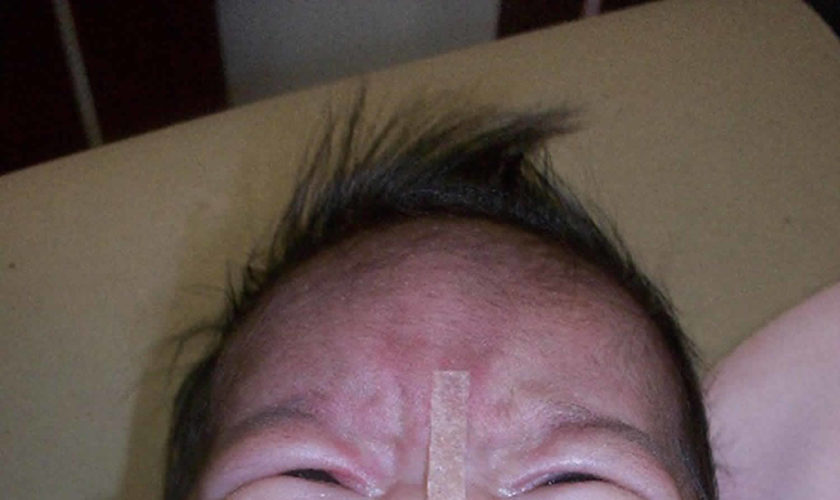
What is trisomy 13
Trisomy 13, also called Patau syndrome, is a serious but rare chromosomal disorder caused by having an additional copy of chromosome 13 in some or all of the body’s cells. Patau syndrome (trisomy 13) is associated with severe intellectual disability and physical abnormalities in many parts of the body. Individuals with trisomy 13 often have heart defects, brain or spinal cord abnormalities, very small or poorly developed eyes (microphthalmia), extra fingers or toes, an opening in the lip (a cleft lip) with or without an opening in the roof of the mouth (a cleft palate), and weak muscle tone (hypotonia). Due to the presence of several life-threatening medical problems, many infants with trisomy 13 die within their first days or weeks of life. Only five percent to 10 percent of children with this condition live past their first year.
Each cell normally contains 23 pairs of chromosomes, which carry the genes you inherit from your parents.
But a baby with trisomy 13 (Patau syndrome) has 3 copies of chromosome 13, instead of 2. This severely disrupts normal development and, in many cases, results in miscarriage, stillbirth, or the baby dying shortly after birth.
Trisomy 13 occurs when extra DNA from chromosome 13 appears in some or all of the body’s cells.
- Trisomy 13: the presence of an extra (third) chromosome 13 in all of the cells.
- Mosaic trisomy: the presence of an extra chromosome 13 in some of the cells.
- Partial trisomy: the presence of a part of an extra chromosome 13 in the cells.
The extra material interferes with normal development.
Most cases of trisomy 13 (Patau syndrome) are not inherited and result from random events during the formation of eggs and sperm in healthy parents. An error in cell division called nondisjunction results in a reproductive cell with an abnormal number of chromosomes. For example, an egg or sperm cell may gain an extra copy of chromosome 13. If one of these atypical reproductive cells contributes to the genetic makeup of a child, the child will have an extra chromosome 13 in each cell of the body.
Translocation trisomy 13 (Patau syndrome) can be inherited. An unaffected person can carry a rearrangement of genetic material between chromosome 13 and another chromosome. These rearrangements are called balanced translocations because there is no extra material from chromosome 13. A person with a balanced translocation involving chromosome 13 has an increased chance of passing extra material from chromosome 13 to their children.
Babies with trisomy 13 (Patau syndrome) grow slowly in the womb and have a low birth weight, along with a number of other serious medical problems.
Trisomy 13 (Patau syndrome) affects about 1 in every 5,000 to 16,000 births 1. Although women of any age can have a child with trisomy 13, the chance of having a child with this condition increases as a woman gets older.
Investigators have also suggested a possible association between preeclampsia and Trisomy 13. Preeclampsia is an abnormal condition of pregnancy characterized by the rapid onset of high blood pressure (hypertension), abnormal amounts of protein in the urine (proteinuria), and/or excessive retention of fluids (edema). According to researchers, the number of cases of preeclampsia appears to be significantly higher in women who are carrying a fetus with Trisomy 13 Syndrome than would be otherwise expected in the general population. In addition, the incidence appears significantly higher than when compared with pregnancies complicated by certain other chromosomal abnormalities (e.g., trisomy 18 [Edwards Syndrome], trisomy 21 [Down Syndrome]). Such researchers suggest the possibility that a gene or genes on fetal chromosome 13 may influence the development of preeclampsia.
In individuals with Trisomy 13 Syndrome, the range and severity of associated symptoms and findings may depend on the specific location of the duplicated (trisomic) portion of chromosome 13, as well as the percentage of cells containing the abnormality. However, in many affected infants and children, such abnormalities may include developmental delays, profound mental retardation, unusually small eyes (microphthalmia), an abnormal groove in the upper lip (cleft lip), incomplete closure of the roof of the mouth (cleft palate), undescended testes (cryptorchidism) in affected males, and extra (supernumerary) fingers and toes (polydactyly). Additional malformations of the head and facial (craniofacial) area may also be present, such as a relatively small head (microcephaly) with a sloping forehead; a broad, flat nose; widely set eyes (ocular hypertelorism); vertical skin folds covering the eyes; inner corners (epicanthal folds); scalp defects; and malformed, low-set ears. Affected infants may also have incomplete development of certain regions of the brain (e.g., the forebrain); kidney (renal) malformations; and structural heart (cardiac) defects at birth (congenital). For example, characteristic heart defects may include an abnormal opening in the partition dividing the upper or lower chambers of the heart (atrial or ventricular septal defects) or persistence of the fetal opening between the two major arteries (aorta, pulmonary artery) emerging from the heart (patent ductus arteriosus). Many infants with Trisomy 13 Syndrome fail to grow and gain weight at the expected rate (failure to thrive) and have severe feeding difficulties, diminished muscle tone (hypotonia), and episodes in which there is temporary cessation of spontaneous berathing (apnea). Life-threatening complications may develop during infancy or early childhood.
Figure 1. Trisomy 13 (Patau’s syndrome)
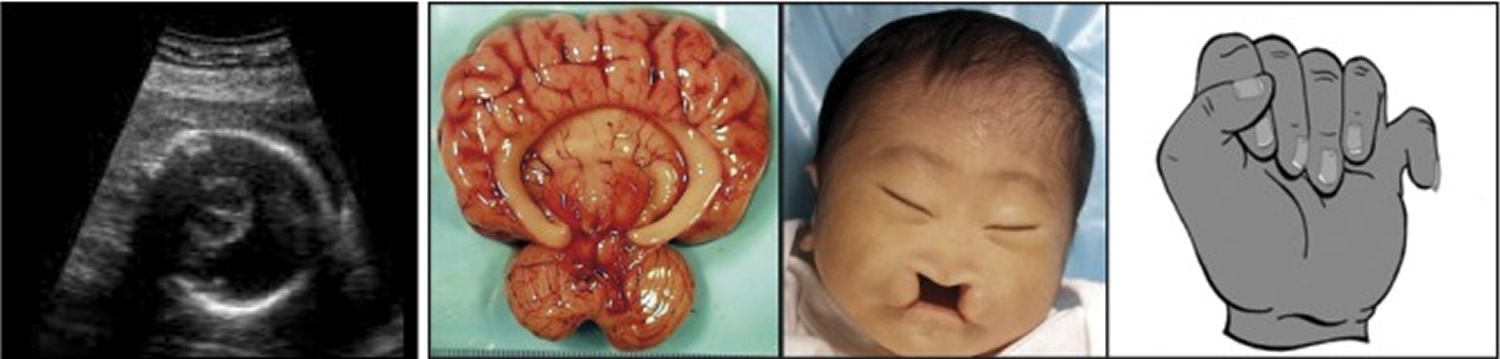
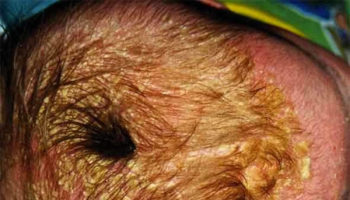
Footnotes: a,b) transverse brain scan in the second trimester showing a monoventricle with fused thalami, typical for alobar holoprosencephaly and the pathology specimen; c) Facial appearance in trisomy 13 with median cleft lip; d) Typical for the hands in trisomy 13 is the postaxial polydactyly
[Source 2]Figure 2. Trisomy 13 (Patau’s syndrome) polydactyly
Trisomy 13 life expectancy
Due to the presence of several life-threatening medical problems, many infants with trisomy 13 (Patau syndrome) die within their first days or weeks of life. More than 9 out of 10 children (over 90%) born with Patau’s syndrome die during the first year. Only 5 to 10% of babies with less severe forms of Patau syndrome, such as partial or mosaic trisomy 13, live for more than a year.
Trisomy 13 symptoms
Babies with Patau syndrome can have a wide range of health problems. Their growth in the womb is often restricted, resulting in a low birth weight, and 80% will be born with severe heart defects.
The brain often doesn’t divide into 2 halves. This is known as holoprosencephaly.
Patau syndrome symptoms include:
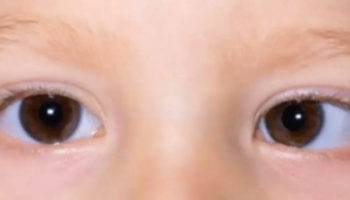
- Absence of 1 or both eyes (anophthalmia)
- An abnormally small eye or eyes (microphthalmia)
- Cleft lip or palate
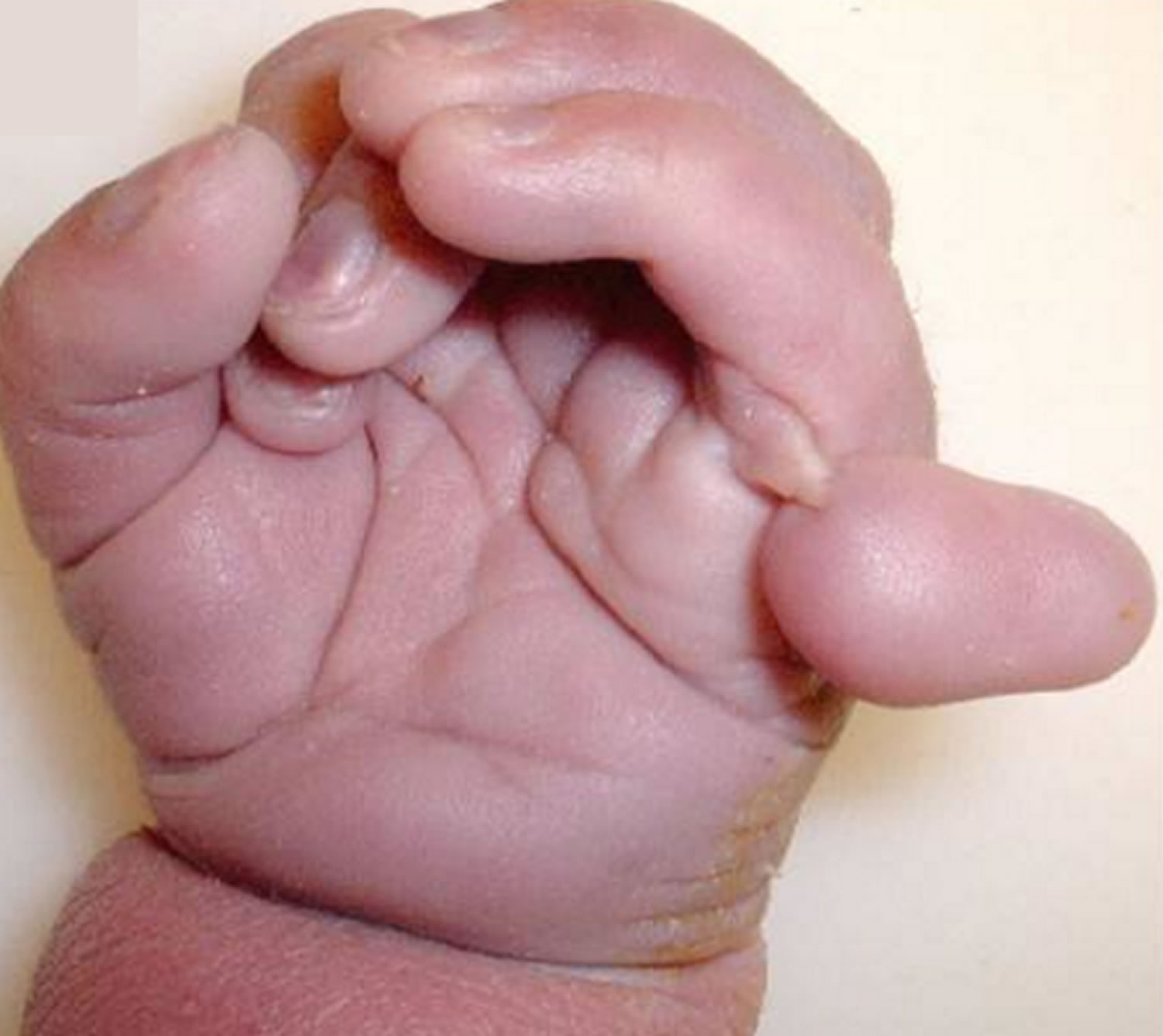
- Clenched hands (with outer fingers on top of the inner fingers)
- Close-set eyes — eyes may actually fuse together into one
- Decreased muscle tone
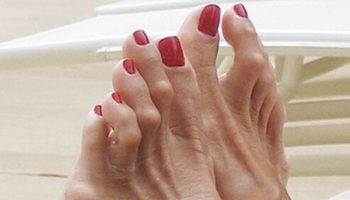
- Extra fingers or toes (polydactyly)
- Hernias: umbilical hernia, inguinal hernia
- Hole, split, or cleft in the iris (coloboma)
- Low-set ears
- Ear malformations and deafness
- Intellectual disability, severe
- Problems with the development of the nasal passages
- Raised, red birthmarks (capillary haemangiomas)
- Scalp defects (missing skin)
- Seizures
- Single palmar crease
- Skeletal (limb) abnormalities
- Skin missing from the scalp (cutis aplasia)
- Small head (microcephaly)
- Small lower jaw (micrognathia)
- Undescended testicle (cryptorchidism)
Patau’s syndrome can also cause other problems, such as:
- an abdominal wall defect where the abdomen doesn’t develop fully in the womb, resulting in the intestines being outside the body, covered only by a membrane – this is known as an exomphalos or omphalocoele
- abnormal cysts in the kidneys
- an abnormally small penis in boys
- an enlarged clitoris in girls
There may also be abnormalities of the hands and feet, such as extra fingers or toes (polydactyly), and a rounded bottom to the feet, known as rocker-bottom feet.
Described features of Patau syndrome are numerous and include:
- congenital heart disease: 50-80%
- hypoplastic left heart syndrome
- ventricular septal defect (VSD)
- central nervous system/head and neck abnormalities: 70%
- spinal anomalies
- spina bifida
- intra uterine growth restriction (IUGR): tends to be early
- abnormal facies: 90%, strong marker
- skeletal abnormalities
- polydactyly: 70% (tends to be post-axial)
- rockerbottom feet
- clenched hands +/- overlapping digits
- abdominal wall abnormalities
- bladder exstrophy
- omphalocoele
- genitourinary anomalies
- cryptorchidism
- cystic renal dysplasia
Associated symptoms and findings may vary in range and severity from case to case. However, Trisomy 13 Syndrome is often characterized by craniofacial, neurologic, heart (cardiac), and/or other defects.
Affected infants typically are unusually small and have feeding difficulties. Various craniofacial malformations are frequently present, such as an abnormally small head (microcephaly) and a sloping forehead; unusual wideness of the soft spots (fontanelles) at the front and back of the skull; incomplete closure of the roof of the mouth (palate); a small jaw; scalp ulceration at the top of the head; and/or low-set, malformed ears. Other characteristics may include a short neck; loose skin folds over the back of the neck; and/or the presence of a benign lesion or birthmark consisting of abnormal clusters of blood vessels (capillary hemangiomas), most frequently on the center of the forehead.
In addition, eye (ocular) abnormalities may include unusually small eyes (microphthalmia); partial absence of ocular tissue from the iris (iris coloboma); abnormal development of the retina (retinal dysplasia); vertical skin folds over the inner corners of the eyes (epicanthal folds); and/or other ocular defects. In addition, the eyebrows may be sparse or absent.
Trisomy 13 Syndrome is also frequently characterized by variable degrees of holoprosencephaly, a condition in which the forebrain fails to divide properly during embryonic development. In those with Trisomy 13 Syndrome, holoprosencephaly may result in various associated, midline facial defects, including closely set eyes (hypotelorism); an abnormal groove in the middle and side of the upper lip (median and lateral cleft lip); abnormalities of the nose; and/or other features. Associated cyclopia has occurred infrequently, characterized by fusion of the eye cavities (orbits) into a single cavity containing one eye.
Affected infants may also have additional abnormalities of the central nervous system (i.e., brain and spinal cord). Holoprosencephaly may be associated with episodes characterized by temporary cessation of spontaneous breathing (apnea) or sudden uncontrolled electrical activity in the brain (seizures). Many infants are thought to be deaf, and profound mental retardation is usually present. In addition, in some cases, additional features may include abnormal tone of voluntary (skeletal) muscles; absence of the band of nerve fibers that joins the two hemispheres of the brain (agenesis of the corpus callosum); underdevelopment of the cerebellum (cerebellar hypoplasia); hydrocephalus; and/or myelomeningocele. Hydrocephalus is a condition in which obstructed flow or impaired absorption of cerebrospinal fluid (CSF) results in an abnormal accumulation of CSF in the skull, usually under increased pressure. CSF is the protective fluid that circulates through the cavities (ventricles) of the brain, the canal containing the spinal cord (spinal canal), and the space between layers of the protective membranes (meninges) surrounding the brain and spinal cord (i.e., subarachnoid space). Myelomeningocele is characterized by protrusion of a membranous sac containing a portion of the spinal cord, its meninges, and CSF through a defect in the spinal column.
About 80 percent of infants with Trisomy 13 Syndrome also have congenital heart defects, such as atrial or ventricular septal defects or patent ductus arteriosus (PDA). In infants with PDA, the channel that is present between the pulmonary artery and the aorta during fetal development fails to close after birth. (The pulmonary artery carries oxygen-depleted blood from the right ventricle to the lungs, where the exchange of oxygen and carbon dioxide occurs. The aorta, the major artery of the body, arises from the left ventricle and supplies oxygen-rich blood to most arteries.) In some cases, other defects may be present involving the pulmonary artery and aorta, certain heart valves, and/or heart chambers. In addition, the heart may be located in the right side of the chest, instead of its normal location in the left side of the chest (dextrocardia).
Kidney (renal) defects may also occur. These may include multiple cysts in the kidneys; abnormal union of the two kidneys at the base (horseshoe kidney); and/or swelling of the kidneys with urine due to blockage or narrowing of the ureters (hydronephrosis), which carry urine into the bladder. Abnormalities of the genitals are also associated with Trisomy 13 Syndrome, including undescended testes (cryptorchidism) and an abnormally formed scrotum in affected males and underdeveloped ovaries and malformed uterus (bicornuate uterus) in affected females.
Infants with Trisomy 13 Syndrome also frequently have certain abnormalities of the hands and feet. These may include more than the normal number of fingers and/or toes (polydactyly); abnormal bending (flexion) and possible overlapping of fingers; and unusually rounded (hyperconvex) nails. The heels of the feet may be abnormally prominent. In addition, Trisomy 13 Syndrome may be associated with abnormal skin ridge patterns (dermatoglyphics), including a single deep crease across the palms of the hands (simian crease).
In some cases, other abnormalities may also be present. Such features may include thin ribs, an underdeveloped pelvis, certain muscle abnormalities, hernias, abnormal development of the pancreas, and/or other anomalies.
Related disorders
Symptoms of the following disorders may be similar to those of Trisomy 13 Syndrome. Comparisons may be useful for a differential diagnosis:
Pseudo-trisomy 13 Syndrome is a rare disorder characterized by holoprosencephaly; associated midline facial abnormalities; extra fingers and/or toes (polydactyly); and/or heart defects, such as atrial or ventricular septal defects. In some cases, additional abnormalities may also be present, including genital defects; absence of the band of nerve fibers joining the two hemispheres of the brain (agenesis of corpus callosum); hydrocephalus; and/or other features. Although symptoms and findings are similar to those potentially associated with Trisomy 13 Syndrome, infants with this disorder do not have an extra chromosome 13 and their chromosomal studies appear normal. Evidence suggests that this disorder may be inherited as an autosomal recessive trait.
There are a number of other disorders, including other chromosomal syndromes, that may be characterized by symptoms and findings similar to those associated with Trisomy 13 Syndrome. Chromosomal testing is necessary to confirm whether a specific chromosomal abnormality is present.
Trisomy 13 causes
Patau’s syndrome happens by chance and isn’t caused by anything the parents have done.
Most cases of the syndrome don’t run in families (they’re not inherited). They occur randomly during conception, when the sperm and egg combine and the foetus starts to develop.
An error occurs when the cells divide, resulting in an additional copy – or part of a copy – of chromosome 13, which severely affects the baby’s development in the womb.
In many cases, the baby dies before reaching full term (miscarriage) or is dead at birth (stillbirth).
In most cases of Patau’s syndrome (75 to 90%), a baby has a whole extra copy of chromosome number 13 in their body’s cells. This is sometimes known as trisomy 13 or simple trisomy 13. The extra genetic material disrupts the normal course of development, causing the characteristic features of trisomy 13 (Patau syndrome).
In 5 to 10% of cases of Patau’s syndrome, genetic material is rearranged (translocated) between chromosome 13 and another chromosome during the formation of reproductive cells (eggs and sperm) or very early in fetal development. This is called a chromosomal translocation. Affected people have two normal copies of chromosome 13, plus an extra copy of chromosome 13 attached to another chromosome. In rare cases, only part of chromosome 13 is present in three copies. The physical signs and symptoms in these cases may be different than those found in full trisomy 13.
In a further 5% of cases with trisomy 13 have an extra copy of chromosome 13 in only some of the body’s cells. In these people, the condition is called mosaic trisomy 13. The severity of mosaic trisomy 13 depends on the type and number of cells that have the extra chromosome. Occasionally, only part of one chromosome 13 is extra (partial trisomy 13). The physical features of mosaic trisomy 13 and partial trisomy 13 are often milder than those of full trisomy 13, resulting in more babies living longer.
In most individuals with Trisomy 13 Syndrome, duplication of chromosome 13 is caused by spontaneous (de novo) errors during the division of reproductive cells in one of the parents (e.g., nondisjunction during meiosis). Evidence suggests that the risk of such errors may increase with advanced parental age. In cases in which only a percentage of cells contains the trisomy 13 abnormality (mosaicism), errors may also occur during cellular division after fertilization (mitosis).
In about 20 percent of affected individuals, trisomy 13 results from a translocation involving chromosome 13 and another chromosome. Translocations occur when regions of certain chromosomes break off and are rearranged, resulting in shifting of genetic material and an altered set of chromosomes. For most individuals with Trisomy 13 Syndrome, such translocations occur spontaneously for unknown reasons (de novo); less commonly, they are transmitted by a parent who is a carrier of a “balanced” translocation. (If a chromosomal rearrangement is balanced–i.e., consists of an altered but balanced set of chromosomes–it is usually harmless to the carrier. However, balanced translocations are sometimes associated with a higher risk of abnormal chromosomal development in the carrier’s offspring. Chromosomal testing may determine whether a parent has a balanced translocation.)
Investigators suggest that certain symptoms and findings associated with Trisomy 13 Syndrome may result from overexpression of developmentally important genes on chromosome 13. For example, the gene that regulates production of an enzyme known as esterase D (ESD) has been located on the long arm (q) of chromosome 13 (13q14.11). Elevated levels of esterase D have been found in the kidney tissues of some affected infants. Further investigations are required to learn more about the specific underlying causes of Trisomy 13 Syndrome and the potential role of esterase D.
Trisomy 13 diagnosis
In some instances, a diagnosis of Trisomy 13 Syndrome may be suggested before birth (prenatally) by specialized tests, such as fetal ultrasonography, amniocentesis, and/or chorionic villus sampling (CVS). During fetal ultrasonography, reflected sound waves create an image of the developing fetus, potentially revealing findings that may suggest a chromosomal disorder or other abnormalities. For example, ultrasound findings that may be suggestive of Trisomy 13 may include holoprosencephaly, polydactyly, and growth retardation.
During amniocentesis, a sample of fluid that surrounds the developing fetus is removed and analyzed, while CVS involves the removal of tissue samples from a portion of the placenta. Chromosomal studies performed on such samples may reveal the presence of an extra chromosome 13.
The diagnosis of Trisomy 13 Syndrome may be made or confirmed after birth (postnatally) by a thorough clinical evaluation, detection of characteristic physical findings, and chromosomal analysis. Testing may also reveal unusual persistence of embryonic and/or fetal hemoglobin in the blood of newborns and infants with Trisomy 13 Syndrome. (Hemoglobin is the oxygen-carrying component of red blood cells.)
For infants diagnosed with the syndrome, careful monitoring and various specialized tests may be conducted to ensure early detection and appropriate management of conditions potentially associated with Trisomy 13 Syndrome.
Screening for Patau syndrome
You’ll be offered a screening test for Patau syndrome – as well as Down syndrome (trisomy 21) and Edwards syndrome (trisomy 18) – from 10 to 14 weeks of pregnancy. The test assesses your chances of having a baby with these syndromes.
The screening test offered at 10 to 14 weeks of pregnancy is called the combined test because it involves a blood test and an ultrasound scan.
If the screening tests show that you have a higher risk of having a baby with Patau’s syndrome, you’ll be offered a diagnostic test to find out for certain whether your baby has the syndrome.
This test will check your baby’s chromosomes in a sample of cells taken from him or her.
Two techniques can be used to obtain the cell sample – amniocentesis or chorionic villus sampling (CVS).
These are invasive tests to remove a sample of tissue or fluid so it can be tested for the presence of the extra copy of chromosome 13.
A newer test has recently been developed where a sample of blood from the mother is taken so that the baby’s DNA found within it can be tested. This is known as non-invasive prenatal testing, and is only available privately.
If you’re not able to have the combined screening test, you’ll be offered a scan that looks for physical abnormalities, including those found in Patau’s syndrome.
This is sometimes called the mid-pregnancy scan and is carried out when you’re between 18 and 21 weeks pregnant.
Genetic testing for parents
Both parents will need to have their chromosomes analyzed if their baby is affected by Patau syndrome caused by a chromosomal translocation.
Genetic testing is carried out to help parents plan for future pregnancies, rather than as part of the decision making process for the current pregnancy.
The test results will allow a more accurate assessment to be made of the likelihood of the syndrome affecting future pregnancies.
Other family members may also be affected and should be tested.
Patau syndrome treatment
There’s no specific treatment for Patau’s syndrome. Treatment varies from child to child and depends on the specific symptoms. As a result of the severe health problems a newborn baby with the syndrome will have, doctors usually focus on minimizing discomfort and ensuring the baby is able to feed.
The treatment of Trisomy 13 Syndrome is directed toward the specific symptoms that are apparent in each individual. Such treatment may require the coordinated efforts of a multidisciplinary team of medical professionals.
In some cases, recommended treatment may include surgical correction of certain abnormalities associated with the disorder. The surgical procedures performed will depend upon the nature and severity of the anatomical abnormalities, their associated symptoms, and other factors.
A supportive team approach for children with this disorder may be of benefit and may include physical therapy, medical, and/or social services. Genetic counseling will also be of benefit for families of children with Trisomy 13 Syndrome. Other treatment for this disorder is symptomatic and supportive.
The organizations listed below are useful further sources of information about Patau’s syndrome and can also provide advice and support:
- Support Organization for Trisomy 18, 13 and Related Disorders (SOFT): https://trisomy.org/
- Hope for Trisomy 13 and 18: https://www.hopefortrisomy13and18.org/
- Trisomy 13. https://ghr.nlm.nih.gov/condition/trisomy-13[↩]
- Witters G, Van Robays J, Willekes C, et al. Trisomy 13, 18, 21, Triploidy and Turner syndrome: the 5T’s. Look at the hands. Facts, Views & Vision in ObGyn. 2011;3(1):15-21. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3991414/[↩]
- Entezami M, Albig M, Knoll U et-al. Ultrasound Diagnosis of Fetal Anomalies. Thieme. (2003) ISBN:1588902129[↩][↩]
- Persistent Stapedial Artery: CT Findings. American Journal of Neuroradiology Sep 2000, 21 (8) 1551-1554 http://www.ajnr.org/content/21/8/1551.long[↩]
- Retinal Dysplasia Mimicking Intraocular Tumor: MR Imaging Findings with Histopathologic Correlation. American Journal of Neuroradiology Oct 2007, 28 (9) 1731-1733; DOI: 10.3174/ajnr.A0635 http://www.ajnr.org/content/28/9/1731.long[↩]
- Nyberg DA, McGahan JP, Pretorius DH. Diagnostic imaging of fetal anomalies. Lippincott Williams & Wilkins. (2003) ISBN:0781732115[↩]
- Papp C, Beke A, Ban Z et-al. Prenatal diagnosis of trisomy 13: analysis of 28 cases. J Ultrasound Med. 2006 Apr;25(4):429-35. https://www.ncbi.nlm.nih.gov/pubmed/16567430[↩]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}