogen storage disease type 4 is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Glycogen storage disease type 4 symptoms
Glycogen storage disease type 4 (Andersen disease) is a multisystem disorder that may affect the liver, voluntary (skeletal) muscles, the heart, the nervous system, and other bodily tissues. Disease nature and course may vary in several aspects, including age at onset, associated symptoms and signs, degree of abnormal glycogen accumulation in various tissues, and specific organs affected.
However, the most common, classic form of the disease is typically characterized by progressive internal scarring (fibrosis) and destruction of liver tissue (cirrhosis), leaving areas of nonfunctioning scar tissue and gradually impaired liver function. In such cases, the disease typically becomes evident during infancy or up to about 18 months of age. Initial symptoms and signs commonly include failure to grow and gain weight at the expected rate (failure to thrive) and abnormal enlargement of the liver and spleen (hepatosplenomegaly). The cirrhosis typically progresses to cause high blood pressure in veins from the spleen and intestines to the liver (portal hypertension); abnormal fluid accumulation in the abdomen (ascites); enlargement of veins in the wall of the esophagus (esophageal varices), which may rupture, resulting in coughing up or vomiting of blood; and liver failure. In some cases, initial symptoms and findings associated with cirrhosis may include yellowish discoloration of the skin, mucous membranes, and whites of the eyes (jaundice); mental confusion; and/or other abnormalities. Rarely, liver cirrhosis associated with glycogen storage disease type 4 (Andersen disease) may also lead to abnormally reduced blood glucose levels (hypoglycemia). In most individuals with classic glycogen storage disease type 4 (Andersen disease), progressive liver disease may lead to liver transplantation or potentially life-threatening complications by approximately age five years. However, some rare cases have also been reported in which affected individuals have nonprogressive liver disease. In some of these cases, mildly affected individuals may not have apparent symptoms (asymptomatic).
Several neuromuscular variants of glycogen storage disease type 4 (Andersen disease) have also been described in the medical literature. Most commonly, there may be primary or isolated muscle involvement beginning in late childhood, with disease of skeletal and/or heart muscle (myopathy and/or cardiomyopathy). Accumulation of abnormal glycogen in skeletal muscle may lead to muscle weakness and fatigue, exercise intolerance, muscle wasting (atrophy), and/or other symptoms and findings. In those with cardiomyopathy, weakening of heart muscle may lead to stretching and enlargement (dilation) of the heart’s lower chambers (ventricles). Dilated cardiomyopathy may gradually lead to weakening of the heart’s pumping action, causing an impaired ability to circulate enough blood to meet the body’s requirements for oxygen (heart failure). Associated symptoms and findings may include fatigue; irritability; feeding difficulties; lack of appetite; failure to thrive; shortness of breath with exertion and eventually at rest; an abnormal accumulation of fluid in body tissues (edema); abnormalities of heart rhythm (arrhythmias); and potentially life-threatening complications in some cases.
A neuromuscular variant has also been reported that is evident at birth. This form may be characterized by generalized edema (hydrops), severely diminished skeletal muscle tone (hypotonia), muscle weakness and atrophy, bending or extension of multiple joints in various fixed postures (contractures), and neurologic involvement, leading to potentially life-threatening complications early in life.
In addition, a rare neuromuscular variant has also been described in adults. This form of the disease, so-called adult polyglucosan body disease, may be characterized by dysfunction of the central and peripheral nervous systems. The central nervous system (CNS) refers to the brain and spinal cord. The peripheral nerves extend from the CNS to muscles, glands, skin, sensory organs, and internal organs. Peripheral nerves include motor nerves; sensory nerves; and nerves of the autonomic nervous system, which are involved in involuntary functions, including regulating blood pressure, temperature, and heart rate. In individuals with adult polyglucosan body disease, associated symptoms and findings may include sensory loss in the legs; progressive muscle weakness of the arms and legs; walking (gait) disturbances; urination difficulties; mild cognitive impairment or dementia; and/or other abnormalities.
Glycogen storage disease type 4 diagnosis
Glycogen storage disease type 4 (Andersen disease) is usually diagnosed or confirmed after birth (postnatally) during infancy or childhood (or, in some cases, adulthood), based upon a thorough clinical evaluation; identification of characteristic physical findings; a complete patient and family history; and the results of various specialized tests. Removal (biopsy) and microscopic examination of small samples of certain tissues (e.g., liver, skeletal muscle, heart, skin, peripheral nerve) may demonstrate abnormal deposition of amylopectin-like materials. However, testing to confirm a diagnosis of glycogen storage disease type 4 (Andersen disease) requires detection of deficient glycogen-branching enzyme activity (indirect enzyme assay), such as in liver tissue, muscle, certain skin cells (cultured fibroblasts), white blood cells (leukocytes), red blood cells (erythrocytes), nerve cells, or other tissues. Reports indicate that, for individuals with adult polyglucosan body disease, peripheral nerve biopsy or evaluation of leukocytes is required for diagnosis, since deficient glycogen-branching enzyme activity is limited to such tissues. In addition, partial glycogen-branching enzyme deficiency may be detected (e.g., in erythrocytes, leukocytes, fibroblasts) in individuals who carry one copy of a mutated gene for glycogen storage disease type 4 (Andersen disease) (heterozygous carriers).
Diagnostic evaluation typically includes various studies to help detect and characterize certain abnormalities that may be associated with the disorder. Such testing may include various laboratory studies (e.g., complete blood count; liver function tests; blood glucose studies; etc.); specialized imaging techniques (e.g., abdominal ultrasound, CT scanning, and/or MRI); testing that records electrical activity in skeletal muscle at rest and during muscle contraction (electromyography [EMG]); studies to help assess cardiac structure and function, such as ultrasound studies of the heart (echocardiography); and/or other tests.
In some cases, a diagnosis of glycogen storage disease type 4 (Andersen disease) may be suggested before birth (prenatally) by specialized tests. These include studies that may detect decreased glycogen-branching enzyme activity in certain fetal cells obtained via amniocentesis or chorionic villus sampling. During amniocentesis, a sample of fluid that surrounds the developing fetus is removed and analyzed, while chorionic villus sampling involves the removal of tissue samples from a portion of the placenta. In addition, if available, DNA mutation analysis may be used in selected cases.
Glycogen storage disease type 4 treatment
The treatment of glycogen storage disease type 4 (Andersen disease) is directed toward the specific symptoms that are apparent in each individual. Such treatment may require the coordinated efforts of a team of medical professionals, such as pediatricians or internists; physicians who diagnose and treat disorders of the digestive tract; neurologists; cardiologists; dietitians; and/or other health care professionals.
Specific therapies are symptomatic and supportive and may include long-term management of cirrhosis and impaired liver function; neuromuscular disease; and/or heart dysfunction. Treatment may commonly require dietary measures to maintain normal levels of glucose in the blood (normoglycemia) and provide sufficient nutritional intake in order to improve liver function and muscular strength. For cases in which there is cardiomyopathy, recommended disease management may include the use of certain medications, such as to treat heart failure and improve cardiac output; surgical intervention; and/or other measures.
In individuals with progressive liver failure, liver transplantation has been conducted and may be effective in some cases. According to reports in the medical literature, following transplantation, some patients may develop progressive accumulation of abnormal glycogen in other organs, such as the heart, leading to potentially life-threatening complications. However, reports indicate that most patients have not had neuromuscular or heart complications (i.e., during follow-up periods of up to 13 years); in addition, in some of these patients, accumulations of glycogen in the heart and skeletal muscle have appeared to diminish following transplantation. However, experts advise that the long-term effectiveness (efficacy) of liver transplantation and its effect on other organ systems remains uncertain in those with glycogen storage disease type 4 (Andersen disease). Thus, further investigation is needed to determine the long-term safety and efficacy of liver transplantation and its effect on disease progression in classic glycogen storage disease type 4 (Andersen disease).
Genetic counseling will be of benefit for affected individuals and family members. Other treatment for this disorder is symptomatic and supportive.
Glycogen storage disease type 5
Glycogen storage disease type 5 also known as McArdle disease or GSD5, is an inherited disorder caused by an inability to break down a complex sugar called glycogen in muscle cells. A lack of glycogen breakdown interferes with the function of muscle cells.
People with glycogen storage disease type 5 typically experience fatigue, muscle pain, and cramps during the first few minutes of exercise (exercise intolerance). Exercise such as weight lifting or jogging usually triggers these symptoms in affected individuals. The discomfort is generally alleviated with rest. If individuals rest after brief exercise and wait for their pain to go away, they can usually resume exercising with little or no discomfort (a characteristic phenomenon known as “second wind”).
Prolonged or intense exercise can cause muscle damage in people with glycogen storage disease type 5. About half of people with glycogen storage disease type 5 experience breakdown of muscle tissue (rhabdomyolysis). In severe episodes, the destruction of muscle tissue releases a protein called myoglobin, which is filtered through the kidneys and released in the urine (myoglobinuria). Myoglobin causes the urine to be red or brown. This protein can also damage the kidneys, and it is estimated that half of those individuals with glycogen storage disease type 5 who have myoglobinuria will develop life-threatening kidney failure.
The signs and symptoms of glycogen storage disease type 5 can vary significantly in affected individuals. The features of this condition typically begin in a person’s teens or twenties, but they can appear anytime from infancy to adulthood. In most people with glycogen storage disease type 5, the muscle weakness worsens over time; however, in about one-third of affected individuals, the muscle weakness is stable. Some people with glycogen storage disease type 5 experience mild symptoms such as poor stamina; others do not experience any symptoms.
Glycogen storage disease type 5 is a rare disorder; however, its prevalence is unknown 16. In the Dallas-Fort Worth area of Texas, where the prevalence of glycogen storage disease type 5 has been studied, the condition is estimated to affect 1 in 100,000 individuals.
Glycogen storage disease type 5 causes
Mutations in the PYGM gene cause glycogen storage disease type 5. The PYGM gene provides instructions for making an enzyme called myophosphorylase. This enzyme is found only in muscle cells, where it breaks down glycogen into a simpler sugar called glucose-1-phosphate. Additional steps convert glucose-1-phosphate into glucose, a simple sugar that is the main energy source for most cells.
PYGM gene mutations prevent myophosphorylase from breaking down glycogen effectively. As a result, muscle cells cannot produce enough energy, so muscles become easily fatigued. Reduced energy production in muscle cells leads to the major features of glycogen storage disease type 5.
Glycogen storage disease type 5 inheritance pattern
Glycogen storage disease type 5 is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Glycogen storage disease type 5 symptoms
Glycogen storage disease type 5 (McArdle disease) is characterized by exercise intolerance. This typically consists in acute crises of early fatigue and muscle stiffness and contractures, especially at the start of the exercise, that usually disappear if exercise is stopped or the intensity is reduced. Symptoms usually present within the first ten years of life, but there is a wide range of clinical onset and severity. Some glycogen storage disease type 5 (McArdle disease) patients have mild symptoms while another form progresses quickly and is apparent shortly after the person is born. Progressively weak muscles, in some individuals, do not manifest until the age of sixty to seventy years old.
Muscles of affected patients usually function normally while at rest or during moderate exercise. Only during strenuous exercise do severe muscle cramps occur. Exercising in the presence of severe pain results in muscle damage (rhabdomyolysis) and myoglobinuria in about 50% of those affected. The myoglobin protein can also damage the kidneys and lead to develop life-threatening kidney failure if not treated promptly.
A unique feature of the disease is the so-called “second wind” phenomenon, which most patients refers to as the ability to resume dynamic, large mass exercise, if they take a brief rest upon the appearance of premature fatigue early in exercise. This “second wind” phenomenon is present in approximately ~90% of people with glycogen storage disease type 5 (McArdle disease).
A severity scale has been developed to describe the variation in clinical features:
- Type 0= asymptomatic or virtually asymptomatic (mild exercise intolerance but not functional limitation in any daily life activity).
- Type 1= exercise intolerance, contractures, myalgia, and limitation of acute strenuous exercise and occasionally in daily life activities; no record of myoglobinuria, no muscle wasting or weakness.
- Type 2= same as 1 and also recurrent exertional myoglobinuria, moderate restriction in exercise and limitation in daily life activities.
- Type 3= same as 2 and also fixed muscle weakness, with or without wasting, and severe limitations on exercise and most daily life activities.
Glycogen storage disease type 5 diagnosis
Traditionally, diagnosis has been based on the inability of the patient to produce lactate during a forearm exercise test, lack of muscle glycogen phosphorylase on muscle biopsy (generally from vastus lateralis or biceps brachialis muscles), and more recently DNA studies to look for mutations in the PYGM gene. Additionally, the measure of plasma creatine kinase (CK) levels as well as the determination of the “second wind” phenomenon help to precisely provide a correct diagnosis. Currently, the diagnosis of glycogen storage disease type 5 (McArdle disease) is mainly based on the molecular analysis of DNA obtained from blood samples. This is a minimally invasive method, and given the accumulated knowledge on the genetics of this disease in different populations, it can be highly targeted. Gene sequencing after PCR amplification is the most frequently utilized technique for screening the different PYGM mutations.
Glycogen storage disease type 5 treatment
At present there is no curative therapy for glycogen storage disease type 5 (McArdle disease), but several different therapeutic approaches have been utilized.
No significant beneficial effects have been reported in glycogen storage disease type 5 (McArdle disease) patients receiving branched chain aminoacids, depot glucagon, dantrolene sodium, verapamil, vitamin B6 or high-dose oral ribose. More controversial results have been obtained for creatine supplementation; low dose supplementation (60 mg/kg/day for 4 weeks) reduced muscle complaints in five of nine patients tested, but higher doses (150 mg/kg/day) actually increased exercise induced myalgia.
However, a beneficial intervention for alleviating exercise intolerance symptoms and protecting the muscle from rhabdomyolysis consists of ensuring that sufficient blood glucose is constantly made available to patients during daytime. This can be achieved by adopting a diet with high proportion (65%) of complex carbohydrates (as those found in vegetables, fruit, cereals, pasta and rice) and low fat (20%). A different strategy could be the ingestion of simple carbohydrates before engaging in a strenuous exercise (75 g of sucrose 30-40 min pre-exercise).
Exercise interventions
glycogen storage disease type 5 (McArdle disease) patients adapt favorably to regular exercise, with a significant increase in VO2 peak after supervised aerobic exercise. In fact, it has been shown that physically active patients are much more likely to improve their clinical course over a four year period compared with their inactive peers.
Investigational therapies
Read-through compounds
The substitution of guanine for thymine at nucleotide 148 of the exon 1 of the PYGM gene is the most common mutation among Caucasians. This variation shows an allele frequency above 50% in the studied cohorts of Caucasian glycogen storage disease type 5 (McArdle disease) patients. This mutation (p.R50X) generates a premature termination codon (PTC), which leads to a self-protective process known as non-sense mediated decay (NMD), which eliminates the majority of messenger RNA transcripts containing this type of mutation. The fact that several compounds might restore protein translation by inducing the ribosome to bypass a PTC (a process known as read-through), provides a promising perspective. However, a preliminary trial with short-term (10 days) gentamycin treatment in glycogen storage disease type 5 (McArdle disease) patients with PTC failed to normalize P31-MRS indicators of myophosphorylase deficiency in the muscle. Further studies need to be performed to further ascertain the potential beneficial effects of read-through compounds treatment in glycogen storage disease type 5 (McArdle disease) patients.
Induced expression of the brain and liver glycogen phosphorylase isoforms in the muscle
The brain and liver isoforms of glycogen phosphorylase are only expressed in muscle tissue in the uterus, in neonates, and in regenerating mature fibers, but not in adult non-regenerating mature fibers, where only the muscle isoform is expressed. Thus, any pharmacologic treatment able to upregulate the expression of the liver and brain isoforms of glycogen phosphorylase in mature muscles might be able to reduce the symptoms of the disease. In fact, treatment of the primary skeletal muscle cultures derived from the McArdle mouse model with sodium valproate induces expression of Pygb mRNA and protein, and consequently reduces the glycogen deposits observed in these cells. Additionally, myophosphorylase positive fibers were identified in five of seven sheep treated with this drug. Currently, a clinical trial sponsored by the University College of London and the University of Copenhagen is being developed:
http://euromacregistry.eu/portal1/content.asp?contentid=1207
Gene therapy
Gene therapy using an adenovirus 5 vector and an adeno-associated virus serotype 2 containing myophosphorylase expression cassettes has been evaluated in the ovine model of glycogen storage disease type 5 (McArdle disease). Intramuscular application of both vectors produced local expression of functional myophosphorylase, limited to the surroundings of the injected site. Additionally, the number of fibers expressing myophosphorylase diminished with time, probably due to an immune response. Further gene-therapy studies in the McArdle animal models need to be performed to determine the potential benefits of this approach in glycogen storage disease type 5 (McArdle disease) patients.
Glycogen storage disease type 6
Glycogen storage disease type 6 also known as Hers disease or GSD6, is an inherited disorder caused by an inability to break down a complex sugar called glycogen in liver cells. A lack of glycogen breakdown interferes with the normal function of the liver.
The signs and symptoms of glycogen storage disease type 6 (Hers disease) typically begin in infancy to early childhood. The first sign is usually an enlarged liver (hepatomegaly). During prolonged periods without food (fasting), affected individuals may have low blood sugar (hypoglycemia) or elevated levels of ketones in the blood (ketosis). Ketones are molecules produced during the breakdown of fats, which occurs when stored sugars are unavailable. Children with glycogen storage disease type 6 (Hers disease) tend to grow slower than their peers, but they often achieve normal height as adults. Some affected children also have mild delays in the development of motor skills, such as sitting, standing, or walking.
The signs and symptoms of glycogen storage disease type 6 (Hers disease) tend to improve with age; most adults with this condition do not have any related health problems.
The exact prevalence of glycogen storage disease type 6 (Hers disease) is unknown 17. At least 11 cases have been reported in the medical literature, although this condition is likely to be underdiagnosed because it can be difficult to detect in children with mild symptoms or adults with no symptoms. glycogen storage disease type 6 (Hers disease) is more common in the Old Older Mennonite population, with an estimated incidence of 1 in 1,000 individuals.
Glycogen storage disease type 6 causes
Mutations in the PYGL gene cause glycogen storage disease type 6 (Hers disease). The PYGL gene provides instructions for making an enzyme called liver glycogen phosphorylase. This enzyme is found only in liver cells, where it breaks down glycogen into a type of sugar called glucose-1-phosphate. Additional steps convert glucose-1-phosphate into glucose, a simple sugar that is the main energy source for most cells in the body.
PYGL gene mutations prevent liver glycogen phosphorylase from breaking down glycogen effectively. Because liver cells cannot break down glycogen into glucose, individuals with glycogen storage disease type 6 (Hers disease) can have hypoglycemia and may use fats for energy, resulting in ketosis. Glycogen accumulates within liver cells, causing these cells to become enlarged and dysfunctional.
Glycogen storage disease type 6 inheritance pattern
Glycogen storage disease type 6 is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Glycogen storage disease type 6 symptoms
Although symptoms of glycogen storage disease type 6 (Hers disease) may not be apparent during childhood, some liver enlargement will be present. Many individuals will have no apparent symptoms (asymptomatic). In general, mild to moderately low blood sugar (hypoglycemia) may be present and can cause symptoms of faintness, weakness, hunger, and nervousness. Diminished muscle tone (hypotonia) and mild muscle weakness may occur in some cases.
Growth rate may be slowed, and enlargement of the liver may occur because of an excess accumulation of glycogen. Glycogen is the stored form of energy derived from carbohydrates. In many cases, the body can adapt to low blood sugar levels and is able to produce energy by other means. Therefore, symptoms may go unnoticed for long periods of time.
Liver enlargement often disappears by puberty and final adult height is often normal. Muscle strength and tone is usually normal by adulthood as well.
Glycogen storage disease type 6 diagnosis
A diagnosis of glycogen storage disease type 6 (Hers disease) is based on a test for activity of the liver phosphorylase enzyme. A small fragment of liver tissue is surgically removed (biopsy) and assayed for the activity of the enzyme. In persons with glycogen storage disease type 6 (Hers disease), this enzyme activity will be reduced.
Glycogen storage disease type 6 treatment
Because symptoms of glycogen storage disease type 6 (Hers disease) are generally mild, the disorder usually requires no treatment other than avoidance of prolonged fasting and monitoring by a physician. In individuals experiencing fasting hypoglycemia, a high carbohydrate diet and frequent feedings may be recommended.
Genetic counseling may be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Glycogen storage disease type 7
Glycogen storage disease type 7 or GSD7, is an inherited disorder caused by an inability to break down a complex sugar called glycogen in muscle cells. A lack of glycogen breakdown interferes with the function of muscle cells.
There are four types of glycogen storage disease type 7. They are differentiated by their signs and symptoms and the age at which symptoms first appear.
The classical form of glycogen storage disease type 7 is the most common form. Its features usually appear in childhood. This form is characterized by muscle pain and cramps, often following moderate exercise; strenuous exercise can lead to nausea and vomiting. During exercise, muscle tissue can be abnormally broken down, releasing a protein called myoglobin. This protein is processed by the kidneys and released in the urine (myoglobinuria). If untreated, myoglobinuria can damage the kidneys and lead to kidney failure. Some people with the classical form of glycogen storage disease type 7 develop high levels of a waste product called uric acid in the blood (hyperuricemia) because the damaged kidneys are unable to remove uric acid effectively. Affected individuals may also have elevated levels of a molecule called bilirubin in the blood that can cause yellowing of the skin and whites of the eyes (jaundice). Individuals with classical glycogen storage disease type 7 often have elevated levels of an enzyme called creatine kinase in their blood. This finding is a common indicator of muscle disease.
Infants with the severe infantile form of glycogen storage disease type 7 have low muscle tone (hypotonia) at birth, which leads to muscle weakness (myopathy) that worsens over time. Affected infants have a weakened and enlarged heart (cardiomyopathy) and difficulty breathing normally. Individuals with this form of glycogen storage disease type 7 usually do not survive past their first year of life.
In the late-onset form of glycogen storage disease type 7, myopathy is typically the only feature. The muscle weakness appears in adulthood, although some individuals have difficulty with sustained exercise starting in childhood. The weakness generally affects the muscles closest to the center of the body (proximal muscles).
The hemolytic form of glycogen storage disease type 7 is characterized by hemolytic anemia, in which red blood cells are broken down (undergo hemolysis) prematurely, causing a shortage of red blood cells (anemia). People with the hemolytic form of glycogen storage disease type 7 do not experience any signs or symptoms of muscle pain or weakness related to the disorder.
Glycogen storage disease type 7 is thought to be a rare condition; more than 100 cases have been described in the scientific literature 18.
Glycogen storage disease type 7 causes
Mutations in the PFKM gene cause glycogen storage disease type 7. This gene provides instructions for making one piece (the PFKM subunit) of an enzyme called phosphofructokinase, which plays a role in the breakdown of glycogen. The phosphofructokinase enzyme is made up of four subunits and is found in a variety of tissues. Different combinations of subunits are found in different tissues. In muscles used for movement (skeletal muscles), the phosphofructokinase enzyme is composed solely of PFKM subunits.
In skeletal muscle, the cells’ main source of energy is stored as glycogen. Glycogen can be broken down rapidly into the simple sugar glucose when energy is needed, for instance to maintain normal blood sugar levels between meals or for energy during exercise. Phosphofructokinase is involved in the sequence of events that breaks down glycogen to provide energy to muscle cells.
PFKM gene mutations result in the production of PFKM subunits that have little or no function. As a result, no functional phosphofructokinase is formed in skeletal muscles, and glycogen cannot be completely broken down. Partially broken down glycogen then builds up in muscle cells. Muscles that do not have access to glycogen as an energy source become weakened and cramped following moderate strain, such as exercise, and in some cases, begin to break down. In other tissues, other subunits that make up the phosphofructokinase enzyme likely compensate for the lack of PFKM subunits, and the enzyme is able to retain some function. This compensation may help explain why other tissues are not affected by PFKM gene mutations. It is unclear why some individuals with glycogen storage disease type 7 are affected with more severe forms of the disorder than others.
Glycogen storage disease type 7 inheritance pattern
Glycogen storage disease type 7 is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Glycogen storage disease type 7 symptoms
The clinical features of glycogen storage disease type 7 are similar to those of glycogen storage disease type 5 with onset of more severe fatigue and muscle pain early in exercise. Glycogen storage disease type 7 usually begins in childhood and is characterized by weakness, pain and stiffness during exercise, sometimes associated with nausea and vomiting and dark, burgundy-colored urine due to the presence of myoglobin (myoglobinuria). Destruction of muscle tissue (rhabdomyolysis) can also occur. A rare form of glycogen storage disease type 7 has been reported in infants that is associated with progressive loss of muscle tone (hypotonia), muscle weakness and death. A late-onset form has been reported in adults who experience only muscle weakness.
Glycogen storage disease type 7 diagnosis
Glycogen storage disease type 7 is diagnosed by a muscle biopsy for measurement of the phosphofructokinase enzyme level or measurement of the phosphofructokinase enzyme level in red blood cells. Molecular genetic testing for the phosphofructokinase gene mutations prevalent in the Ashkenazi Jewish population are available on a research basis.
Glycogen storage disease type 7 treatment
Strenuous exercise should be avoided to prevent muscle pain and cramps. Consumption of carbohydrates should be avoided because this can exacerbate exercise intolerance.
Genetic counseling is recommended for affected individuals and their families.
Glycogen storage disease type 9
Glycogen storage disease type 9 also known as GSD 9, is a condition caused by the inability to break down a complex sugar called glycogen. The different forms of the condition can affect glycogen breakdown in liver cells or muscle cells or sometimes both. A lack of glycogen breakdown interferes with the normal function of the affected tissue.
When glycogen storage disease type 9 affects the liver, the signs and symptoms typically begin in early childhood. The initial features are usually an enlarged liver (hepatomegaly) and slow growth. Affected children are often shorter than normal. During prolonged periods without food (fasting), affected individuals may have low blood sugar (hypoglycemia) or elevated levels of ketones in the blood (ketosis). Ketones are molecules produced during the breakdown of fats, which occurs when stored sugars are unavailable. Affected children may have delayed development of motor skills, such as sitting, standing, or walking, and some have mild muscle weakness. Puberty is delayed in some adolescents with glycogen storage disease type 9. In the form of the condition that affects the liver, the signs and symptoms usually improve with age. Typically, individuals catch up developmentally, and adults reach normal height. However, some affected individuals have a buildup of scar tissue (fibrosis) in the liver, which can rarely progress to irreversible liver disease (cirrhosis).
glycogen storage disease type 9 can affect muscle tissue, although this form of the condition is very rare and not well understood. The features of this form of the condition can appear anytime from childhood to adulthood. Affected individuals may experience fatigue, muscle pain, and cramps, especially during exercise (exercise intolerance). Most affected individuals have muscle weakness that worsens over time. glycogen storage disease type 9 can cause myoglobinuria, which occurs when muscle tissue breaks down abnormally and releases a protein called myoglobin that is excreted in the urine. Myoglobinuria can cause the urine to be red or brown.
In a small number of people with glycogen storage disease type 9, the liver and muscles are both affected. These individuals develop a combination of the features described above, although the muscle problems are usually mild.
Glycogen storage disease type 9 that affects the liver is estimated to occur in 1 in 100,000 people 19. The forms of the disease that affect muscles or both muscles and liver are much less common, although the prevalence is unknown.
Glycogen storage disease type 9 causes
Mutations in the PHKA1, PHKA2, PHKB, or PHKG2 genes are known to cause glycogen storage disease type 9. These genes provide instructions for making pieces (subunits) of an enzyme called phosphorylase b kinase. The enzyme is made up of 16 subunits, four each of the alpha, beta, gamma, and delta subunits. At least two different versions of phosphorylase b kinase are formed from the subunits: one is most abundant in liver cells and the other in muscle cells.
The PHKA1 and PHKA2 genes provide instructions for making alpha subunits of phosphorylase b kinase. The protein produced from the PHKA1 gene is a subunit of the muscle enzyme, while the protein produced from the PHKA2 gene is part of the liver enzyme. The PHKB gene provides instructions for making the beta subunit, which is found in both the muscle and the liver. The PHKG2 gene provides instructions for making the gamma subunit of the liver enzyme.
Whether in the liver or the muscles, phosphorylase b kinase plays an important role in providing energy for cells. The main source of cellular energy is a simple sugar called glucose. Glucose is stored in muscle and liver cells in a form called glycogen. Glycogen can be broken down rapidly when glucose is needed, for instance to maintain normal levels of glucose in the blood between meals or for energy during exercise. Phosphorylase b kinase turns on (activates) the enzyme that breaks down glycogen.
Although the effects of gene mutations on the respective protein subunits are unknown, mutations in the PHKA1, PHKA2, PHKB, and PHKG2 genes reduce the activity of phosphorylase b kinase in liver or muscle cells and in blood cells. Reduction of this enzyme’s function impairs glycogen breakdown. As a result, glycogen accumulates in and damages cells, and glucose is not available for energy. Glycogen accumulation in the liver leads to hepatomegaly, and the liver’s inability to break down glycogen for glucose contributes to hypoglycemia and ketosis. Reduced energy production in muscle cells leads to muscle weakness, pain, and cramping.
Glycogen storage disease type 9 inheritance pattern
Glycogen storage disease type 9 can have different inheritance patterns depending on the genetic cause of the condition.
When caused by mutations in the PHKA1 or PHKA2 gene, glycogen storage disease type 9 is inherited in an X-linked recessive pattern. These genes are located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. However, some women with one altered copy of the PHKA2 gene have signs and symptoms of glycogen storage disease type 9, such as mild hepatomegaly or short stature in childhood. These features are usually mild but can be more severe in rare cases. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
When the condition is caused by mutations in the PHKB or PHKG2 gene, it is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Glycogen storage disease type 9 symptoms
Glycogen storage disease type 9 is caused by deficiency of the enzyme phosphorylase kinase. The specific symptoms present, severity and prognosis can vary depending upon the subtype and the areas of the body affected. The symptoms and severity can vary even among individuals with the same mutation. In addition, some subtypes have only been reported in a handful of individuals, which prevents physicians from developing a complete picture of associated symptoms and prognosis. Therefore, it is important to note that affected individuals may not have all of the symptoms discussed below. Affected individuals should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis. Individuals with the liver form of glycogen storage disease type 9 have a wide range of clinical symptoms ranging from less severe to more severe hepatic manifestations of the disease. Natural history studies are necessary to understand completely the long-term course and prognosis of glycogen storage disease type 9.
Glycogen storage disease type 9a
Glycogen storage disease type 9a is the most common subtype of glycogen storage disease type 9, and is caused by the deficiency of phosphorylase kinase in the liver. It accounts for approximately 75% of affected individuals and is also known as X-linked liver glycogenesis or PHKA2-related phosphorylase kinase deficiency. Affected individuals often develop an enlarged liver (hepatomegaly), low blood glucose levels (hypoglycemia) and high levels of blood ketones during fasting, and growth delays. Some children have delays in motor development. Hypoglycemia can develop after fasting overnight, after shorter periods of fasting, or if food intake is reduced during illness. Symptoms of hypoglycemia include shakiness, irritability, unexplained fatigue, headache, pale skin, and rapid heartbeat. Hypoglycemia can result in the body burning fat for energy in which causes high levels of ketones in the body (hyperketosis). Hyperketotic hypoglycemia can be associated with nausea and vomiting. Although hypoglycemia can be considered mild symptoms could be masked because of the body’s ability to lower levels of blood glucose than in unaffected individuals. Hypoglycemia can also be very severe and may recur. Growth delays can be pronounced during childhood, but most children show catch-up growth and ultimately reach a normal adult height. Diminished muscle tone (hypotonia) and muscle weakness may also be seen during early childhood. Puberty may be delayed. Increased levels of different lipids such as cholesterol (hypercholesterolemia) and triglycerides (hypertriglyceridemia) may be seen in blood of some affected individuals.
Although glycogen storage disease type 9a has, historically, been considered a benign (mild) disorder, this notion is being currently dispelled with reports of patients with severe symptoms. It is being increasingly recognized that there is a broad range in the severity of symptoms. Some people have few or no problems with hypoglycemia while others have severe and recurrent hypoglycemia. There have been reports in the medical literature of cases in which scar tissue has developed within the liver (fibrosis) and, in some children may develop irreversible scarring of the liver (cirrhosis).
Glycogen storage disease type 9b
This subtype of the disorder is characterized by phosphorylase kinase deficiency of the liver and the muscle. It is also known as PHKB-related phosphorylase kinase deficiency. The symptoms are similar to those in people with glycogen storage disease type 9a. Children with glycogen storage disease type 9b can develop an enlarged liver (hepatomegaly), hypoglycemia, diminished muscle tone (hypotonia), muscle weakness, and growth delays that can result in childhood short stature. Despite the deficiency of PhK in muscle as well as liver, muscle weakness is not always reported in people with this subtype.
Glycogen storage disease type 9c
This subtype of glycogen storage disease type 9 is characterized by phosphorylase kinase deficiency of the liver. It is also known as PHKG2-related phosphorylase kinase deficiency. The symptoms are similar to those in people with glycogen storage disease type 9a and glycogen storage disease type 9b, but tend to be severe. Like glycogen storage disease type 9a and glycogen storage disease type 9b, this form of the disorder is characterized by an enlarged liver, hypoglycemia, hypotonia and delays in motor development in some children, and growth delays in childhood. Most individuals attain a normal adult height.. Some children may develop recurrent episodes of low blood glucose levels (hypoglycemia). This can result in the body burning fat for energy resulting in high levels of ketones in the body (hyperketosis). Hyperketotic hypoglycemia may only occur after prolonged fasting, such as overnight or during an illness if food intake is reduced, and can be associated with nausea and vomiting. Benign tumors of the liver, also known as hepatic adenomas may be seen in some individuals. Affected individuals may present with a wide range of disease symptoms. Understanding of this disease continues to evolve as more cases come to light.
In some cases of glycogen storage disease type 9c, more serious complications can occur such as the development scar tissue (fibrosis) within the liver as well as degeneration, inflammation and scarring of the liver (cirrhosis). The risk of these complications appears to be greater in glycogen storage disease type 9c than in other forms of the disorder. Liver transplantation may be needed for survival in some patients who have severe liver damage.
Glycogen storage disease type 9d
This extremely rare form of the disorder is characterized by phosphorylase kinase deficiency of the muscle. The liver is not affected. Affected individuals may develop progressive muscle weakness, muscle degeneration (atrophy), muscle cramps, abnormal muscle pain (myalgia) that occurs following exercise (exercise-induced muscle pain), abnormal muscle stiffness following exercise and rust colored urine due to excretion of myoglobin, a muscle protein (myoglobinuria). In general, affected individuals cannot exercise at normally accepted levels (exercise intolerance). The onset of symptoms can occur in childhood or adulthood; most patients have adult onset. Notably, some individuals with phosphorylase kinase deficiency in muscle do not have any obvious symptoms. This form is also known as PHKA1-related phosphorylase kinase deficiency.
Glycogen storage disease type 9 diagnosis
A diagnosis of glycogen storage disease type 9 is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests.
Clinical testing and workup
The diagnosis of the liver form of glycogen storage disease type 9 is often first suspected from symptoms, such as hepatomegaly and growth delay, and abnormalities on routine laboratory tests including elevated liver transaminases, and elevations of cholesterol and triglyceride levels. Some children may present with seizures caused by low glucose levels. However, these findings are nonspecific and more specialized enzyme and genetic tests are needed to diagnose glycogen storage disease type 9. These tests include an enzyme assay that measures the activity of phosphorylase kinase in red blood cells (erythrocytes) or in liver tissue. However, normal phosphorylase kinase activity does not exclude a diagnosis (samples from some affected individuals have had normal activity when tested).
Individuals with symptoms of muscle PhK activity can have elevated creatine kinase level in blood but the presentation is similar to many other muscle disorders, and measurement of phosphorylase kinase activity in a muscle sample is needed to further investigate the diagnosis.
Molecular genetic testing can confirm a diagnosis of glycogen storage disease type 9. Molecular genetic testing can detect mutations in specific genes known to cause glycogen storage disease type 9 but, like the enzyme test, is available only as a diagnostic service at specialized laboratories.
Prenatal diagnosis for at-risk pregnancies allows prior identification of risk in families with affected individuals. Evaluation of family members at risk may be done by carrier testing.
Glycogen storage disease type 9 treatment
The treatment of glycogen storage disease type 9 is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, liver specialists (hepatologists), pediatric gastroenterologists, nutritionists, physical therapists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may be of benefit for affected individuals and their families.
There are no dietary restrictions associated with glycogen storage disease type 9, although ingestion of simple sugars should be limited. A high-protein, complex carbohydrate diet is recommended. Prolonged fasting should be avoided. Frequent, small meals that can be supplemented with uncooked cornstarch are recommended to avoid hypoglycemia. Some individuals may need to have a bedtime snack or cornstarch to prevent nighttime development of hypoglycemia. Some individuals will only require cornstarch supplementation before bedtime. If hypoglycemia or ketosis develops, affected individuals can be treated with Polycose® (glucose polymer powder) or fruit juice. Some individuals may be unable to tolerate oral therapy with Polycose® or fruit juice and may require glucose to be delivered through an IV line. If the muscles are affected, physical therapy may be recommended. Vigorous exercise should be avoided and drugs that can damage muscle tissue (such as statins) should be taken after consultation with a physician.
Monitoring of blood glucose and ketone levels periodically as well as during periods of stress is necessary. Follow-up of liver involvement may be done by checking liver enzyme levels such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase and gamma glutaryl transferase (GGT) and abdominal ultrasound/MRI every 6-12 months or as clinically relevant.
Prognosis is considered generally good for the X-linked and certain autosomal forms of the disease. However, at this time, more severe presentations such as liver fibrosis and cirrhosis are being reported, even in the X-linked form. Further research is needed to completely understand long-term complications of the disease progression into adulthood.
If affected individuals require general anesthesia, precautions against malignant hyperthermia should be taken. Malignant hyperthermia is a disorder characterized by an abnormal and potentially life-threatening response to muscle relaxants and general anesthesia drugs.
Extra rare glycogen storage diseases
Glycogen storage disease type 13
Glycogen storage disease type 13 (GSD13) also known as beta-enolase deficiency, is an inherited disease of the muscles. The muscles of an affected individual are not able to produce enough energy to function properly, causing muscle weakness and pain. Glycogen storage disease type 13 is caused by changes (mutations) in the ENO3 gene and is inherited in an autosomal recessive pattern 20. Glycogen storage disease type 13 was first reported in 2001 21.
Patients so far described have a small amount of residual enzyme activity (5-20%) and their symptoms appear milder than McArdle disease. These can include exercise intolerance, myalgia, contracture, and generalised muscle weakness after exercise. Creatine kinase (CK) levels may be normal at baseline, but raised after intense activity.
With so few patients identified to date, the clinical picture and the age of onset for glycogen storage disease type 13 has yet to be fully understood. Investigations may be triggered by an episode of rhabdomyolysis rather than presentation to a family physician due to day to day symptoms. Next-generation sequencing may well start to show this condition to be more common than currently thought.
Glycogen storage disease type 13 causes
Glycogen storage disease type 13 (GSD13) is caused by changes (mutations) in the ENO3 gene. Glycogen is a substance that is stored in muscle tissue and is used as an important source of energy for the muscles during movement and exercise. The ENO3 gene makes a chemical called enolase, which is an enzyme that helps the muscles use glycogen for energy. In glycogen storage disease type 13, the ENO3 genes do not work properly such that the body cannot make enolase, and as a result, the muscles do not have enough energy to work properly 20. Glycogen storage disease type 13 was first reported in 2001 21.
Glycogen storage disease type 13 signs and symptoms
Glycogen storage disease type 13 causes muscle pain (myalgia). Individuals with glycogen storage disease type 13 also experience exercise intolerance, which means they have difficulty exercising because they may have muscle weakness and tire easily 20.
Is hypoglycemia associated with glycogen storage disease type 13?
Unfortunately, because glycogen storage disease type 13 is very rare, there is limited information about the associated features of this condition. However, the first individual known to have glycogen storage disease type 13 did not have hypoglycemia 20.
Glycogen storage disease type 13 diagnosis
Glycogen storage disease type 13 is diagnosed by taking a sample of muscle tissue (muscle biopsy) to determine if there is enough of the chemical enolase working in the muscle cells. Genetic testing can also be done to look for changes (mutations) in the ENO3 gene 20.
Glycogen storage disease type 13 treatment
Maintain aerobic fitness and avoid anaerobic activity.
Danon disease
Danon disease is a condition characterized by weakening of the heart muscle (cardiomyopathy); weakening of the muscles used for movement, called skeletal muscles, (myopathy); and intellectual disability. Males with Danon disease usually develop the condition earlier than females and are more severely affected. Signs and symptoms begin in childhood or adolescence in most affected males and in early adulthood in most affected females. Affected males, on average, live to age 19, while affected females live to an average age of 34.
Cardiomyopathy is the most common symptom of Danon disease and occurs in all males with the condition. Most affected men have hypertrophic cardiomyopathy, which is a thickening of the heart muscle that may make it harder for the heart to pump blood. Other affected males have dilated cardiomyopathy, which is a condition that weakens and enlarges the heart, preventing it from pumping blood efficiently. Some affected men with hypertrophic cardiomyopathy later develop dilated cardiomyopathy. Either type of cardiomyopathy can lead to heart failure and premature death. Most women with Danon disease also develop cardiomyopathy; of the women who have this feature, about half have hypertrophic cardiomyopathy, and the other half have dilated cardiomyopathy.
Affected individuals can have other heart-related signs and symptoms, including a sensation of fluttering or pounding in the chest (palpitations), an abnormal heartbeat (arrhythmia), or chest pain. Many affected individuals have abnormalities of the electrical signals that control the heartbeat (conduction abnormalities). People with Danon disease are often affected by a specific conduction abnormality known as cardiac preexcitation. The type of cardiac preexcitation most often seen in people with Danon disease is called the Wolff-Parkinson-White syndrome pattern.
Skeletal myopathy occurs in most men with Danon disease and about half of affected women. The weakness typically occurs in the muscles of the upper arms, shoulders, neck, and upper thighs. Many males with Danon disease have elevated levels of an enzyme called creatine kinase in their blood, which often indicates muscle disease.
Most men with Danon disease, but only a small percentage of affected women, have intellectual disability. If present, the disability is usually mild.
There can be other signs and symptoms of the condition in addition to the three characteristic features. Several affected individuals have had gastrointestinal disease, breathing problems, or visual abnormalities.
Danon disease is a rare condition, but the exact prevalence is unknown 22.
Danon disease causes
Danon disease is caused by mutations in the LAMP2 gene. The LAMP2 gene provides instructions for making a protein called lysosomal associated membrane protein-2 (LAMP-2), which, as its name suggests, is found in the membrane of cellular structures called lysosomes. Lysosomes are compartments in the cell that digest and recycle materials. The role the LAMP-2 protein plays in the lysosome is unclear. Some researchers think the LAMP-2 protein may help transport cellular materials or digestive enzymes into the lysosome. The transport of cellular materials into lysosomes requires the formation of cellular structures called autophagic vacuoles (or autophagosomes), which then attach (fuse) to lysosomes. The LAMP-2 protein may be involved in the fusion between autophagic vacuoles and lysosomes.
Mutations in the LAMP2 gene lead to the production of very little or no LAMP-2 protein, which may impair the process of transporting cellular material into the lysosome. Some studies have shown that in cells without the LAMP-2 protein, fusion between autophagic vacuoles and lysosomes occurs more slowly, which may lead to the accumulation of autophagic vacuoles. People with Danon disease have an abnormally large number of autophagic vacuoles in their muscle cells. It is possible that this accumulation leads to breakdown of the muscle cells, causing the muscle weakness seen in Danon disease.
Danon disease inheritance pattern
Danon disease is inherited in an X-linked dominant pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. In most cases, males experience more severe symptoms of the disorder than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Danon disease symptoms
Symptoms of Danon disease vary from person to person and depend on gender. Boys usually show early signs of muscle problems (difficulty sitting or walking) and motor skills may be awkward or delayed. Intellectual disability is usually noticed by parents and/or teachers and can be quite mild. The development of heart disease can lead to further fatigue and shortness of breath. Visual complaints are also prevalent with serious color vision disturbances and near-complete loss of retinal pigment in some patients.
In general, young girls may have no symptoms and will report normal muscle strength and have normal intellect. As females age, symptoms of heart disease can begin to develop. Muscle symptoms are reported by some girls and women but overt findings of frank muscle weakness are usually absent. Visual complaints may also be reported in women and can be an early feature of the disease, although manifestations are less severe than in men.
Clinical researchers believe that the skeletal muscle involvement in Danon disease preferentially involves the muscles of the back, shoulder, upper legs and the neck muscles. These are the proximal muscles; that is, those closest to the center of the body. Symptoms of weakness in these muscles can include back pain and difficulty raising one’s arms over the head, getting out of a chair, or walking up steps. In a young boy, these problems may be suggested by problems meeting motor milestones (sitting, crawling, and walking, running). An experienced neurologist can recognize the extent of muscle disease by performing a physical examination. Increasingly it is apparent that for some patients the muscle disease progresses over time and some older males may require assistance devices (walkers, wheelchairs).
The diseased heart muscle (cardiomyopathy) can lead to a thickened, stiff heart (hypertrophic cardiomyopathy) or to an enlarged heart (dilated cardiomyopathy). Hypertrophic cardiomyopathy is more common in males (approximately 90% hypertrophic and 10% dilated), whereas females are more apt to show features of dilated cardiomyopathy (approximately 50% hypertrophic and 50% dilated). Sometimes the cardiomyopathy can be the first sign of disease in male children. In both instances, problems with heart function and symptoms of heart failure (shortness of breath, fatigue, fluid gain) can occur. Death from the heart disease seems to more occur frequently in males, especially as they reach the second and third decades of life. Heart transplantation has been performed successfully and can greatly improve symptoms and extend life. Implantable cardiac defibrillators are used to manage heart arrhythmias and should probably be considered at the first symptoms of arrhythmia.
The extent of intellectual disability in affected males has been described in some epidemiological studies. The majority of boys will be mildly affected cognitively, usually allowing them to achieve the ability to read, hold jobs, form relationships, and live independently. Furthermore, providing education and learning support may help some boys improve their intellectual functioning. In women, intellect appears to be normal, although very little information in the literature addresses this question.
Less prevalent symptoms also include liver and lung involvement, although these have not been studied extensively and might be secondary to muscle involvement (e.g. serum liver enzyme elevation and respiratory muscle weakness). Some speculation also exists on psychiatric disease, with some case reports detailing depression, psychosis, suicidal ideation, and attention-deficit hyperactivity disorder in Danon disease patients. However, it is unclear if psychiatric episodes are related to Danon disease.
Males with Danon disease typically have abnormalities on certain laboratory tests. The creatine kinase level in the blood is often elevated, and is a reflection of ongoing muscle damage. The creatine kinase is usually elevated in males, but can be normal in some females who have Danon disease. Abnormalities in liver enzyme tests are common in males; in some boys, these are mistakenly interpreted as a sign of primary liver disease rather than a reflection of skeletal muscle dysfunction; frank liver dysfunction has not been well-described in Danon disease. The electrocardiogram (ECG), which measures electrical impulses made by the heart, is often abnormal. This abnormality in conduction and electrical impulse is also known as an arrhythmia. Frequently, an arrhythmia called Wolff-Parkinson-White syndrome or a pre-excitation syndrome will be seen on the ECG. An examination of the retina by an experienced eye doctor (ophthalmologist) will often detect changes in the pigment of the retina. This can be a useful sign in women, as the retinal changes appear to precede other symptoms of the disease in some females.
Danon disease diagnosis
Because Danon disease is rare and unfamiliar to most physicians, diagnosis is difficult and takes substantial time. The diagnosis is suggested on the basis of a family history compatible with X-linked dominant inheritance and symptoms in affected relatives (cardiomyopathy, skeletal myopathy, intellectual disability, Wolff-Parkinson White, etc.). Skeletal muscle biopsy is done in some males in an effort to determine the cause of muscle weakness. If, in the course of examining the biopsy materials, glycogen buildup and/or empty spaces appear in the cells of the muscle tissue (vacuolization), Danon disease must be considered. This also holds true for the analysis of a heart biopsy. A muscle biopsy that yields evidence of glycogen build-up and empty spaces in the muscle cells are key signs and indications that a diagnosis of Danon disease is a high probability.
It is important to recognize that, in early stages of Danon disease, and probably also in women, the muscle biopsy can be non-specific. Thus, a normal or non-specific muscle biopsy does not exclude Danon disease. If other features of Danon disease are present, a non-diagnostic muscle biopsy should not discourage more definitive genetic testing. Patients who appear to have Pompe disease (based on muscle biopsy for instance) but have normal acid maltase activity, should be evaluated for Danon disease. Unexplained hypertrophic cardiomyopathy in males is probably due to Danon disease in some people.
Antibodies to the LAMP-2 protein are available and tissue staining (of a muscle biopsy) for the absence of LAMP-2 protein is another potential, but not widely available, diagnostic approach. LAMP-2 antibody testing is likely to be normal in women with Danon disease and if done should be interpreted with caution due to the possibility of a false-negative result.
Genetic testing of the LAMP2 gene is currently the gold standard for diagnosis and is available in specialized genetics laboratories. Most genetic mutations causing Danon disease predict reduced levels or even absence of the LAMP2 gene product, the LAMP-2 protein. Although the sensitivity of LAMP2 genetic testing is not known at this time, it is the best that is available. The noninvasive nature of DNA-based testing and the inclusion of LAMP2 gene testing in hypertrophic cardiomyopathy genetic diagnostic panels favor this method as the most common route to diagnosis.
Danon disease treatment
The treatment of Danon disease is directed toward the specific symptoms that are apparent in each individual. It requires a team that should include a primary care physician as well as several specialists, including a cardiologist, neurologist, ophthalmologist, geneticist, genetic counselor, rehabilitation physician, educational specialist, and physical therapist. Currently there is no specific therapy that is known to slow the underlying biological problems caused by LAMP-2 protein deficiency.
The severity of cardiomyopathy is the major prognostic factor. Imaging studies including echocardiography and cardiac magnetic resonance can assess heart function, extent of hypertrophy, and degree of cardiac fibrosis (formation of scar tissue on the heart). Medications for heart disease should be given when indicated by clinical signs and symptoms. The rapid progression of the cardiomyopathy in some males necessitates prompt consideration for heart transplantation. Early involvement of electrophysiology to study the electrical conduction system of the heart is warranted in patients with arrhythmias. A device called a Holter monitor can be used to continuously record the electrical impulses of the heart. For symptomatic arrhythmias, early implantation of a cardioverter-defibrillator may be appropriate. Cardiac ablation therapy, which is a technique utilized to destroy the abnormal focus in the heart generating the irregular rhythm, can also be performed. As the disease can progress rapidly in males, consideration for early defibrillator implantation and evaluation for cardiac transplantation are appropriate in males as cardiomyopathy progresses.
Assessment of muscle strength, especially the proximal muscles of the shoulder, neck, and legs, should be performed regularly. Physical therapy can be helpful in maintaining muscle strength and flexibility. Intellectual disability should be screened for in males and appropriate educational interventions applied as needed. Regular eye examinations, to track the development and progression of retinal disease, should be considered. Biological relatives who are at risk for Danon disease should be evaluated by a physician for early signs of disease. At a minimum, evaluation of such relatives should include a medical history, physical examination (attention to cardiac, neurological, and ocular exams), creatine kinase testing, ECG, and echocardiogram.
Genetic consultation and counseling is recommended for all patients and families so that inheritance and reproductive risks are clearly communicated.
Lafora disease
Lafora disease has recently been widely accepted as being a glycogen storage disease 23. It is an ultra-rare, progressive, autosomal recessive neurodegenerative disorder.
Lafora disease is caused by loss-of-function mutations in either the laforin gene (EPM2A) or malin gene (EPM2B). It is associated with gradual accumulation of Lafora bodies, aggregates of poorly branched, hyperphosphorylated, insoluble glycogen also known as polyglusan.
References- Hicks J, Wartchow E, Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011 Oct;35(5):183-96.
- Glycogen storage diseases. https://agsd.org.uk/all-about-gsd/gsd-variants/von-gierke-disease-gsd1/gsd1-section1-tab1/
- Glycogen storage disease.. https://en.wikipedia.org/wiki/Glycogen_storage_disease
- Kannourakis G. Glycogen storage disease. Semin. Hematol. 2002 Apr;39(2):103-6.
- Stone WL, Basit H, Adil A. Glycogen Storage Disease. [Updated 2019 Jun 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459277
- Parikh NS, Ahlawat R. StatPearls [Internet]. StatPearls Publishing; Treasure Island (FL): Jan 13, 2019. Glycogen Storage Disease Type I (Von Gierke Disease).
- Dagli A, Sentner CP, Weinstein DA. Glycogen Storage Disease Type III. 2010 Mar 9 [Updated 2016 Dec 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26372
- Chan J, Desai AK, Kazi ZB, Corey K, Austin S, Hobson-Webb LD, Case LE, Jones HN, Kishnani PS. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017 Mar;120(3):163-172.
- Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis. 2017 May 25;2(1-2):1-71.
- Glycogen storage disease type 0. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-0
- Glycogen storage disease type 1. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-i
- Pompe disease. https://ghr.nlm.nih.gov/condition/pompe-disease
- Glycogen storage disease type 3. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-iii
- Glycogen Storage Disease Type 3. https://rarediseases.org/rare-diseases/forbes-disease/
- Glycogen storage disease type 4. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-iv#statistics
- Glycogen storage disease type 5. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-v
- Glycogen storage disease type 6. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-vi
- Glycogen storage disease type 7. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-vii
- Glycogen storage disease type 9. https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-ix
- Comi GP, Fortunato F, Lucchiari S, Bordoni A, Prelle A, Jann S, Keller A, Ciscato P, Galbiati S, Chiveri L, Torrente Y, Scarlato G, Bresolin N. Beta-enolase deficiency, a new metabolic myopathy of distal glycolysis. Annals of Neurology. 2001; 50:202-207. http://www.ncbi.nlm.nih.gov/pubmed/11506403
- Glycogen storage disease type 13. https://agsd.org.uk/all-about-gsd/gsd-variants/other-gsds/gsd13/
- Danon disease. https://ghr.nlm.nih.gov/condition/danon-disease
- Lafora Disease. https://agsd.org.uk/all-about-gsd/gsd-variants/other-gsds/lafora-disease/





{kind=link}