
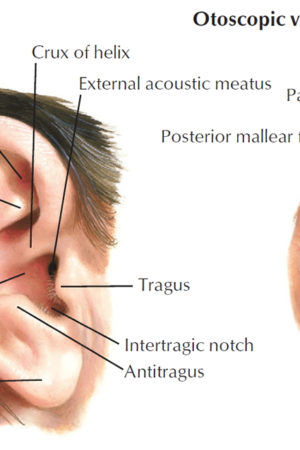
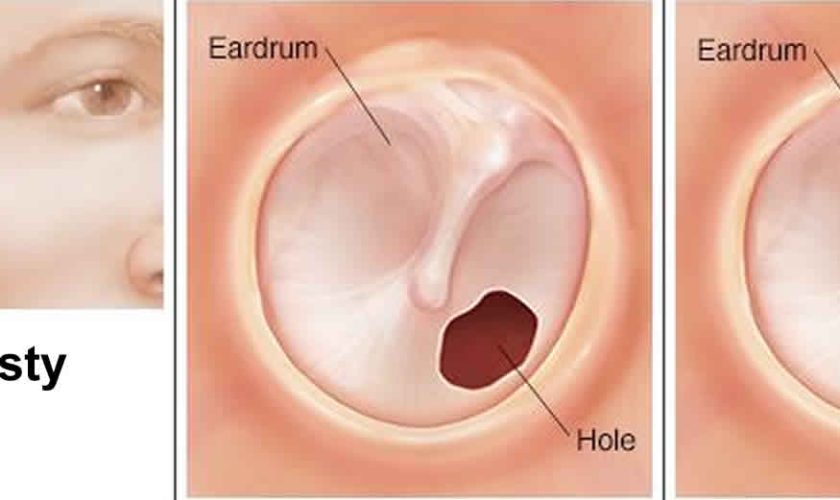
Myringoplasty
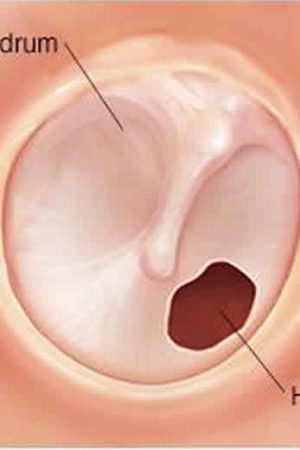
Myringoplasty also called tympanoplasty is microsurgical technique to reconstruct a ruptured or perforated eardrum (tympanic membrane) with the placement of a graft, either medial or lateral to the tympanic membrane annulus, often using the patient’s own tissues. The goal of this surgical procedure is not only to close the perforation but also to improve hearing. The success of the operation depends on the ability to eradicate disease from the middle ear (eg, inflamed granulation tissue and cholesteatoma). Various techniques have been developed and refined, and a number of grafting materials are available. Both the lateral and medial grafting techniques are detailed below.
Myringoplasty can be used for small perforations, such as nonhealing tympanic membranes after pressure-equalizing tube extrusion or traumatic perforations. The technique involves freshening the edges of the perforation to promote healing and placing a carefully trimmed graft lateral to the defect 1. Grafting materials for myringoplasty include fat, Gelfilm, Gelfoam, AlloDerm, and cigarette paper. Gelfoam can also be placed as packing in the middle ear to support the graft.
Myringoplasty is a safe and effective outpatient procedure used to both eradicate disease from the middle ear and restore hearing and middle ear function 2. Your child will need to stay in the hospital overnight. A number of surgical approaches and grafting techniques are available for use by the surgeon. Paramount to success are the preoperative assessment, good hemostasis intraoperatively, and thoughtful surgical planning with careful placement of the graft.
When planning myringoplasty, the surgeon must consider the location of the perforation (marginal versus central), and size (total versus subtotal). Areas of myringosclerosis and tympanosclerosis should be noted. Important comorbidities worth noting include craniofacial disorders and underlying environmental allergies or chronic allergic rhinitis. Critical factors that make myringoplasty less successful include adhesive otitis media, severe eustachian tube dysfunction with either perforation of the contralateral ear or ongoing intermittent otorrhea, cholesteatoma, and previous surgical repair 3.
Myringoplasty key points
- A myringoplasty is an operation to fix a hole in the eardrum.
- The operation usually takes about two to three hours.
- Your child will sleep and feel no pain during the operation.
- After the operation, your child will have to stay overnight in the hospital.
- While your child gets better at home, there are some things your child should not do.
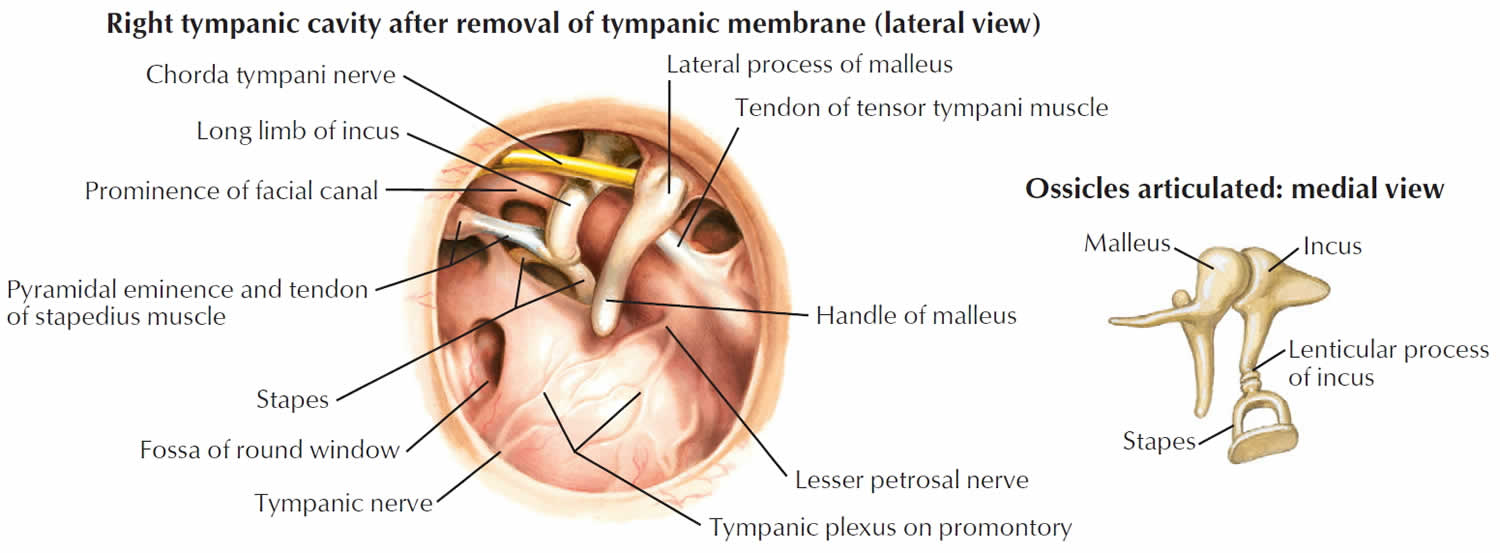
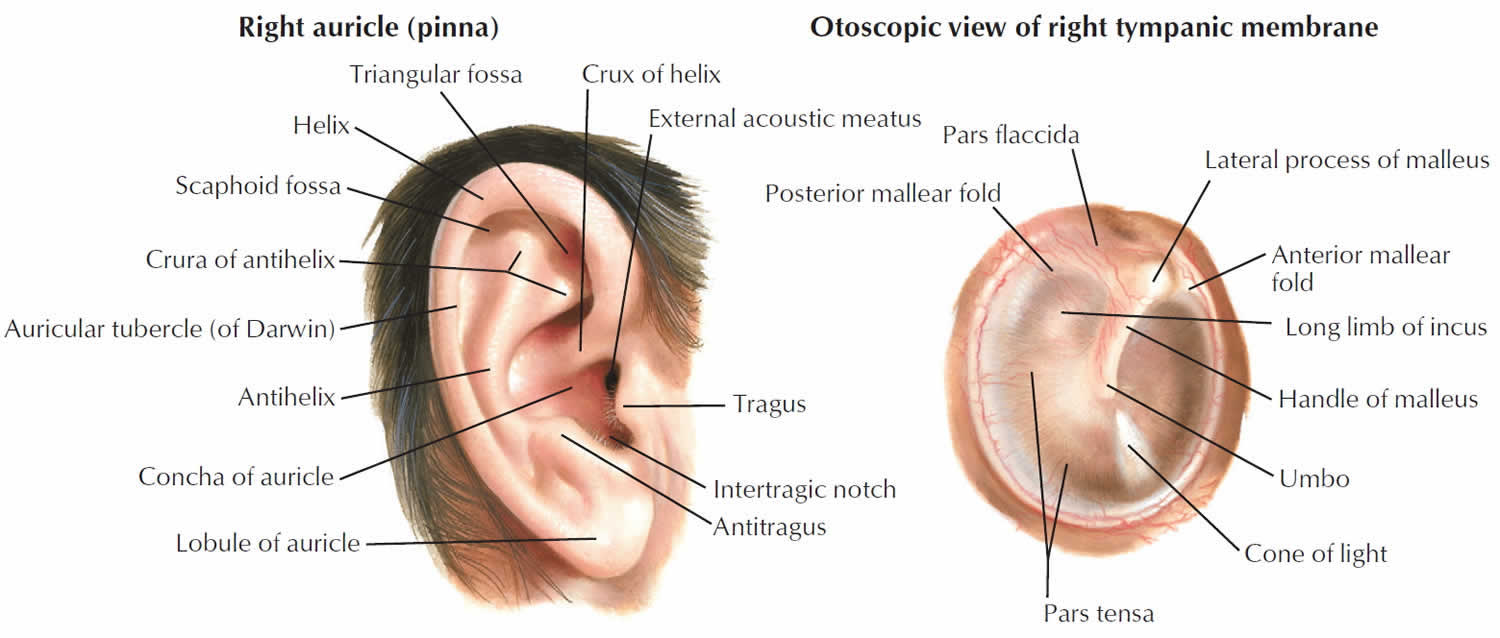
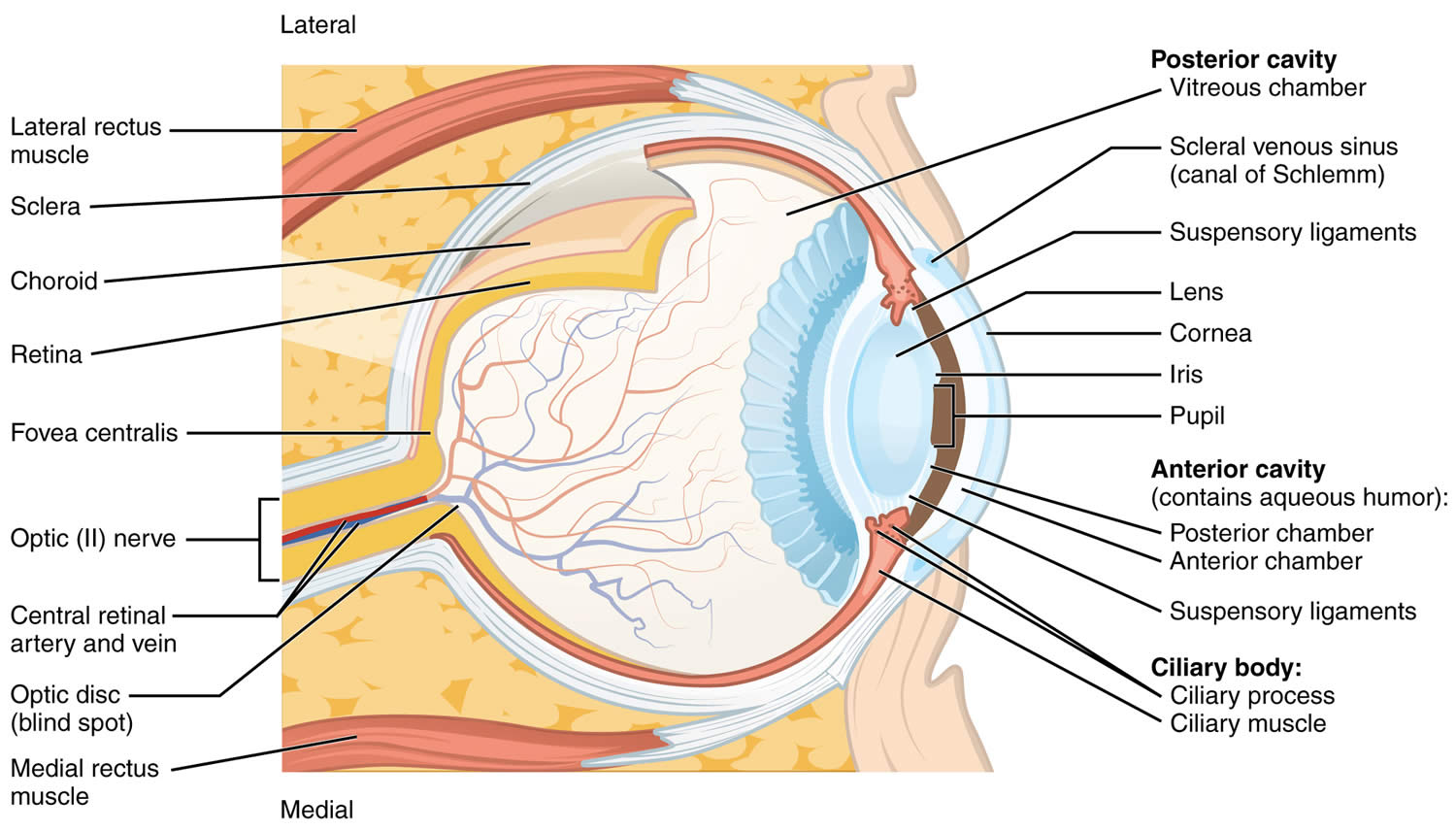
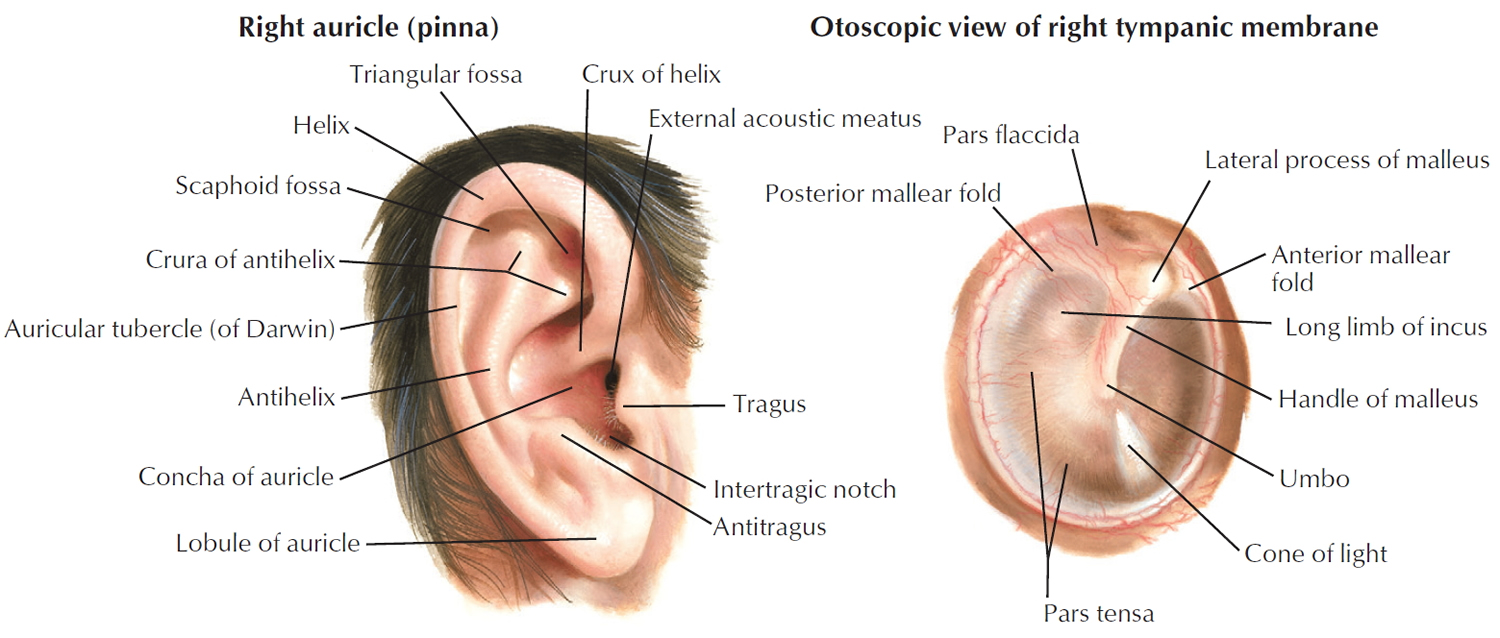
Tympanic membrane anatomy
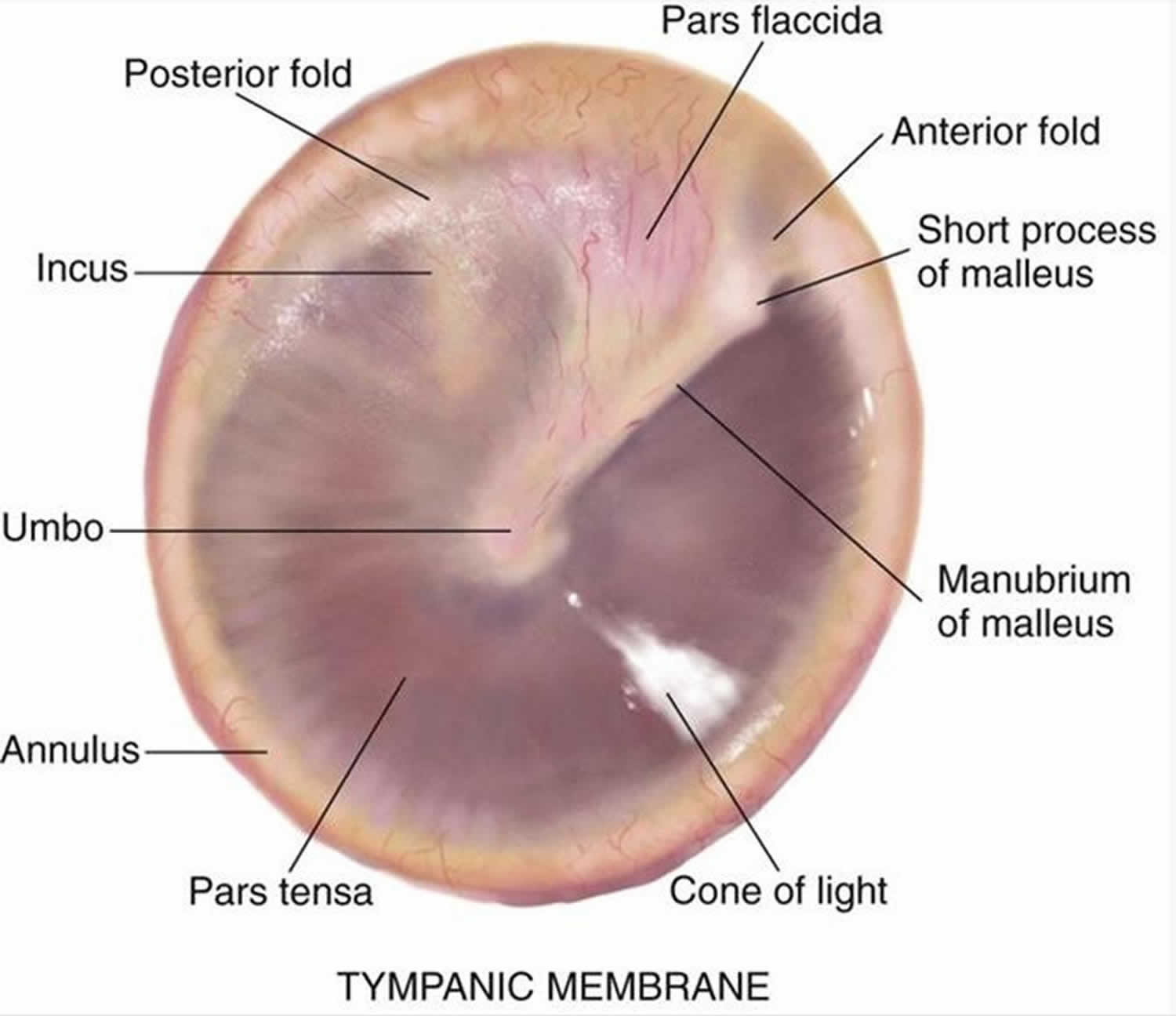
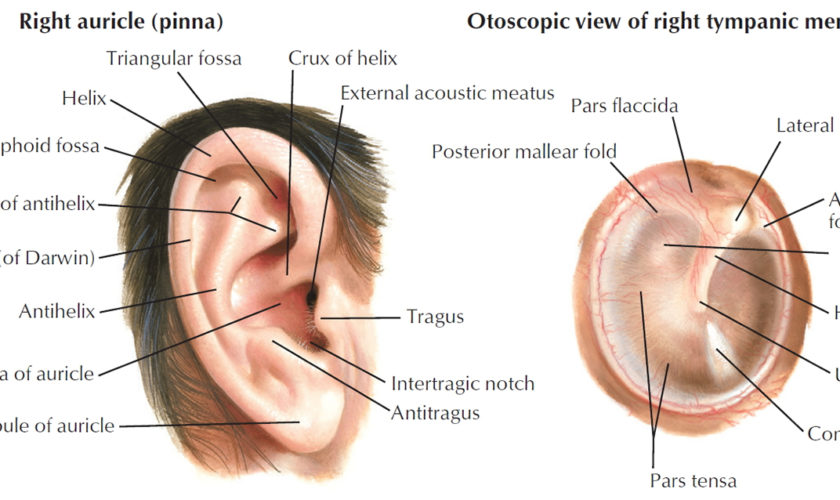
The eardrum also called tympanic membrane is a thin layer of tissue that vibrates in response to sound. An understanding of the tympanic membrane anatomy is critical to successful repair. Myringoplasty (tympanoplasty) procedure mandates an understanding of the layers. The tympanic membrane typically consists of the following 3 layers:
- Lateral epithelial layer
- Middle fibrous layer
- Medial mucosal layer
The outer epithelial layer is composed of stratified squamous epithelium, which is continuous with the skin of the external auditory canal. This is significant because in-growth of this outer epithelial portion through the perforation can result in an epithelial cyst called an acquired cholesteatoma. Untreated, this cyst then releases destructive enzymes that can enlarge the size of the perforation and ultimately cause ossicular erosion. The lateral grafting technique that is discussed later in this text requires that this entire epithelial layer be stripped from the drum remnant prior to placement of the graft so as to avoid iatrogenic cholesteatoma formation.
The middle fibrous layer is composed of connective tissue consisting of outer radial fibers and inner circular fibers. It provides strength to the drum. A healed perforation is also commonly deficient of this middle fibrous layer. The epithelial and endothelial layers regenerate creating a “dimeric” membrane. This miscalculation can be corrected when carefully examined under binocular microscopy. Because this middle layer is absent in the pars flaccida superiorly, the posterior-superior aspect of the drum can be drawn inward toward the middle ear as a retraction pocket.
The inner layer of the tympanic membrane consists of simple cuboidal and columnar epithelium cells. This layer is identical to the mucosal lining of the rest of the middle ear mucosal tissue and is considered to be critical to ensure healing of tympanic membrane perforations, and the surgeon often abrades or rasps the undersurface of the tympanic membrane remnant to stimulate regrowth.
Annulus
The peripheral edge of the tympanic membrane is rimmed by a dense fibrous layer called the annulus, which is essentially a thickening of the pars tensa. Successful elevation of the annulus is critical for medial grafting technique. The annulus is deficient superiorly at the “12 o’clock” location. This area is the notch of Rivinus and can guide the surgeon to a natural plane to elevate the annulus.
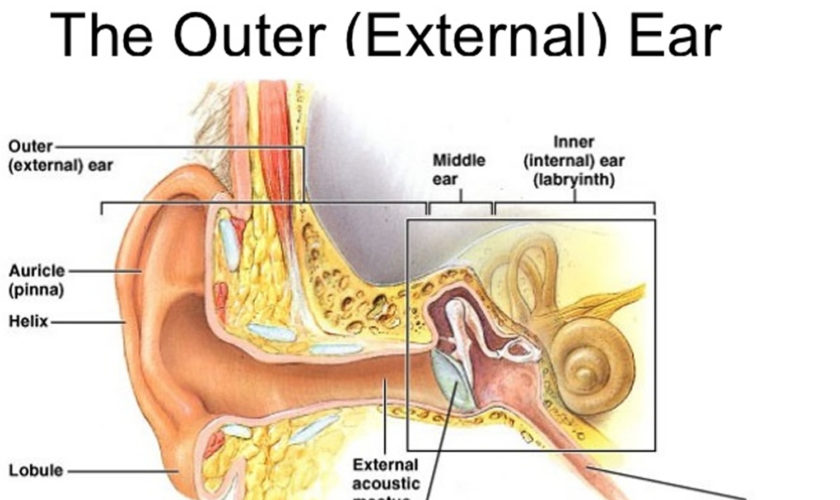
Ear canal
The ear canal has bone in the medial component (inner one-third). The lateral portion, which extends into the pinna, is composed of cartilage. The boney/cartilaginous interface is located at the medial two-thirds junction. Most incisions that are made to raise a tympanomeatal flap or perform either an endaural or transcanal approaches are made at this location as well. The superiorly placed vascular strip is another critical area within the ear canal. This region is demarcated by the tympanosquamous suture line superiorly and the tympanomastoid junction line inferiorly. Canal incisions are often made along these junctions.
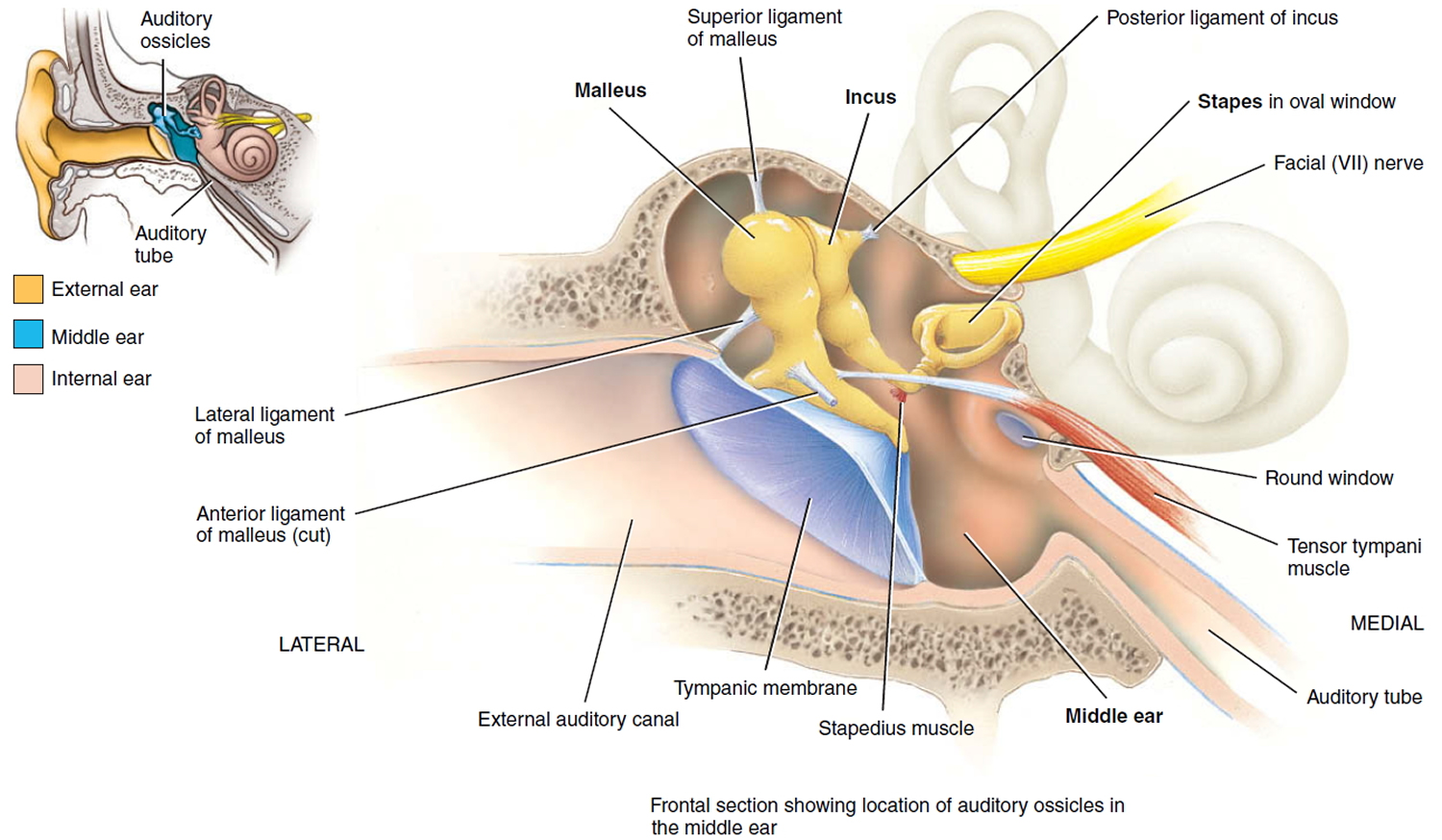
Middle ear
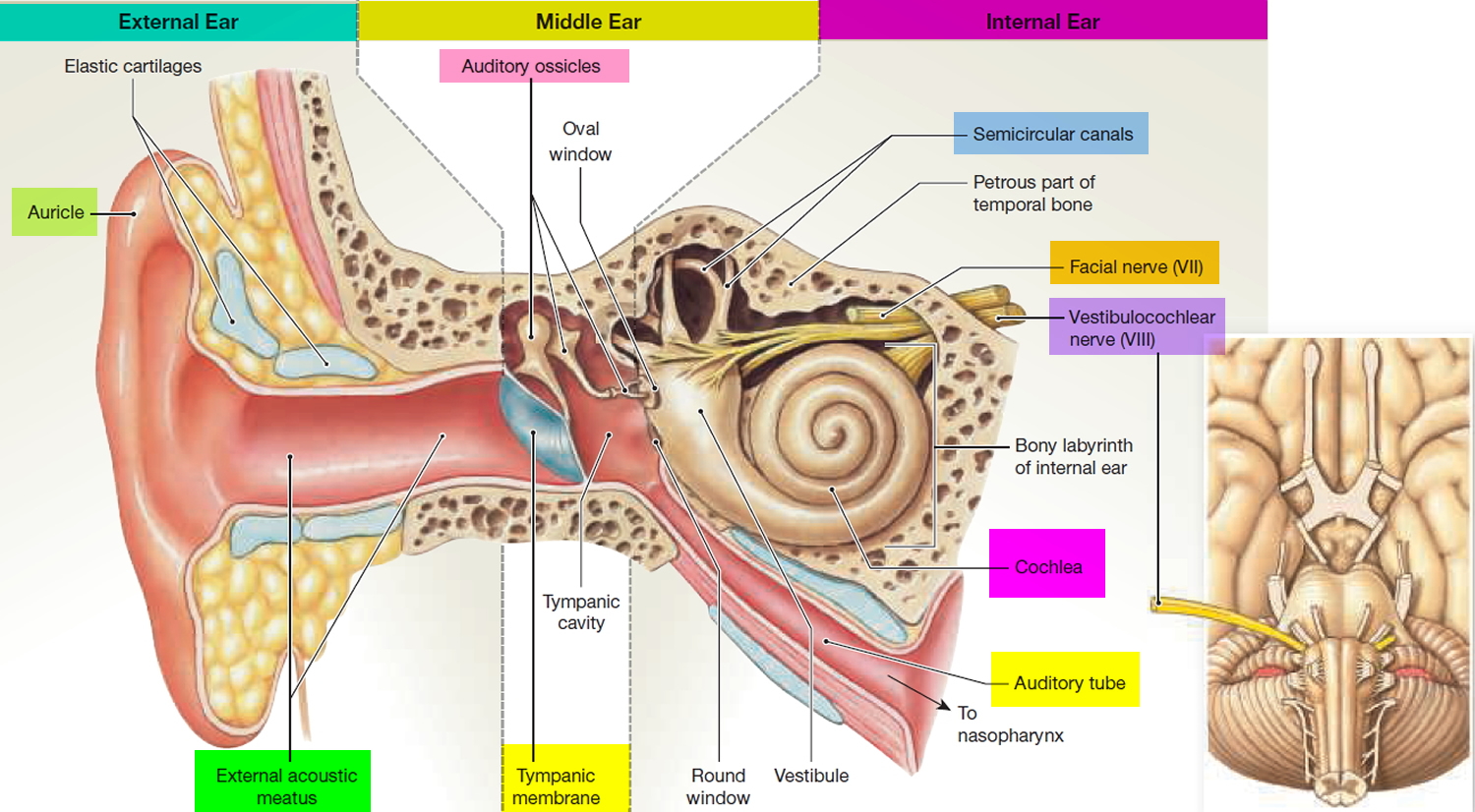
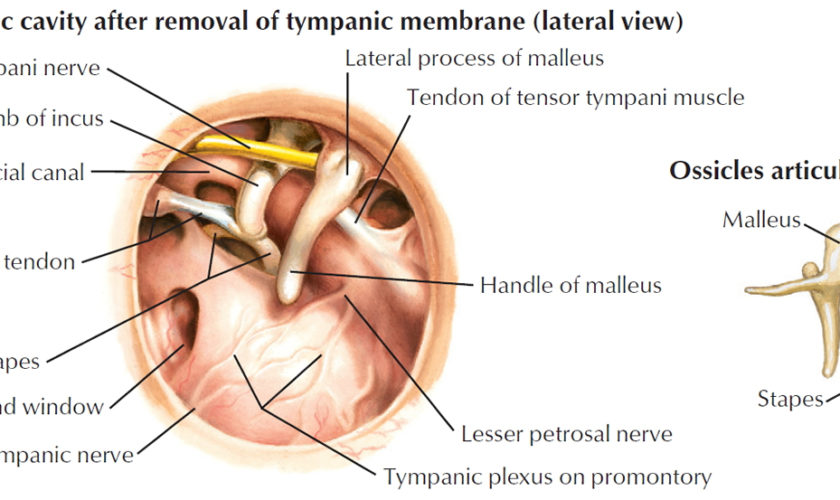
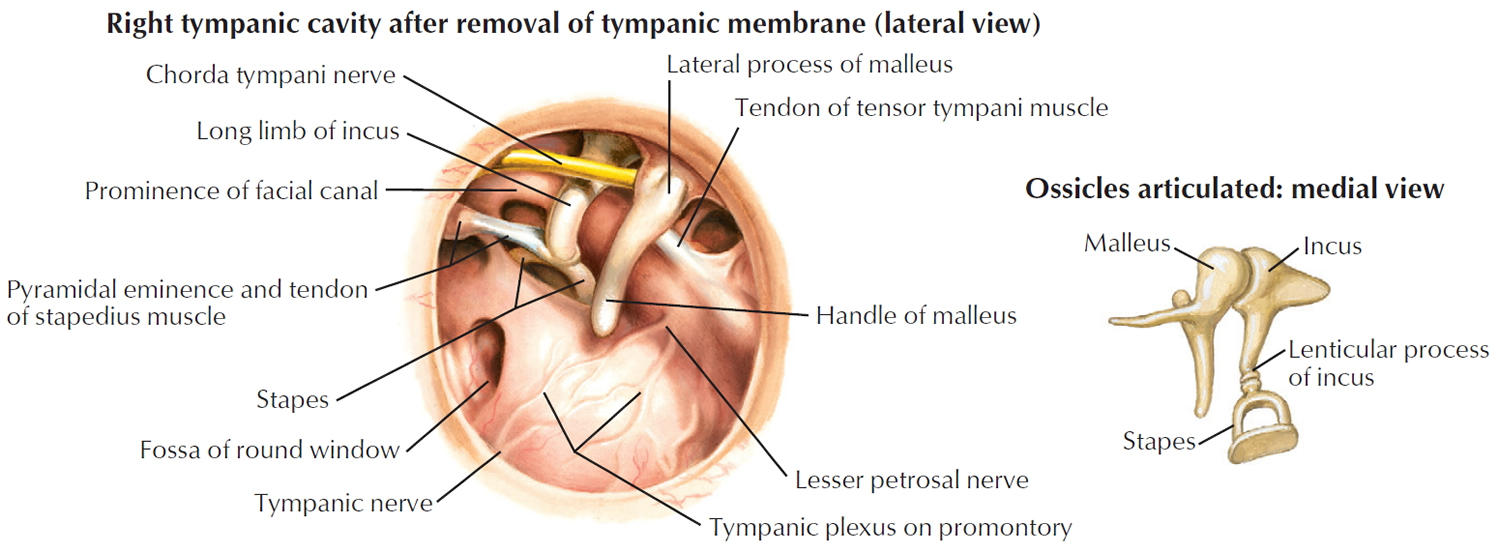
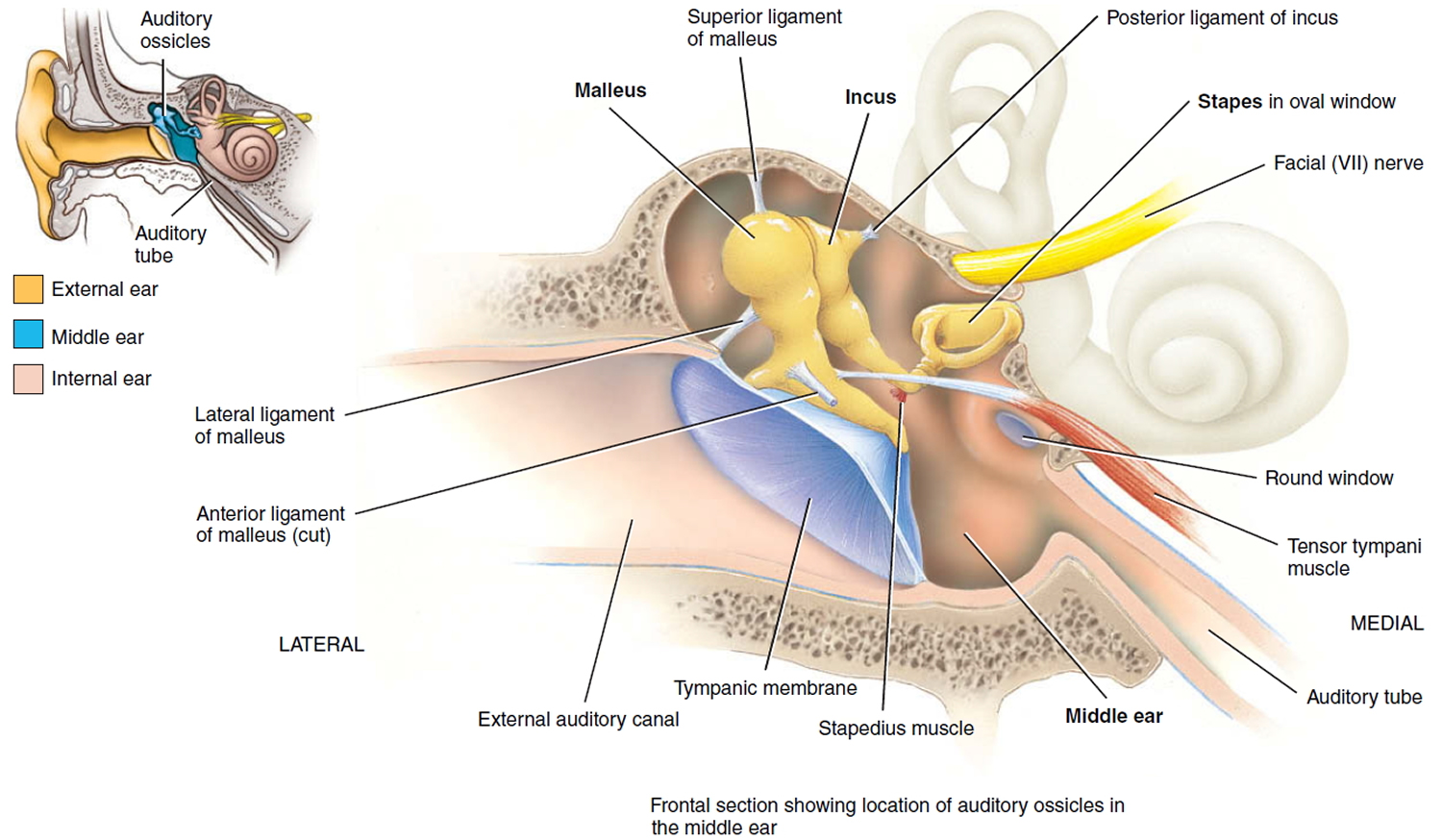
The middle ear is an air-filled space bordered by the bony labyrinth of the inner ear medially, the tympanic membrane laterally, and the cranium superiorly. This space contains the ossicles, nerves (facial nerve, chorda tympani, Jacobsen nerve), small muscles (stapedius and tensor tympani), ligaments, and blood vessels. The petrous portion of the internal carotid artery and the internal jugular vein, which are both in proximity to the middle ear space, can be dehiscent and should be noted on any preoperative imaging. Rarely, middle ear pathology can involve these structures.
In order for successful grafting of the tympanic membrane to improve hearing, an intact ossicular chain must be present. The malleus transmits energy from the tympanic membrane to the incus, which itself is connected to the stapes superstructure resting on the oval window. Diarthrodial joints connect the 3 ossicles and allow the transmission of acoustic energy from the tympanic membrane to the inner ear. The incudostapedial joint is the most fragile and, hence, has the highest likelihood to require repair.
Mastoid
The middle ear communicates with the mastoid air cells via the mastoid antrum. The temporal bone air cells are usually pneumatized by 3 years of age. However, the air cells can remain underdeveloped and sclerotic in patients with persistent eustachian tube dysfunction. A poorly pneumatized or fluid-filled mastoid bone predisposes a patient to require a more extensive tympanomastoidectomy to improve the chances of successful graft placement.
Eustachian tube
The eustachian tube connects the middle ear with the nasopharynx and allows pressure equilibration in the middle ear. Enlarged adenoids or biofilms within this lymphoid tissue are hypothesized to predispose a patient to persistent middle ear disease. This bony-cartilaginous tube is approximately 45° from the horizontal in adults but only 10° from horizontal in infants. In addition, the infant eustachian tube is about 50% of the adult length.
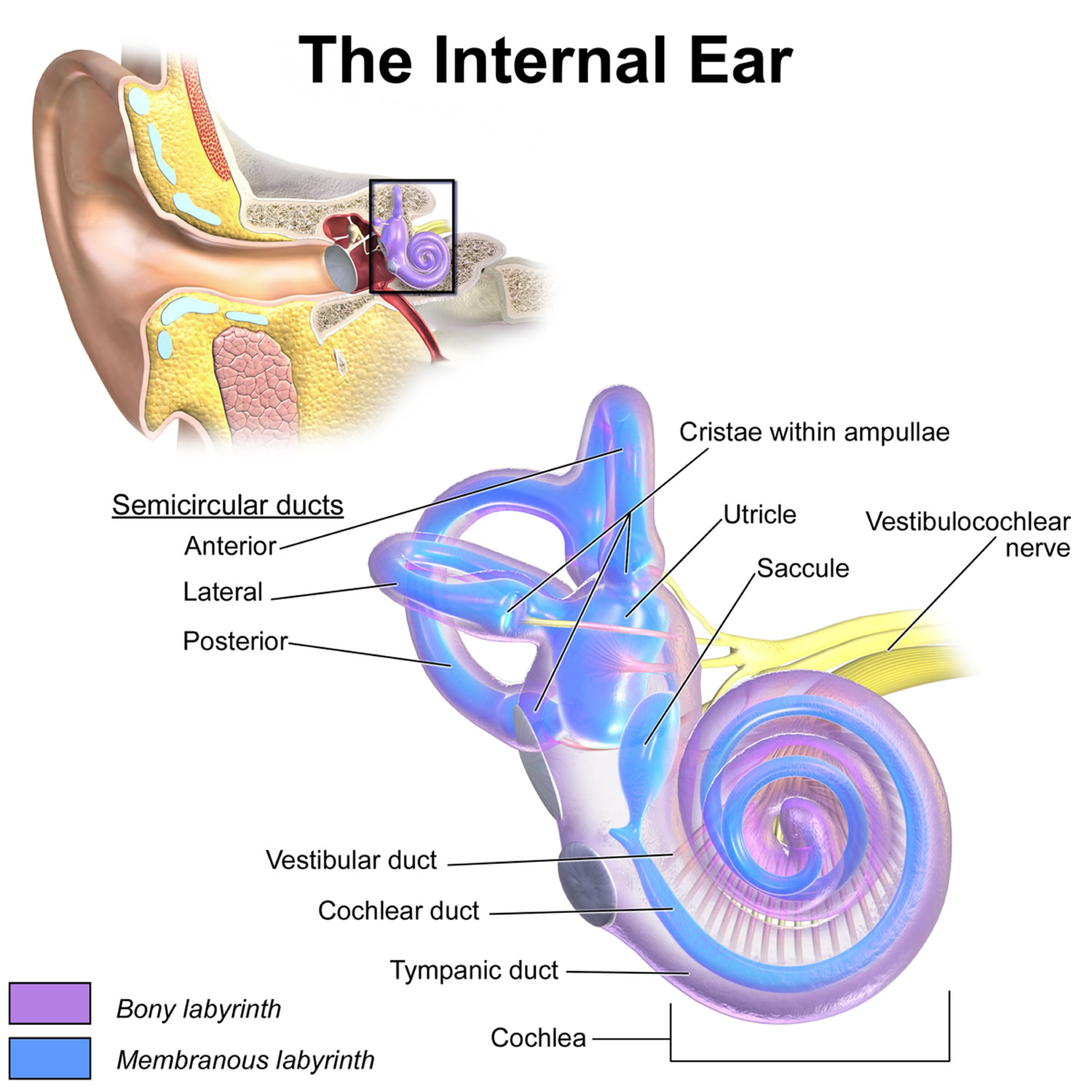
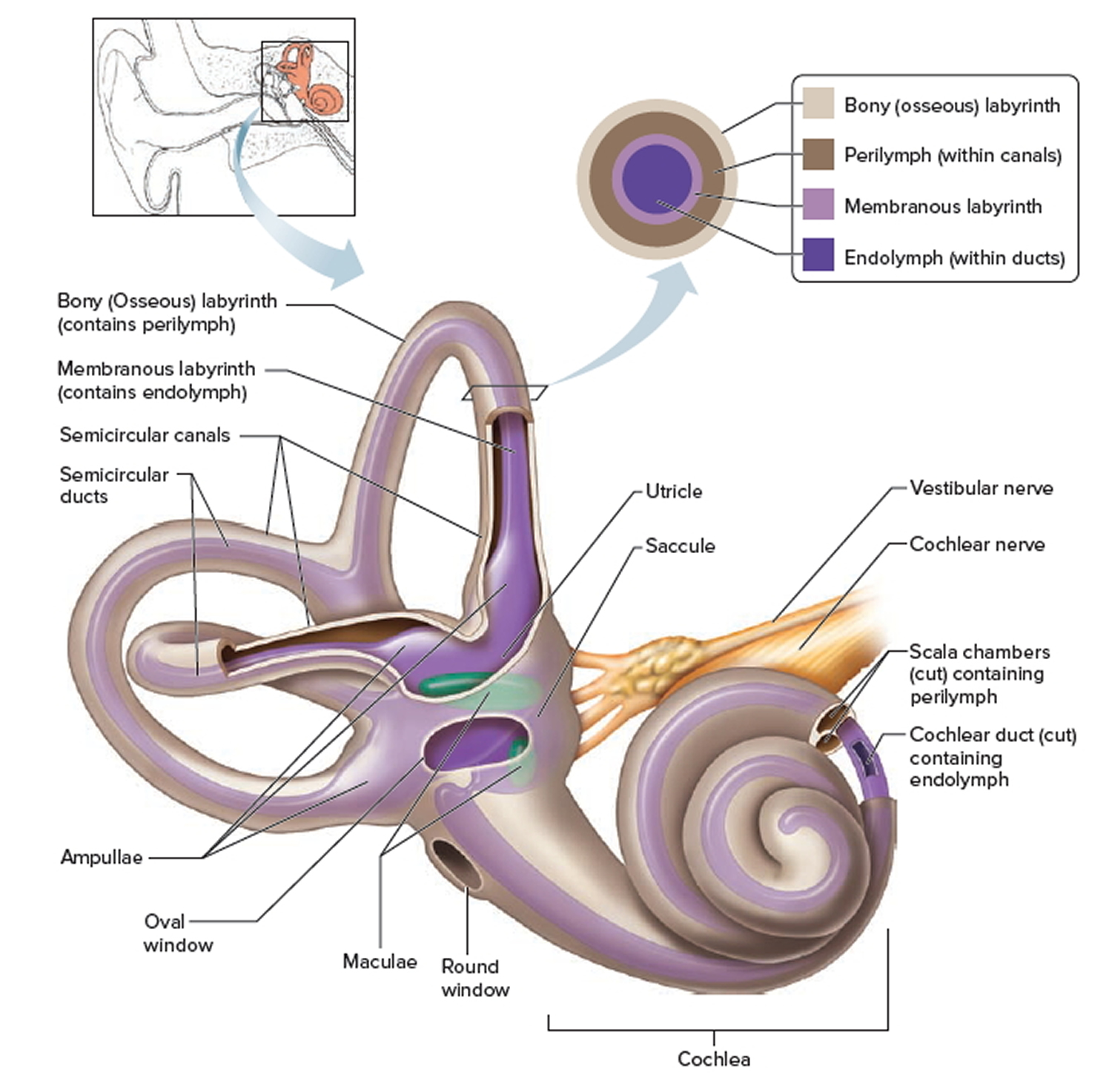
Inner ear
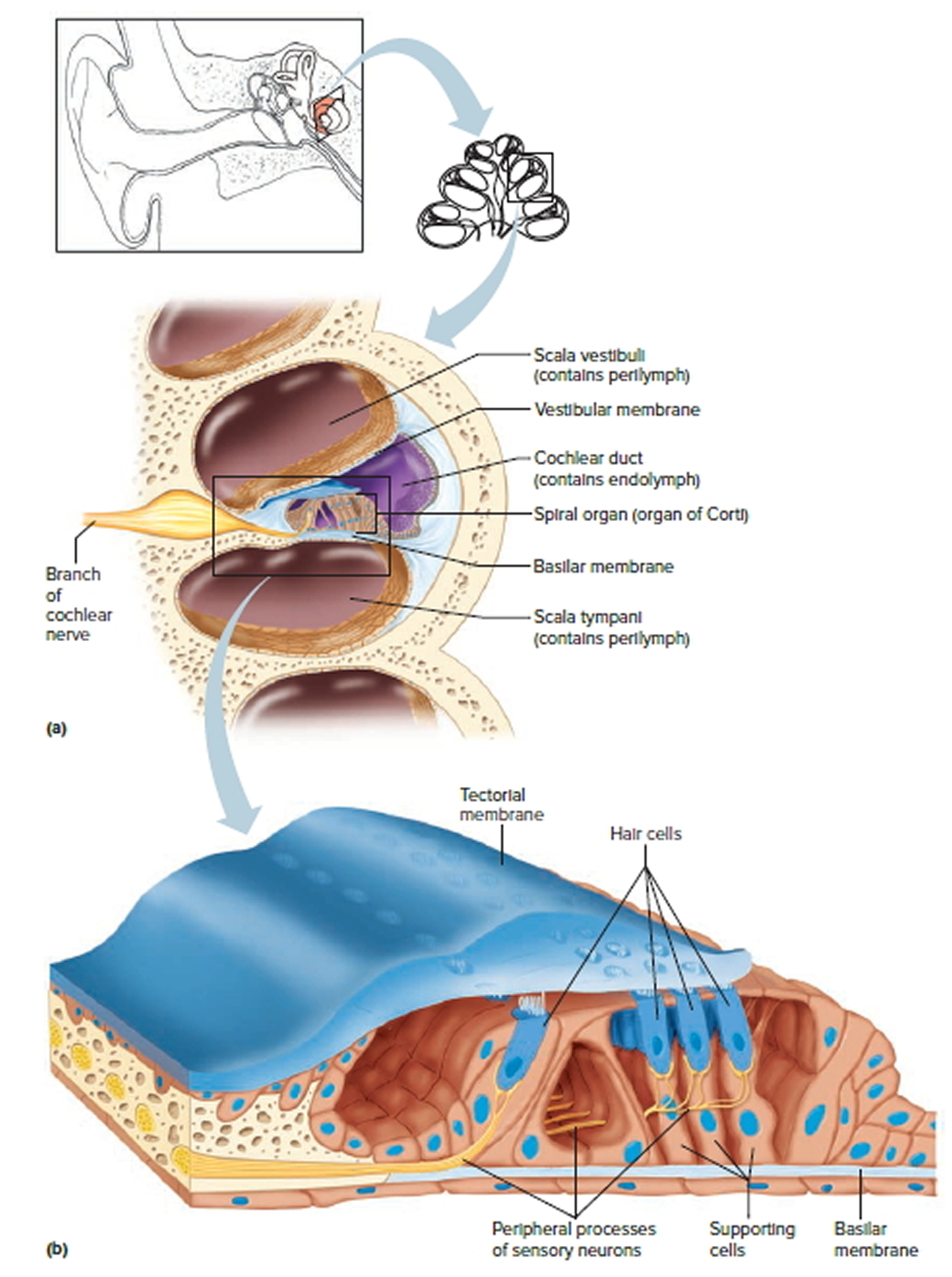
The inner ear is composed of the cochlea, which is the end-organ for hearing, and the vestibular organs. The vestibular organs include the utricle, saccule, and the 3 semicircular canals and are involved in balance.
Figure 1. Ear anatomy
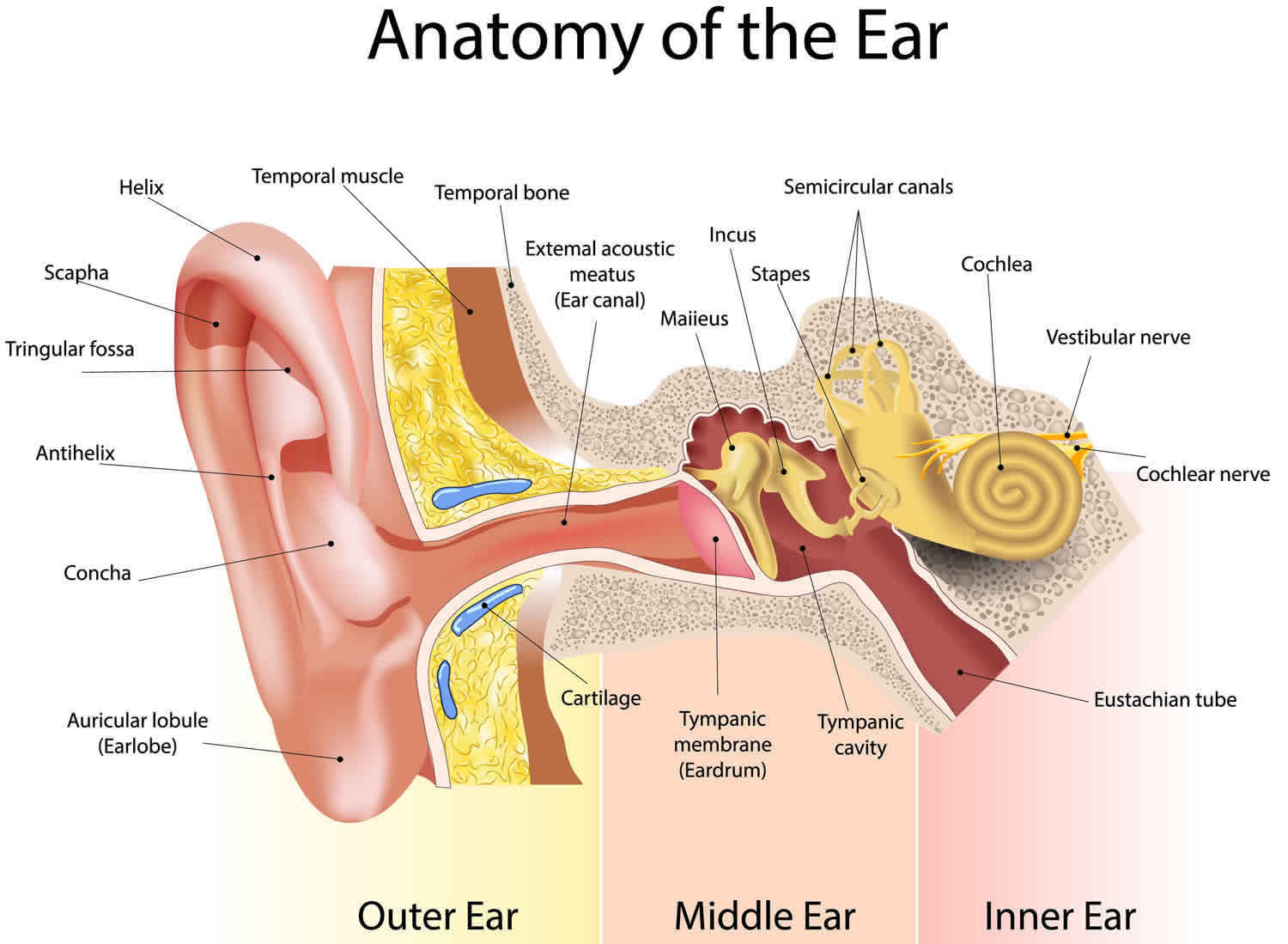
Figure 2. Tympanic membrane anatomy (right ear)
Myringoplasty surgery
Myringoplasty (tympanoplasty) is an outpatient procedure for adults and for most children. The operation takes about two to three hours. Your child will need to stay in the hospital overnight.
Your child will sleep and feel no pain during the operation. Just before your child has the operation, they will be given a sleep medicine. This is called a general anesthetic. This means that your child will sleep and feel no pain during the operation.
The ear nose and throat (ENT) doctor will take a tiny piece of tissue from an area around the ear. This is done by making a cut behind your child’s ear. The piece of ear tissue is then used to fix the hole in your child’s eardrum. Your child will have dissolvable stitches behind the ear and gauze packing in the ear to absorb any fluid.
Various techniques and grafting materials can be used. Which approach is used depends on the size and location of the perforation, the presence or absence of cholesteatoma or granulation tissue, the status of the ossicles and mastoid, other anatomical considerations (eg, narrow external auditory canals), as well as the surgeon’s preference and expertise 4.
Examining the middle ear and ossicles and removing any elements of adhesions or cholesteatoma is critical. The chosen approach should provide optimal visualization of the perforation and tympanic membrane. One should be careful not to disrupt an intact and mobile ossicular chain if the hearing loss is only low-frequency conductive, as is often the case with hearing loss secondary to a perforation 5.
Before the operation
Evaluation of the patient for myringoplasty involves a detailed history and physical examination. Important aspects of the history include the duration of the perforation, severity of otalgia, otorrhea, hearing loss, vertigo, previous attempts at repair, otitis media, and otitis externa. The number and frequency of infections (including time of most recent infection) provide insight into the severity of disease. Past otologic surgical history is critical and should include any history of tympanostomy tubes (also called myringotomy tubes, ventilation tubes, or pressure equalization (PE) tubes) and details of any prior tympanoplasties (including approaches, grafts used, outcomes).
Prior to considering surgery in any patient, acute and chronic infections should be controlled using ototopical, oral, and/or intravenous antibiotics or antifungals, if indicated. Ototopical drops with steroids may also be needed if granulation tissue or aural polyps are visualized to improve inflammation. Ideally, an ear should be “dry” for 3-4 months before surgery is performed to enhance the chance of success. Individuals who undergo surgery must keep the operated ear dry for a period of several weeks or until the graft has healed. Operating on an actively infected ear is contraindicated.
Physical examination
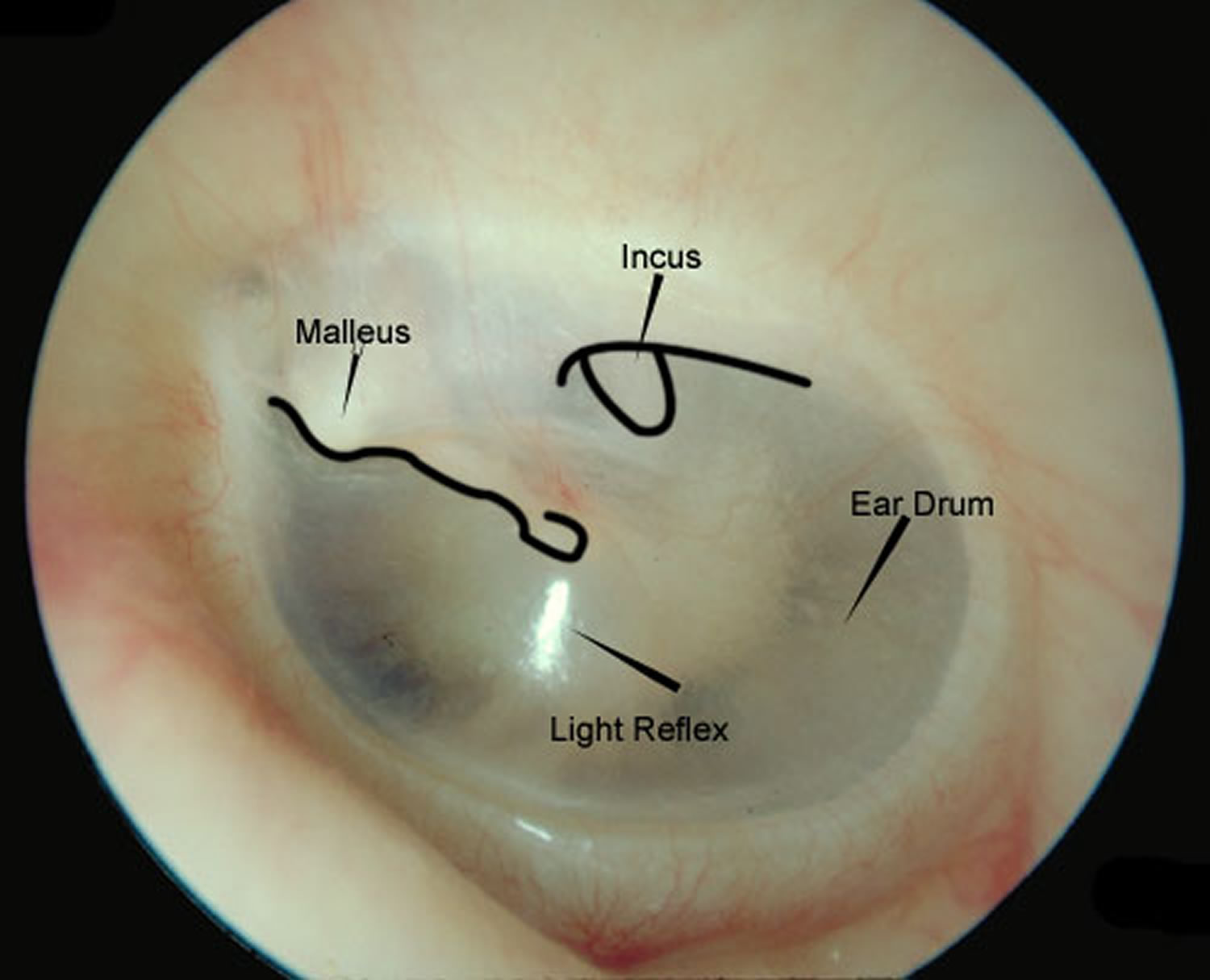
The physical examination should focus on pneumatic otoscopy and otomicroscopy. In the office, tuning forks can give an important assessment of hearing; in older patients, tuning forks can be used to confirm audiometric findings. Additional assessments include documentation of facial nerve functionality and inspection for previous incisions. Although a small amount of cerumen is tolerated in the routine otoscopic examination, obstructive cerumen should be removed when evaluating a patient for myringoplasty in order to provide an unimpeded view of the entire tympanic membrane.
Monomeric (or more accurately, dimeric) areas may appear as perforations until inspected more closely under microscopy. Retraction pockets should be closely inspected for accumulation of squamous debris. Considering the status of the contralateral ear when considering repair of a tympanic membrane perforation is essential. The ear with more significant hearing loss is usually operated on first if bilateral perforations exist.
Audiometric testing
Audiometry should be performed preoperatively in all surgical candidates. Tympanometry can add useful information in younger children who are difficult to properly examine. The primary reason for audiometric analysis is to establish the degree of conductive hearing loss. Perforations usually cause low-frequency conductive loss 6. If underlying ossicular discontinuity exists and is not addressed during surgery, then postoperative hearing can be worse despite an intact neo-tympanum. One should consider ossicular involvement if the conductive hearing loss is flat across all frequencies or greater than 35 db. Finally, the presence and degree of any sensorineural hearing loss should be documented preoperatively.
Radiographic testing
Computed tomography (CT) and magnetic resonance imaging (MRI) is not essential but may be indicated in patients in whom a concern for cochlear, labyrinthine, or intracranial pathology exists. Other patients who might be considered for preoperative imaging include patients with a history of facial palsy, children with craniofacial abnormalities, and revision cases in which the anatomy may be distorted.
Pre-operative imaging assists the surgeon in preoperatively identifying pathology and planning surgery. CT scan should be ordered when concern exists for cholesteatoma and in patients with previous mastoid surgery, otalgia, or vertigo. MRI is beneficial for delineating the integrity of the dura as well as detecting small retrocochlear lesions such as acoustic neuromas.
Grafting materials
Various materials exist for use for tympanic membrane grafting. True temporalis fascia is the most common graft because of its ease of harvest and its abundant availability, even in revision cases. Some surgeons prefers loose areolar fascia (also known as “fool’s fascia”) and prefer to save the true fascia for revision cases. Also, the “fool’s fascia” is considered by some to be more pliable, have less donor site morbidity, and to be more transparent after healing. It is available via the same postauricular incision that can be used for tympanoplasty, or a separate incision can be made in or beyond the postauricular hairline if a transcanal or endaural technique is used. A mild amount of donor site morbidity occurs, with postoperative pain over the temporalis muscle being the most common symptom.
- The postauricular incision is marked and injected with lidocaine with epinephrine.
- Dissection is carried down onto the fascia (loose areolar /true temporalis).
- The graft is harvested.
- Muscle is removed from the fascia graft, and the graft is then set on the back table for later use.
Cartilage is available to be harvested easily from either the tragus or the conchal bowl, if a post-auricular approach is being used. Tragal cartilage is harvested with perichondrium attached via a small incision on the internal surface of the tragus 7. This graft is an appropriate size and carries very little donor site morbidity. In addition, the perichondrium can be reflected to stabilize the graft. Conchal cartilage also carries no additional significant morbidity. Other grafting materials include lobular fat, periosteum, perichondrium, vein, and AlloDerm.
Myringoplasty procedure
Transcanal approach
The transcanal approach is especially good for small posterior perforations, but can be used for medium-sized perforations if the anterior tympanic membrane is easily visualized. This technique can be challenging for significant anterior perforations, narrow/stenotic ear canals, or individuals with a significant anterior canal bulge. Inspecting the perforation prior to preparing the patient and determining that at least a 5-mm speculum can be placed is important. Canalplasty can be used to improve visualization if slightly limited.
Medial graft
When performing a transcanal medial tympanoplasty procedure, the following steps are followed:
- The ear canal is suctioned and surgical Betadine used during the surgical prep is removed.
- The external auditory canal is cleaned and injected with 1% lidocaine with 1:100,000 epinephrine or 0.5% lidocaine with 1:200,000 epinephrine, primarily for vasoconstriction to optimize visualization during the procedure.
- The edge of the perforation is dissected and removed using a sharp pick and cup forceps; this “postage-stamping” and “freshening” of the perforation is critical to ensure that the graft incorporates into the native tympanic membrane remnant.
- Next, a tympanomeatal flap is created. It is customized based on the location of the perforation and surgeon’s preference. The flap design should be such that it can be easily and atraumatically raised and the undersurface of tympanic membrane perforation can be readily accessed. A medially-based tympanomeatal flap is usually created with radial incisions at 12 o’clock and 6 o’clock (ie, superiorly and inferiorly) that either connect directly or via a semilunar incision in the posterior canal just medial to the bony-cartilaginous junction.
- The tympanomeatal flap is raised medially with a round knife. To avoid traumatic tearing, take great care not to suction on the flap.
- When the annulus is reached, the tympanotomy is made such that the instrument of choice (eg, round knife, gimmick, sickle knife, pick) lifts the annulus while hugging the bony groove from which the fibrous annulus can be dissected. The fibrous annulus is then dissected circumferentially with care not to injure the ossicles, the chorda tympani nerve, or residual drum. The flap is then positioned, usually anteriorly, such that the perforation is exposed.
- A canalplasty of the posterior or anterior external auditory canal can be performed to optimize visualization. Take care not to injure the facial nerve or temporomandibular joint.
- If indicated, the middle ear and ossicles are inspected and palpated to confirm ossicular continuity. Middle ear disease (granulation tissue, tympanosclerosis, adhesions, cholesteatoma) is completely removed. Removing hypertrophic middle ear mucosa with either a McCabe dissector or Duckbill elevator, particularly mucosa abutting the fibrous annulus in anterior tympanic membrane perforations, is important.
- Ossicular reconstruction can be performed if necessary, followed by grafting of the perforation. Elevating the tympanic membrane remnant off the long process of the malleus with a sickle knife may be necessary. This allows both closer inspection of the ossicles and better placement of the graft.
- The middle ear must be carefully packed with the surgeon’s preferred material – either Gelfilm, Gelfoam, or Surgicel. This is often soaked in either oxymetazoline, antibiotic ear drops, or diluted epinephrine (1:10,000). Packing the mesotympanum and hypotympanum is important, although excess packing should be avoided near the ossicles so as to prevent adhesions.
- The graft is trimmed on the back table. The graft should adequately cover the entire defect. Hemostasis is critical to intraoperative visualization and successful placement of the graft. The graft should be well supported so as to avoid shifting or displacement.
- Some surgeons advocate that nitrous oxide anesthetic be switched off at this point because this particular agent has a tendency to accumulate in spaces such as the middle ear and can potentially dislodge the graft.
- The tympanomeatal flap is laid back down over the graft, and the posterior canal skin edges are laid flat. Pieces of Gelfoam, Surgicel, or antibiotic ointment are placed along the tympanic membrane and graft and layered laterally to cover the canal incisions.
- Antibiotic ointment is placed in the lateral canal, and either Vaseline-coated or antibiotic-coated sculpted cotton ball is placed in the external auditory meatus.
- An optional Glasscock or mastoid pressure dressing is placed at the end of the case, particularly if a mastoidectomy has been performed.
Endaural approach
The endaural technique is useful with many perforations, especially when a small atticotomy is anticipated (when improved access to and visualization of the epitympanum is needed). Many of the steps involved in the transcanal technique are similarly performed in the endaural tympanoplasty as well. When performing an endaural medial tympanoplasty procedure, the following steps are followed:
- The canal is prepared as detailed above, but the injection may continue laterally to the lateral external auditory canal and tragus.
- An incision is made at 12 o’clock and extended superolaterally between the tragus and helical root.
- A medially-based tympanomeatal flap is raised, and the middle ear is entered with the same care as described previously. The tympanic membrane is freed superiorly and inferiorly.
- Middle ear work is carried out as indicated. Atticotomy can be performed using a small curette or drill if access into the epitympanum is needed.
- Grafting is performed and the tympanomeatal flap is laid back down. Gelfoam is placed along the tympanic membrane and fills the canal. Antibiotic ointment and a cotton ball are used laterally.
Postauricular approach
The postauricular technique is the most commonly performed approach for either revision tympanoplasties or those in which a mastoidectomy is anticipated. This technique offers the best visualization of the anterior tympanic membrane and is preferred for large anterior perforations. In addition, it can be combined with mastoidectomy if disease is found in the mastoid that requires the surgeon’s attention. A basic outline of the procedure follows:
- The canal is prepared in a similar fashion to the transcanal technique.
- Radial and horizontal canal incisions are made as described previously, and the canal is packed with cotton soaked in oxymetazoline or epinephrine.
- A postauricular incision is marked 5 mm posterior to the auricular crease in a curvilinear fashion, extending form the mastoid tip to the temporal line. The incision is injected with 1% lidocaine with either 1:100,000 epinephrine or 0.5% lidocaine with 1:200,000 epinephrine. The incision is carried down through the skin and subcutaneous tissue with care not to enter the ear canal. When the temporalis fascia is reached, a graft can be harvested using a Freer elevator and scissors. If multiple previous grafts have been harvested, either tissue from the contralateral ear or AlloDerm can be used as well.
- A periosteal incision is made in a “T” or “7” fashion, and the periosteum is raised into the lateral ear canal until the previously-made canal incisions are reached. The cotton in the ear canal is removed.
- A rubber Penrose drain can be inserted to retract the lateral canal and auricle anteriorly. Self-retaining retractors such as Weitlaner or Perkins retractors are used to provide further exposure. The perforation is visualized and prepared.
- The tympanomeatal flap is raised medially and the middle ear is entered as described previously. Do not suction on the flap.
- Canalplasty can be performed and middle ear work is carried out as indicated, including ossicular reconstruction. The perforation is grafted, and the tympanomeatal flap is laid back down with Gelfoam layered lateral to the tympanic membrane.
- The auricle and lateral ear canal are relaxed and the postauricular incision is closed in a layered fashion. The remainder of the ear canal is packed with Gelfoam and antibiotic ointment. A pressure dressing is applied to prevent a postauricular hematoma.
Grafting technique
Although variations exist, 2 primary grafting techniques exist: medial grafting (or underlay) and lateral grafting (or overlay). These terms refer to the position of the graft in relation to the fibrous annulus, not to the malleus or tympanic remnant.
The medial grafting technique is performed as described previously. The primary advantage of the medial graft technique is that it is quicker and easier to perform than lateral grafting. It also carries a high success rate (approximately 90% in experienced hands). The biggest disadvantage is its limited exposure and poor utility for larger perforations and its difficulty with repair of near-total perforations.
Advantages of the lateral graft technique include wide exposure and versatility for larger perforations and for any needed ossicular reconstruction. Disadvantages include the requirement of a higher technical skill level, a longer operative time, slower healing rate, and the risk of blunting and lateralization of the graft. The lateral graft technique is championed by the some doctors as a technique more suited for total drum replacement. The basic steps involved in lateral grafting are described as follows:
- Lateral (overlay) tympanoplasty is performed through the previously-described postauricular incision. Important differences exist in the canal incisions. In this procedure, a vascular strip is created by making radial incisions at about 2 o’clock and 5 o’clock. These incisions are connected medially just lateral to the annulus on the posterior canal wall and laterally just medial to the bony-cartilaginous junction along the anterior canal wall.
- The skin of the anterior external auditory canal is raised medially. When the annulus is reached, squamous epithelium is raised off of the tympanic remnant, and the canal skin is removed in continuity with the remnant skin and stored in saline solution. This maneuver is done with a cupped forceps.
- Bony canalplasty can be performed anteriorly to ensure visualization of the entire annulus. Protecting the flap with a portion of trimmed Silastic as a shield is helpful. Care must be taken not to enter the glenoid fossa, which risks injuring the temporomandibular joint (TMJ) and causing prolapse into the ear canal.
- Antibiotic-soaked Gelfoam is packed into the middle ear to support the tympanic remnant. The fascia graft is placed medial to the malleus and draped onto the posterior canal wall for stabilization. If possible, the graft should not extend onto the anterior canal wall in an effort to prevent blunting of the graft.
- The canal skin/tympanic remnant is returned and placed lateral to the graft and carefully positioned. Gelfoam is then packed tightly into the anterior aspect of the medial canal to prevent blunting, and the vascular strip is laid back down, covering the lateral extension of the fascia graft to improve its blood supply. Antibiotic-soaked Gelfoam is then packed into the rest of the external auditory canal.
- The postauricular incision is closed in layers, and antibiotic ointment is placed on the incision and in the lateral canal. A cotton ball is placed in the external auditory meatus, and a mastoid pressure dressing is applied.
Myringoplasty recovery
After the operation, your child’s surgical team will take your child to the recovery room, also called the Post Anesthetic Care Unit (PACU). This is where your child will wake up. Your child will stay in PACU for about one hour. Your child’s surgical team will then move your child to a room on the nursing unit.
Your child’s surgical team will give your child fluids through a tube in their arm, called an IV, until they are able to drink easily. Your child will have a gauze bandage around their head, which will be taken off the day after the operation.
Postoperative hearing should be immediately assessed in the recovery room with a tuning fork. If a pressure dressing is applied, it should be removed on the first or second postoperative day depending on surgeon preference.
Although the ear must be kept otherwise dry, patients are allowed to wash their hair, while keeping a cotton ball with Vaseline in the canal for dry ear measures. Pain is usually managed with acetaminophen and/or ibuprofen. Narcotics such as hydrocodone (Vicodin or Norco) are usually prescribed when a post-auricular approach is used. Oral antibiotics are the surgeon’s preference and can be given for 5-7 days. A cotton ball is replaced in the external auditory meatus as needed for bleeding or drainage. Ototopical drops are typically administered postoperatively for 7-21 days after surgery and continued until the first postoperative visit.
At the first postoperative visit (3-4 weeks after surgery), the ear is examined under the microscope, and any canal packing or residual antibiotic is removed. At this time, a good assessment can be made as to the healing and neovascularization of the graft. Granulation tissue at the tympanomeatal flap is addressed. Ototopical drops are continued as the graft continues to heal. Postoperative audiometric testing is delayed until healing is complete (typically 6-12 weeks). Follow-up visits are scheduled to ensure complete proper healing and restoration of hearing.
Medications that may be prescribed after surgery
- Pain control: Acetaminophen (Tylenol) liquid solution may be given. Some children will requireprescription pain medication. Pain may be worse during evening; some children should be given medication at night.
- NO ibuprofen (Motrin or Advil) or aspirin for twoweeks after surgeryunless otherwiseinstructed by physician.
- Antibiotic eardrops may be prescribed twoweeks after surgery. Give drops at room temperature.
- Antibiotics may be prescribed for 7 to 10 days.
Call your child’s ENT doctor, your family doctor or your local medical clinic right away if your child has any of these signs after going home:
- Fever greater than 101º F (38.5 °C)
- Severe ear pain, pain that gets worse or pain noted more than 7 days after surgery
- Excessive drainage or blood leaking from the ear
- Swelling, redness or drainage from incision site
- Dizziness that lasts for more than one week
- Vomiting (throwing up) that does not stop
- The packing falls out of the ear
If it is an emergency or if you are concerned about your child’s condition, do not wait. Take your child to the closest emergency department.
Special precautions after ear surgery
- No nose blowing for two weeks. Sneeze with an open mouth.
- Water precautions: Keep ear canal dry for the first twoweeks;place cotton ball coated with Vaseline in the ear(s) when bathing. Hair may be washed twodays after surgery. The sutures may get wet but the ear canal should stay dry. No swimming for usually 4 to 6 weeks. The physician will advise you when the ear can get wet.
- Wound and suture line care: A large dressing is usually applied after surgery and should be left in place for one-twodays. After the dressing is removed (at your appointment or at home as instructed by your doctor), clean the incisionwith hydrogen peroxide and apply bacitracin ointment. Use Q-tips or cotton balls to clean the incision. Wash your hands before and after cleaning the incision. Apply a cotton ball to the outside of the ear canal if drainage is present.
- Keep the incision protected from the sun for 6 to 12 months, keep covered or apply sunscreen.
Taking care of your child at home
Please follow these steps at home to help your child get better:
- Your child may have a small gauze bandage over their ear. Please keep this bandage on for one or two days after going home.
- Do not let the cut behind your child’s ear get wet. Do not get any water in the ear. Your child can have a bath, but take care not to pull on the ear or get it wet if you need to wash their hair.
- Do not let your child play contact sports like hockey or soccer until the ENT doctor says it is OK.
- Do not let your child go swimming until the ENT doctor says it is OK.
- Do not let your child play a musical instrument that you blow in until the ENT doctor says it is OK.
- Do not let your child blow their nose. Have them cough or sneeze with their mouth open.
- Your child may return to school or day care when your ENT doctor says it is OK. Usually, this will be one week after the operation.
Pain management at home
Follow these instructions when your child goes home after the procedure.
You may give your child medicine for pain.
You may receive a prescription for pain medication before you leave the hospital. Follow the dosage instructions given to you by the pharmacist. Although these prescription pain medications can be beneficial, they are also potentially very dangerous if not used properly.
When using these medications, if you notice any changes in either breathing or level of drowsiness that concern you, stop the medication and seek medical attention. If your child is unresponsive, call your local emergency services number immediately.
Do not give your child over-the-counter medicine that may have a sedative effect (makes people sleepy) while giving the prescription for pain medicine. Examples of these medicines are decongestants and antihistamines. Discuss these medications with your pharmacist.
You may give your child acetaminophen if they have pain. Give the dose printed on the bottle for your child’s age. Do not give your child ibuprofen or acetylsalicylic acid for two weeks after the surgery. These medications could increase your child’s risk of bleeding after the operation. Check with the nurse or doctor first before giving these medicines to your child.
Myringoplasty recovery time
Routine activities may be resumed in 2-5 days. Most children return to school in 3 to 5 days if eating and sleeping well and pain-free. Vigorous exercise, heavy lifting and physical activities should be avoided for 2 weeks. No swimming until advised by your doctor, typically in 4-6 weeks.
Follow-up care
A follow-up appointment with the ENT doctor
The ENT unit will make a follow-up appointment with the doctor for your child. If everything is normal during the appointment, the doctor will:
- Check your child’s ear to see how it is healing.
- Take out the packing from your child’s ear.
- Tell you when your child can start to play sports again.
Myringoplasty complications
Common complaints after surgery:
- Nausea and vomiting may occur for the first 24 to 48 hours.
- Pain: Mild to moderate ear pain and/or pain at theincision site for 3 to 5 daysis expected.
- Fever: A low-grade fever may be observed several days.
- Ear drainage after surgery. Packing material is placed in the ear canal; sometimes there is clear, pink, or bloody drainage from the ear for 3-5 days. This may also occurwhen ear drops are started.
- Dizziness or unsteadiness: Dizziness is common for several days.
- Decreased hearing in the operated ear for several weeks.
Complications of the surgery include recurrence of the perforation, tympanic membrane retraction, otorrhea, cholesteatoma development, persistence or worsening of any conductive hearing loss, sensorineural hearing loss (rare), and taste disturbances. Post-auricular incisions are at risk for hematoma, and a mastoid pressure dressing is recommended for the first postoperative night. Outcomes can be optimized by a proper and detailed preoperative assessment and the careful construction of an effective surgical plan.
The graft can fail because of infection, failure to pack the graft securely in place, technical error, failure to clear mastoid and middle ear disease, and because of a concurrent undetected cholesteatoma. Excising all tympanosclerosis at the edge of the perforation so as to allow vascularized perimeters to incorporate the graft is critical.
Myringoplasty outcomes
The indications and outcomes vary depending on the specific clinical problem. Success rates of tympanic membrane closure vary greatly in the literature (35-98%) but are usually greater than 80% and depend largely on the size and location of the perforation, surgical technique, and overall health of the middle ear 8.
- Aggarwal R, Saeed SR, Green KJ. Myringoplasty. J Laryngol Otol. 2006 Jun. 120(6):429-32.[↩]
- Webb B, Chang CYJ. Efficacy of Tympanoplasty without mastoidectomy for Chronic Superative Otitis Media. Arch of Otolaryngol Head and Neck Surg. 2008/11. 1155-1158.[↩]
- Chang CYJ. Chronic Disorders of the Middle Ear and Mastoid (Tympanic Membrane Perforations and Cholesteatoma. Mitchell RB. Pediatric Otolaryngology for the Clinician. New York, NY: Springer; 2009.[↩]
- Wright D, Safranek S. Treatment of otitis media with perforated tympanic membrane. Am Fam Physician. 2009 Apr 15. 79(8):650, 654.[↩]
- Luetje III CM. Reconstruction of the Tympanic Membrane and Ossicular Chain. Bailey BJ. Head & Neck Surgery – Otolaryngology. 4th Edition. Philadelphia, PA: Lippincott Williams & Wilkins; 2006.[↩]
- Wasson JD, Papadimitriou CE, Pau H. Myringoplasty: impact of perforation size on closure and audiological improvement. J Laryngol Otol. 2009 Sep. 123(9):973-7.[↩]
- Dornhoffer JL. Cartilage tympanoplasty. Otolaryngol Clin North Am. 2006 Dec. 39(6):1161-76.[↩]
- Hardman J, Muzaffar J, Nankivell P, Coulson C. Tympanoplasty for Chronic Tympanic Membrane Perforation in Children: Systematic Review and Meta-analysis. Otol Neurotol. 2015 Jun. 36 (5):796-804.[↩]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}