
What is Ehlers Danlos syndrome
Ehlers-Danlos syndrome is a group of genetic collagen disorders that affect connective tissues supporting the skin, bones, blood vessels, and many other organs and tissues throughout your body. Defects in connective tissues cause the signs and symptoms of these conditions, which range from mildly loose joints to life-threatening complications.
The various forms of Ehlers-Danlos syndrome have been classified in several different ways. Originally, 11 forms of Ehlers-Danlos syndrome were named using Roman numerals to indicate the types (type I, type II, and so on). In 1997, researchers proposed a simpler classification (the Villefranche nomenclature) that reduced the number of types to six and gave them descriptive names based on their major features. In 2017, the classification was updated to include rare forms of Ehlers-Danlos syndrome that were discovered more recently. The 2017 classification describes 13 types of Ehlers-Danlos syndrome.
The combined prevalence of all types of Ehlers-Danlos syndrome appears to be at least 1 in 5,000 individuals worldwide. It affects both men and women with no predisposition to race or ethnicity. The hypermobile and classical forms are most common; the hypermobile type may affect as many as 1 in 5,000 to 20,000 people, while the classical type probably occurs in 1 in 20,000 to 40,000 people. Other forms of Ehlers-Danlos syndrome are rare, often with only a few cases or affected families described in the medical literature. There may be a family history – for instance in vascular Ehlers-Danlos syndrome sudden death in a close relative. Progress on the Human Genome Project has provided valuable information regarding the actual genes involved. Ehlers-Danlos syndrome is usually diagnosed in younger patients as typical features such as joint laxity, skin fragility and scarring tendencies are recognisable from early childhood.
An unusually large range of joint movement (hypermobility) occurs in most forms of Ehlers-Danlos syndrome, and it is a hallmark feature of the hypermobile type. Infants and children with hypermobility often have weak muscle tone (hypotonia), which can delay the development of motor skills such as sitting, standing, and walking. The loose joints are unstable and prone to dislocation, chronic pain and early-onset arthritis. In the arthrochalasia type of Ehlers-Danlos syndrome, infants have hypermobility and dislocations of both hips at birth.
Many people with the Ehlers-Danlos syndromes have soft, velvety skin that is highly stretchy (elastic) and fragile. Affected individuals tend to bruise easily, and some types of the condition also cause abnormal scarring. People with the classical form of Ehlers-Danlos syndrome experience wounds that split open with little bleeding and leave scars that widen over time to create characteristic “cigarette paper” scars. The dermatosparaxis type of the disorder is characterized by loose skin that sags and wrinkles, and extra (redundant) folds of skin may be present.
Some forms of Ehlers-Danlos syndrome, notably the vascular type and to a lesser extent the kyphoscoliotic, classical, and classical-like types, can cause unpredictable tearing (rupture) of blood vessels, leading to internal bleeding and other potentially life-threatening complications. The vascular type of Ehlers-Danlos syndrome is also associated with an increased risk of organ rupture, including tearing of the intestine and rupture of the uterus during pregnancy.
Other types of Ehlers-Danlos syndrome have additional signs and symptoms. The cardiac-valvular type causes severe problems with the valves that control the movement of blood through the heart. People with the kyphoscoliotic type experience severe curvature of the spine that worsens over time and can interfere with breathing by restricting lung expansion. A type of Ehlers-Danlos syndrome called brittle cornea syndrome is characterized by thinness of the clear covering of the eye (the cornea) and other eye abnormalities. The spondylodysplastic type features short stature and skeletal abnormalities such as abnormally curved (bowed) limbs. Abnormalities of muscles, including hypotonia and permanently bent joints (contractures), are among the characteristic signs of the musculocontractural and myopathic forms of Ehlers-Danlos syndrome. The periodontal type causes abnormalities of the teeth and gums.
If you have Ehlers-Danlos syndrome, you will need to be careful when doing activities that can put stress on your joints and increase the risk of injury, such as contact sports.
There is no specific treatment for Ehlers-Danlos syndrome, but symptoms can be managed through:
- medications to ease paid and reduce blood pressure
- physical therapy such as exercises or physical braces to keep joints as stable and strong as possible.
If you have Ehlers-Danlos syndrome, you may see a range of health professionals to help you manage your condition, including physiotherapists, occupational therapists, psychologists, rheumatologists and genetic counselors.
Ehlers Danlos syndrome types
Figure 1. Ehlers Danlos syndrome types
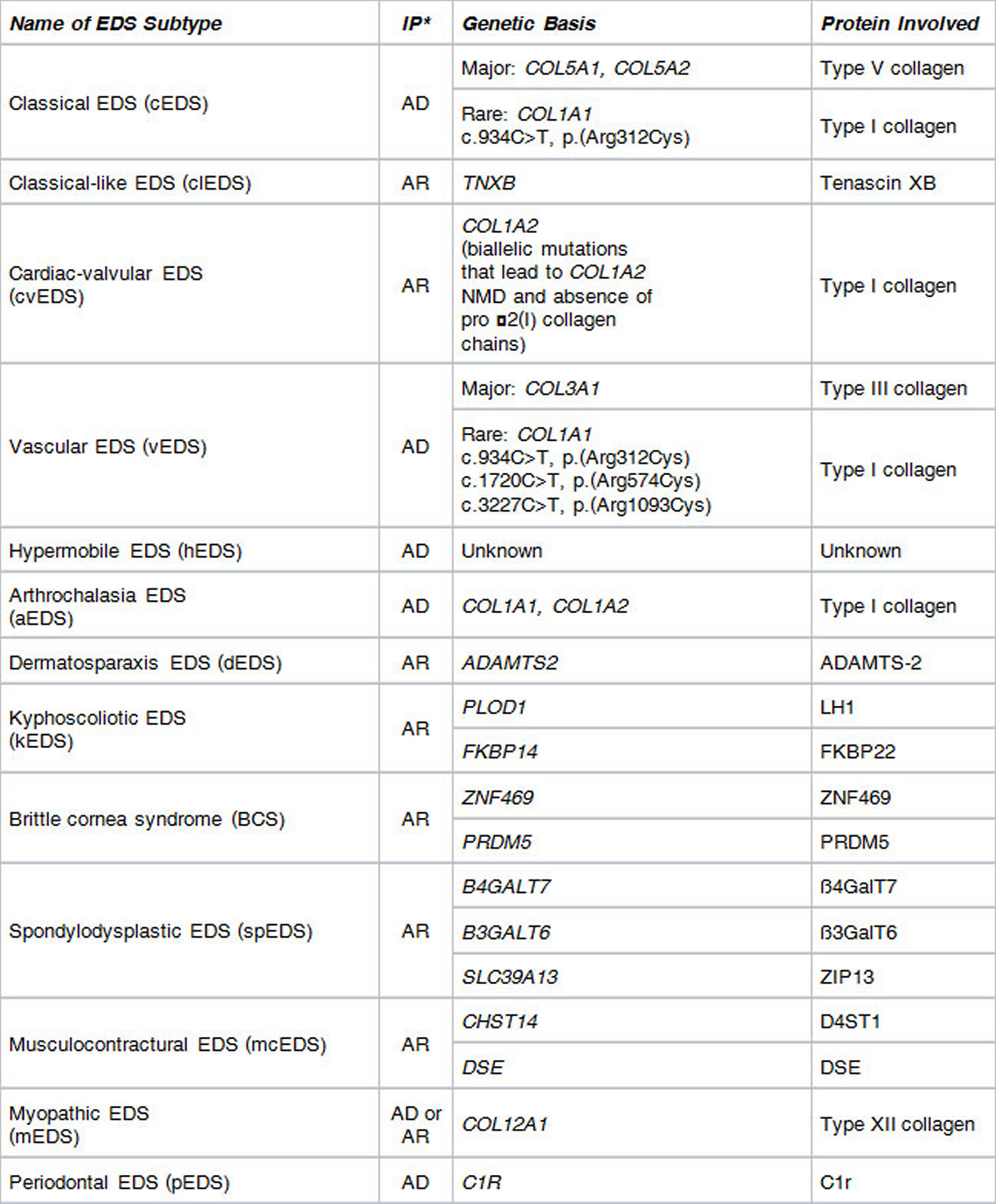
* Inheritance Pattern: AD = autosomal dominant; AR = autosomal recessive
[Source 1]Classical Ehlers-Danlos syndrome
Major criteria are:
- Skin hyperextensibility and atrophic scarring; and
- Generalized joint hypermobility.
There are nine minor criteria. Minimal clinical standards suggesting classical Ehlers-Danlos syndrome are the first major criterion plus either the second major criterion or at least three minor criteria.
A final diagnosis requires confirmation by molecular testing. More than 90% of those with classical Ehlers-Danlos syndrome have a heterozygous mutation in one of the genes encoding type V collagen (COL5A1 and COL5A2). Rarely, specific mutations in the genes encoding type I collagen can be associated with the characteristics of classical Ehlers-Danlos syndrome. Classical Ehlers-Danlos syndrome is inherited in the autosomal dominant pattern.
Skin is hyperextensible if it can be stretched over a standardized cut off in the following areas: 1.5 cm for the distal part of the forearms and the dorsum of the hands; 3 cm for neck, elbow and knees; 1 cm on the volar surface of the hand (palm).
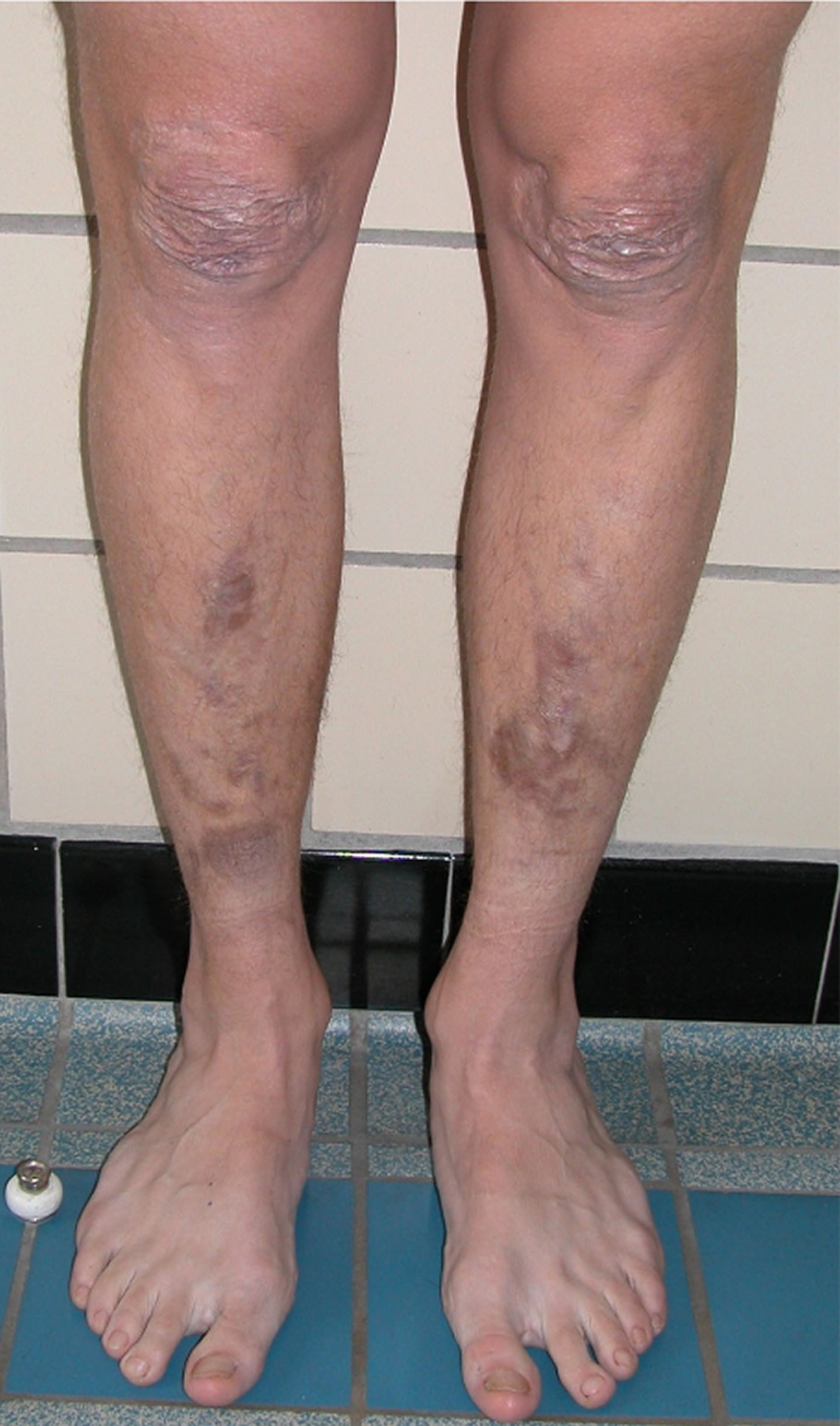
Abnormal scarring can range in severity. Most with classical Ehlers-Danlos syndrome have extensive atrophic scars at a number of sites. A minority are more mildly affected. The relevance of surgical scars should be considered with caution in classical EDS, they can appear normal in patients with classical EDS if well managed. Atrophic surgical scars can be found in the general population due to mechanical factors and site of the incision.
Joint hypermobility is evaluated according to the Beighton score; a Beighton score of >5 is considered positive for the presence of generalized joint hypermobility. Since joint hypermobility decreases with age, patients with a Beighton score <5/9 may be considered positive based on their historical observations.
Figure 2. Classical Ehlers-Danlos Syndrome with hyperextensible skin
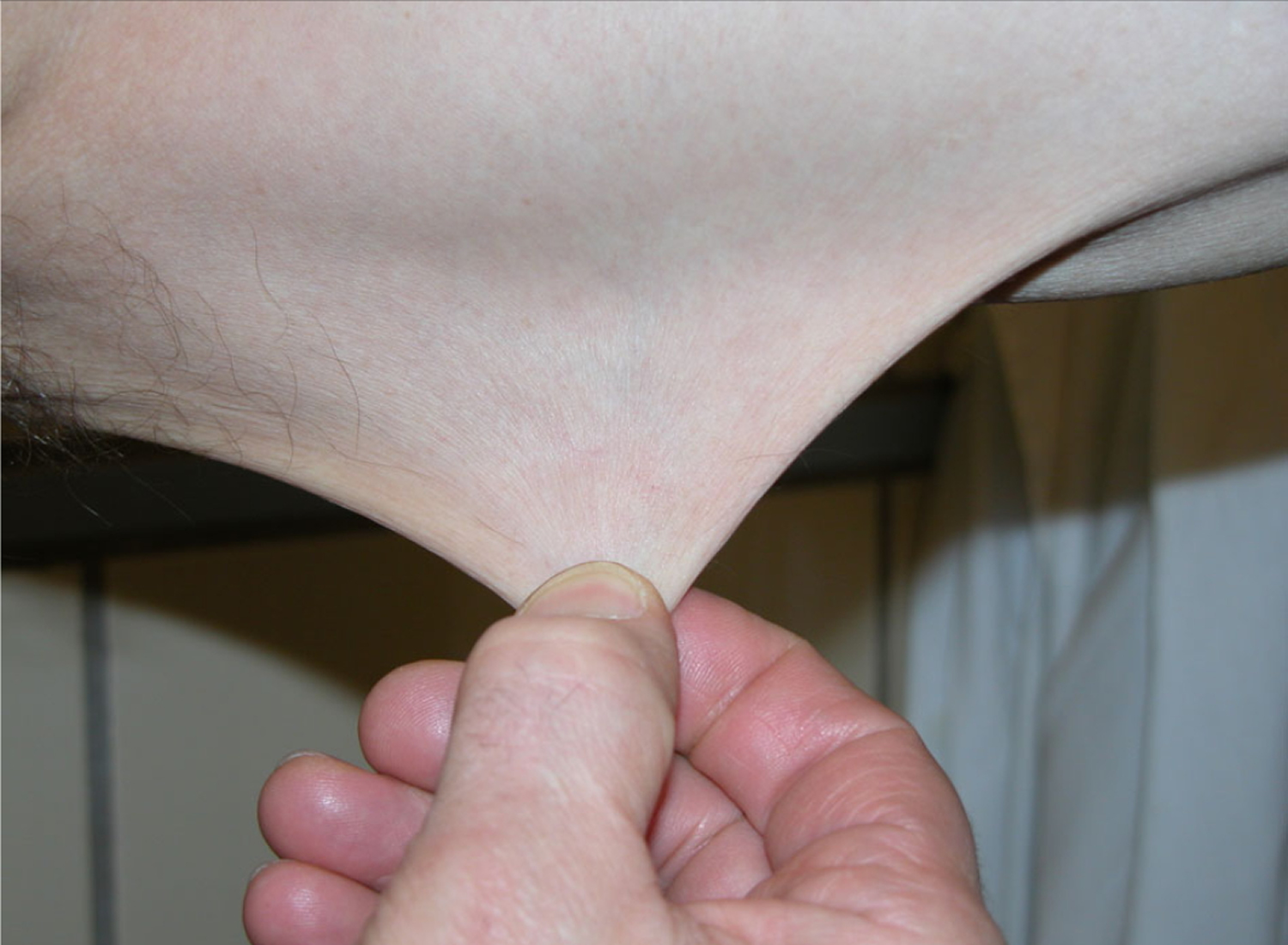
Figure 3. Classical Ehlers-Danlos syndrome atrophic scarring
Classical-like Ehlers-Danlos syndrome (clEDS)
Major criteria are:
- Skin hyperextensibility with velvety skin texture and absence of atrophic scarring;
- Generalized joint hypermobility with or without recurrent dislocations (most often shoulder and ankle); and
- Easily bruised skin or spontaneous ecchymoses (discolorations of the skin resulting from bleeding underneath).
There are seven minor criteria. Minimal clinical standards suggesting classical-like Ehlers-Danlos syndrome are all three major criteria plus a family history compatible with autosomal recessive transmission.
A final diagnosis requires molecular testing; classical-like Ehlers-Danlos syndrome is caused by a complete lack of Tenascin XB (due to biallelic TNXB mutations, that lead to nonsense-mediated mRNA decay, or biallelic deletion of TNXB). TNXB is the only gene associated with clEDS. Classical-like Ehlers-Danlos syndrome is inherited in the autosomal recessive pattern.
Note: skin hyperextensibility and joint hypermobility are defined as in classical Ehlers-Danlos syndrome.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Cardiac-valvular Ehlers-Danlos syndrome (cvEDS)
Major criteria are:
- Severe progressive cardiac-valvular problems (aortic valve, mitral valve);
- Skin involvement: skin hyperextensibility, atrophic scars, thin skin, easy bruising; and
- Joint hypermobility (generalized or restricted to small joints).
There are four minor criteria. Minimal clinical standards suggesting cardiac-valvular Ehlers-Danlos syndrome are the first major criterion plus a family history compatible with autosomal recessive transmission, and either one other major criterion or at least two minor criteria.
A final diagnosis requires confirmation by molecular testing; cardiac-valvular Ehlers-Danlos syndrome is caused by a complete lack of the proa2-chain of type I collagen due to biallelic COL1A2 mutations, that lead to nonsense-mediated mRNA decay. COL1A2 is the only gene associated with cardiac-valvular Ehlers-Danlos syndrome. Cardiac-valvular Ehlers-Danlos syndrome is inherited in the autosomal recessive pattern.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Vascular Ehlers-Danlos syndrome (vEDS)
Major criteria are:
- Family history of vascular Ehlers-Danlos syndrome with documented causative variant in COL3A1;
- Arterial rupture at a young age;
- Spontaneous sigmoid colon perforation in the absence of known diverticular disease or other bowel pathology;
- Uterine rupture during the third trimester in the absence of previous C-section and/or severe peripartum perineum tears; and
- Carotid-cavernous sinus fistula formation in the absence of trauma.
There are twelve minor criteria. Minimal clinical standards suggesting vascular Ehlers-Danlos syndrome diagnostic studies should be performed are: a family history of the disorder, arterial rupture or dissection in individuals less than 40 years of age; unexplained sigmoid colon rupture: or spontaneous pneumothorax in the presence of other features consistent with vascular Ehlers-Danlos syndrome. Testing for vascular Ehlers-Danlos syndrome should also be considered in the presence of a combination of the other “minor” criteria.
A final diagnosis requires confirmation by molecular testing. Patients with vascular Ehlers-Danlos syndrome typically have a heterozygous mutation in the COL3A1 gene, with the rare exception of specific heterozygous arginine-to-cysteine substitution mutations in COL1A1 that are also associated with vascular fragility and mimic COL3A1-vEDS. In very rare instances, biallelic pathogenic variants in COL3A1 may be identified. Vascular Ehlers-Danlos syndrome is inherited in the autosomal dominant pattern.
For more information, please see Diagnosis, natural history, and management in vascular Ehlers–Danlos syndrome 3.
Hypermobile Ehlers-Danlos syndrome
Hypermobile type Ehlers-Danlos syndrome (hEDS) is the most common subtype of the Ehlers-Danlos syndromes and possibly the most common of all hereditary disorders of connective tissue. Among the population of Ehlers-Danlos syndrome population of 1 in 5,000 people, 80–90% of which could be cases of hypermobile Ehlers-Danlos syndrome. Accurate estimates for the occurrence of hypermobile Ehlers-Danlos syndrome are lacking. High estimates under the previous classification system give figures suggesting up to 2 million people in the UK, 10 million in the USA, 17 million in Europe, and 255 million worldwide have hypermobile Ehlers-Danlos syndrome. The new classification of hypermobile Ehlers-Danlos syndrome is more selective, so figures will be lower than previously reported. Hypermobile Ehlers-Danlos syndrome was previously known as Ehlers Danlos syndrome type 3. For more information please go here: (Hypermobile Ehlers–Danlos syndrome (a.k.a. Ehlers–Danlos syndrome Type III and Ehlers–Danlos syndrome hypermobility type): Clinical description and natural history. https://www.ehlers-danlos.com/pdf/2017-FINAL-AJMG-PDFs/Tinkle_et_al-2017-American_Journal_of_Medical_Genetics_Part_C-_Seminars_in_Medical_Genetics.pdf)
The diagnosis of hypermobile Ehlers-Danlos syndrome (hEDS) remains clinical; there is no molecular, genetic cause yet identified, so there is no test available for almost all with hypermobile Ehlers-Danlos syndrome. At present, no single gene mutation causing hypermobile Ehlers-Danlos syndrome has been identified. Hypermobile type Ehlers-Danlos syndrome is likely to be caused by many different genetic changes. With the stricter classification of hypermobile type Ehlers-Danlos syndrome and more in-depth genetic studies, additional hypermobile Ehlers-Danlos syndrome-related genes should be identified. Identification of genetic causes for hypermobile Ehlers-Danlos syndrome may lead to it being further broken down into specific named hypermobile Ehlers-Danlos syndrome subtypes, or perhaps lead to hypermobile Ehlers-Danlos syndrome being redefined or replaced by multiple new specific Ehlers-Danlos syndrome types, in a manner similar to how classical-like EDS (clEDS) has been defined as a new Ehlers-Danlos syndrome type due to the genetic cause being identified.
hEDS has a roughly 50% chance of being passed on to each child (autosomal dominance), but other patterns of inheritance may explain this disorder in certain families. hEDS inheritance is somewhat difficult to analyze, as hEDS may be mild during much of life, or compared to close relatives with the disorder, or may even appear to “skip” a generation. This may be in part because JH is heavily influenced by age, gender, and weight. The reason for a perceived excess of females remains poorly understood, but may come down to sex hormones having a greater influence upon JH. The best way to describe hEDS is as an autosomal dominant disorder influenced by age and gender, with symptoms more common in females.
There is a clinical spectrum ranging from asymptomatic joint hypermobility, through “non-syndromic” hypermobility with secondary manifestations, to hypermobile Ehlers-Danlos syndrome.
A diagnosis of hypermobile Ehlers-Danlos syndrome should be assigned only in those who meet all of the criteria, which should help research efforts to discover the underlying genetic cause(s) which, in turn, may help clinical management. As this is a clinical diagnosis, it’s important to be relatively confident that the diagnosis is not instead one of the many other disorders of connective tissue. Hypermobile Ehlers-Danlos syndrome is inherited in the autosomal dominant pattern.
The clinical diagnosis of hypermobile Ehlers-Danlos syndrome needs the simultaneous presence of criteria 1 and 2 and 3. This is a complex set of criteria, and there is much more detail than presented in this overview.
- Generalized joint hypermobility and
- Two or more of the following features must be present (A & B, A & C, B & C, or A & B & C):
- Feature A—systemic manifestations of a more generalized connective tissue disorder (a total of five out of twelve must be present)
- Feature B—positive family history, with one or more first degree relatives independently meeting the current diagnostic criteria for hypermobile Ehlers-Danlos syndrome
- Feature C—musculoskeletal complications (must have at least one of three); and
- All these prerequisites must be met: absence of unusual skin fragility, exclusion of other heritable and acquired connective tissue disorders including autoimmune rheumatologic conditions, and exclusion of alternative diagnoses that may also include joint hypermobility by means of hypotonia and/or connective tissue laxity.
There is a range of conditions which can accompany hypermobile Ehlers-Danlos syndrome, although there is not enough data for them to become diagnostic criteria. While they’re associated with hypermobile Ehlers-Danlos syndrome, they’re not proven to be the result of hypermobile Ehlers-Danlos syndrome and they’re not specific enough to be criteria for diagnosis. Some of these include sleep disturbance, fatigue, postural orthostatic tachycardia, functional gastrointestinal disorders, dysautonomia, anxiety, and depression. These conditions may be more debilitating the joint symptoms; they often impair daily life, and they should be considered and treated.
Figure 4. Hypermobile Ehlers-Danlos syndrome diagnostic criteria

What is Hypermobility spectrum disorders?
Hypermobility spectrum disorders are a group of conditions related to joint hypermobility. Hypermobility spectrum disorders are intended to be diagnosed after other possible answers are excluded, such as any of the Ehlers-Danlos syndromes including hypermobile Ehlers-Danlos syndromes (hEDS). Hypermobility spectrum disorders, just like hypermobile Ehlers-Danlos syndromes, can have significant effects on your health. Whatever the problems that arise, whatever the diagnosis, it is important that these effects are managed appropriately and that each person is treated as an individual. Hypermobility spectrum disorders and hypermobile Ehlers-Danlos syndromes can be equal in severity, but more importantly, both need similar management, validation, and care.
What happens to people with hypermobile Ehlers-Danlos syndrome?
If a person has hypermobile Ehlers-Danlos syndrome, it will likely affect them in different ways throughout their lifetime, and the person may be diagnosed with many other conditions known to occur in those with hypermobile Ehlers-Danlos syndrome. For example, three disease phases were proposed in a 2010 study: a “hypermobility” phase, a “pain” phase, and a “stiffness” phase. Alternatively, existing studies have led to speculation that there is a natural transition from hypermobile Ehlers-Danlos syndrome to generalized joint hypermobility with age.
Existing studies show that children with hypermobile Ehlers-Danlos syndrome who experience pain will be more likely to have pain limited to lower limbs (e.g., “growing pains”) and pain caused by repetitive tasks such as handwriting in the school setting. Children with hypermobile Ehlers-Danlos syndrome may have poor coordination. The “pain” phase is often accompanied by diagnosis with fibromyalgia or other long-term (chronic) pain conditions and perhaps chronic fatigue, typically starting in the second to fourth decade and accompanied by chronic pain, headaches, digestive system disorders, among others. The “stiffness” phase is seen in only a few persons, and, unfortunately for them, the symptoms of the “pain” phase may persist and escalate, functionality may overall be significantly reduced.
Pain
Yes, hypermobile Ehlers-Danlos syndrome CAN cause significant pain!
Uneducated doctors all-too-often make emphatic statements such as, “Ehlers-Danlos syndrome cannot cause pain!” In fact, while hypermobile Ehlers-Danlos syndrome does not necessarily cause significant pain for every person meeting diagnostic criteria, many with hypermobile Ehlers-Danlos syndrome will develop significant pain for some portion of their lives. Any doctor who invalidates the fact that hypermobile Ehlers-Danlos syndrome can cause significant pain may have a profoundly negative impact, not only upon the success of the relationship between clinician and patient, but also upon the quality of life of those with hypermobile Ehlers-Danlos syndrome.
Health practitioners need to understand that the occurrence of significant pain for many persons with hypermobile Ehlers-Danlos syndrome is well-supported in the literature and has logically proposed or proven cause. In fact, joint/muscle pain in two or more limbs (recurring daily, ≥ 3 months) and long-term widespread pain (≥ 3 months) are specifically considered as a part of the criteria for diagnosis of hypermobile Ehlers-Danlos syndrome.
Skin and Connective Tissue
People with hypermobile Ehlers-Danlos syndrome do NOT have to have profoundly stretchy skin!
Most notably, in hypermobile Ehlers-Danlos syndrome, the degree of softness, stretchiness, fragility, bruisability, and poor wound healing of skin differs from “normal” subjects but is mild in comparison to other types of Ehlers-Danlos syndrome. Clinicians who are not up to date on the classification of Ehlers-Danlos syndrome types often expect that all Ehlers-Danlos syndrome types demonstrate severe skin changes, such as those observed in persons with classical Ehlers-Danlos syndrome and vascular Ehlers-Danlos syndrome. Mild skin stretchiness (rather than severe) is clearly considered as a systemic manifestation in the criteria for clinical diagnosis of hypermobile Ehlers-Danlos syndrome.
Stretch marks are not inevitable in hypermobile Ehlers-Danlos syndrome, however, they often appear in persons with hypermobile Ehlers-Danlos syndrome during adolescent growth spurts and are not necessarily due to rapid weight gain. The absence of stretch marks should not argue against a diagnosis of hypermobile Ehlers-Danlos syndrome. Other tissues which may fail in hypermobile Ehlers-Danlos syndrome include the protective coverings around organs. Weakness in this connective tissue in hypermobile Ehlers-Danlos syndrome often results in hernia (tissues or organs pushing through). Hernias may also be more likely in persons with hypermobile Ehlers-Danlos syndrome who undergo abdominal surgery, such as laparotomy or C-section.
Fatigue
Chronic fatigue is one of the most common complications among persons with hypermobile Ehlers-Danlos syndrome.
Chronic, debilitating fatigue is common in hypermobile Ehlers-Danlos syndrome, and such fatigue has significant impact on mental and physical function and ultimately on quality of life. Those with hypermobile Ehlers-Danlos syndrome will often meet the criteria for chronic fatigue syndrome (CFS). Under no circumstances should a diagnosis of CFS to a person who also meets criteria for hypermobile Ehlers-Danlos syndrome mean that hypermobile Ehlers-Danlos syndrome should be disregarded. hypermobile Ehlers-Danlos syndrome is considered to be a specific cause of chronic fatigue, while chronic fatigue syndrome is considered a syndrome with unknown cause; in this case the diagnosis of chronic fatigue syndrome would more appropriately called into question.
Cardiovascular
While conditions of the heart and blood vessels can occur with hypermobile Ehlers-Danlos syndrome, they are not usually life-threatening, but deserve individual consideration.
Heart and blood vessel conditions occurring in hypermobile Ehlers-Danlos syndrome include heart valve and vessel dysfunction including mitral valve prolapse (MVP) and aortic root dilation. Problems with blood pressure and heart rate can also occur including postural orthostatic tachycardia syndrome (POTS), neurally-mediated hypotension and orthostatic intolerance. Those having blood pressure changes may also experience near-fainting or fainting episodes.
Gastrointestinal Disorders
Digestive system problems occur with high frequency in hypermobile Ehlers-Danlos syndrome.
As many as 75% of people with hypermobile Ehlers-Danlos syndrome are likely to encounter problems with the function of their digestive system in their lifetime. Problems can occur anywhere along the digestive system, including the mouth and throat (e.g., chewing and swallowing) in addition to the functions of the rest of the gastrointestinal tract. Disorders include poor movement of material along the tract, reflux, heartburn, abdominal pain, bloating, irritable bowel, diarrhea, constipation, or incontinence. Structural problems occur at a higher rate in those with hypermobile Ehlers-Danlos syndrome than in the general population including hernias, internal organ displacement, and rectal prolapse. The relationship between all structural abnormalities and hypermobile Ehlers-Danlos syndrome requires further study.
Dysautonomia
“Fight or Flight” and “Rest and Digest” function poorly for a majority with hypermobile Ehlers-Danlos syndrome.
An extensive body of literature clearly indicates frequent problems with involuntary bodily functions (dysautonomia) in hypermobile Ehlers-Danlos syndrome. Problems with involuntary body functions can lead to fatigue, dizziness, fainting, memory changes, poor concentration, reduced sweat production, changes in gut movement, bladder dysfunction, and/or certain psychological traits. Dysautonomia can be a devastating manifestation of hypermobile Ehlers-Danlos syndrome, and, for some patients, it affects their quality of life more profoundly than joint instability, pain, or any other part of the disorder.
Bone Density
Existing evidence does not clearly indicate that hypermobile Ehlers-Danlos syndrome is expected to cause low bone density or increased risk for fragility fractures. Carefully designed research is needed.
While existing studies have suggested a link between various Ehlers-Danlos syndrome types and conditions such as osteoporosis (low bone density) and osteopenia (low bone mineralization), those with Ehlers-Danlos syndrome often have a significantly reduced level of activity, and, rather than any alteration in bone density or mineralization being directly attributable to Ehlers-Danlos syndrome, alteration in bone density or mineralization should be considered to be a direct result of reduced activity. Bone health should not be ignored in hypermobile Ehlers-Danlos syndrome.
Osteoarthritis
Hypermobile Ehlers-Danlos syndrome increases the susceptibility of joints to osteoarthritis.
Osteoarthritis (a breakdown of joints) has been described in the literature as a possible long-term consequence of joint hypermobility for decades. Since joint hypermobility is a hallmark feature of hypermobile Ehlers-Danlos syndrome, and joint hypermobility is recognized to cause osteoarthritis, then hypermobile Ehlers-Danlos syndrome would logically be expected to predispose many of those with the disorder to osteoarthritis. By the same token, osteoarthritis is by no means universal for all persons with joint hypermobility, so it should NOT be expected to be inevitable for all people with hypermobile Ehlers-Danlos syndrome.
Headaches
Hypermobile Ehlers-Danlos syndrome may lead to severe or debilitating headaches for a multitude of reasons.
Headaches in general, as well as migraines in particular, are known to occur more frequently in persons with Ehlers-Danlos syndrome than in the general population. Headaches in hypermobile Ehlers-Danlos syndrome are considered to have many causes, with potential mechanisms including instability, strain, or muscle spasms in the neck, jaw joint dysfunction, and/or medication side effects. The headaches vary in type and severity among all persons with hypermobile Ehlers-Danlos syndrome who have headaches. Some find headaches to be the most disabling manifestation of hypermobile Ehlers-Danlos syndrome.
Temporomandibular Joint and Dental Issues
Dentists, Orthodontists, and Oral Surgeons may be the first to consider a diagnosis of hypermobile Ehlers-Danlos syndrome.
A hypermobile jaw joint is more likely to show dysfunction than a non-hypermobile jaw. Those with hypermobile Ehlers-Danlos syndrome and jaw joint dysfunction often have sounds, locking, dislocation, grinding teeth, and headaches in the temple.
Mouth manifestations of hypermobile Ehlers-Danlos syndrome may include fragile gums which bleed easily, gum recession, gum disease, small or absent frenula (small connective flaps between the front gums and lips), pointed and deep teeth, shortened roots, abnormal enamel, tooth fractures, ineffective dental anesthesia, and rapid orthodontic correction (and rapid return without orthodontia).
Spine
It is difficult at times to determine whether a person with hypermobile Ehlers-Danlos syndrome would benefit from surgery, and, even when surgery is necessary, it doesn’t always solve everything.
People with hypermobile Ehlers-Danlos syndrome may have neck pain, difficulty walking, numbness and tingling of the hands and feet, dizziness, swallowing difficulties, and changes in speech. These people are more likely to have signs of looseness or instability around the head and neck. In many cases, these symptoms are not entirely attributable to head and neck dysfunction: symptoms may still persist after successful surgery.
Abnormal spine curvature is common in people with hypermobile Ehlers-Danlos syndrome, in a large part due to a combination of structural and functional abnormalities in the supporting tissues of the spine. Conservative management will ideally allow avoidance of surgery.
Gynecologic Issues
Women with hypermobile Ehlers-Danlos syndrome may experience heavy periods or painful intercourse.
Pelvic Dysfunction
It is unclear whether hypermobile Ehlers-Danlos syndrome alone predisposes women to pelvic dysfunction or how much of a role childbirth plays.
While the existing literature suggests that pelvic floor problems including urinary incontinence or uterine, rectal, or bladder prolapse are common to hypermobile Ehlers-Danlos syndrome, many studies did not control for childbirth history, and included various EDS types.
Pregnancy and Childbirth
No studies to date recommend against pregnancy and childbirth based simply upon a diagnosis of hypermobile Ehlers-Danlos syndrome.
While some studies of women with Ehlers-Danlos syndrome type 3/Ehlers–Danlos syndrome Hypermobility Type and joint hypermobility syndrome suggest increased rates of infertility, pregnancy losses, and premature births, other studies did not. In the case of studies regarding premature birth, the study group included women with Classic Ehlers-Danlos syndrome. One of the most thorough studies prior to the current Ehlers-Danlos syndrome classifications suggested similar measures of fertility.
When it comes to how pregnancy affects symptoms of hypermobile Ehlers-Danlos syndrome, it goes like this: some get worse, some don’t change, and some get better during the pregnancy. When it comes to labor, rapid labor is thought to occur in more than 1/3 of deliveries. The most considerable complications related to labor and delivery thought to occur at a significant rate in women with hypermobile Ehlers-Danlos syndrome include bleeding during or after birth, as well as abnormal scarring from C-section or episiotomy. In general, no studies to date recommend against pregnancy and childbirth based simply upon a diagnosis of hypermobile Ehlers-Danlos syndrome in the absence of any other factors.
Urinary System
Hypermobile Ehlers-Danlos syndrome may predispose children to urinary incontinence, urinary tract infections, vesicoureteral reflux, and voiding dysfunction.
Sleep Disturbance
Patients with hypermobile Ehlers-Danlos syndrome may feel particularly tired.
People with hypermobile Ehlers-Danlos syndrome often experience significant sleep deprivation. Going without healthy, restorative sleep can lead to any combination of significant health problems such as impaired immune response, poor muscle coordination, and higher perception of pain, as well as problems with memory and thought processes (e.g. “brain fog”), moodiness, depression, and anxiety. Some with hypermobile Ehlers-Danlos syndrome carry formal diagnoses of additional conditions such as restless leg syndrome or sleep apnea.
Mast Cell Activation Disorder
Current understanding of mast cell activation syndrome (MCAS) in hypermobile Ehlers-Danlos syndrome is limited.
Mast cell activation syndrome is an immune condition that can create allergy-like symptoms, up to and including anaphylaxis. More research is needed to see whether MCAS is a condition that occurs with greater frequency in hypermobile Ehlers-Danlos syndrome than in the general population and how it affects a person with hypermobile Ehlers-Danlos syndrome and the management.
Psychiatric
Hypermobile Ehlers-Danlos syndrome is not in the psyche, it is in the connective tissue, but it can affect the psyche.
Many with hypermobile Ehlers-Danlos syndrome are assigned psychiatric diagnoses or frankly ignored when clinicians fail to recognize that they meet diagnostic criteria for hypermobile Ehlers-Danlos syndrome. Clearly, this leads to a failure in attempts to effectively care for the person with hypermobile Ehlers-Danlos syndrome. It is important to realize that psychological conditions (e.g. anxiety or depression) are common in chronic conditions including hypermobile Ehlers-Danlos syndrome. Ignoring significant coexisting psychological problems will lead to suboptimal treatment.
Quality of Life
Studies to date consistently suggest that hypermobile Ehlers-Danlos syndrome adversely affects quality of life.
Carefully designed studies have clearly demonstrated that quality of life is often measured or reported as lower in people with Ehlers-Danlos syndrome than in the general population — in particular, people with Ehlers-Danlos syndrome and associated secondary issues such as digestive system disorders, anxiety, depression, physical pain.
Management of hypermobile Ehlers-Danlos syndrome
Recognizing hypermobile Ehlers-Danlos syndrome is only half of the battle. Diagnosis alone is not enough. As with any disorder, effective management of hypermobile Ehlers-Danlos syndrome includes recognizing its complexity. A body of evidence-based standards of care exists and includes the efforts of multiple healthcare practitioners. Management of hypermobile Ehlers-Danlos syndrome must follow established standards of care and include treatment of both immediate and long-term issues as well as focusing on preventative care.
Key points for management of conditions in hypermobile Ehlers-Danlos syndrome include:
- The approach should be holistic focusing on the complications, the desire(s) of the patient, quality of life and functionality, as well as psychological aspects.
- Results should not be expected overnight: It often takes months of routine toning exercise to stop deterioration, and it may take years before substantial reduction in pain is recognized. Fatigue, like pain, often responds to treatment such as exercise therapy but only very slowly.
- Use of multiple medications together, physical therapy, and complementary medicine is often more effective than as-needed use of one or two medications at a time. Some patients who continue to struggle to cope with their pain may need a broad pain management program. The overall goal should be to maintain adequate control of pain to a tolerable level, not to completely eliminate it.
- Management of psychiatric issues should include consideration of counseling and cognitive behavioral therapy, in addition to consideration of techniques such as distraction, hypnosis, and careful consideration of drug therapy.
Arthrochalasia Ehlers-Danlos syndrome (aEDS)
Major criteria are:
- Congenital bilateral hip dislocation;
- Severe generalized joint hypermobility, with multiple dislocations/subluxations; and
- Skin hyperextensibility.
There are five minor criteria. Minimal criteria for arthrochalasia Ehlers-Danlos syndrome are congenital bilateral hip dislocation (major criterion 1) plus either: skin hyperextensibility (major criterion 3); or severe generalized joint hypermobility (major criterion 2) with at least two minor criteria.
A final diagnosis requires confirmation by molecular testing; arthrochalasia Ehlers-Danlos syndrome is caused by heterozygous mutations in either COL1A1 or COL1A2, that cause entire or partial loss of exon 6 of the respective gene. No other genes are associated with arthrochalasia Ehlers-Danlos syndrome. Absence of a causative mutation in COL1A1 or COL1A2 that leads to complete or partial deletion of the exon 6 of either gene excludes the diagnosis of arthrochalasia Ehlers-Danlos syndrome. Arthrochalasia Ehlers-Danlos syndrome is inherited in the autosomal dominant pattern.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Dermatosparaxis Ehlers-Danlos syndrome (dEDS)
There are nine major criteria and eleven minor criteria. Minimal criteria suggestive of dermatosparaxis Ehlers-Danlos syndrome include the two major criteria of extreme skin fragility and characteristic craniofacial features, plus either: one other major criterion, or three minor criteria.
A final diagnosis requires confirmation by molecular testing; dermatosparaxis Ehlers-Danlos syndrome is caused by biallelic mutations in ADAMTS2. It is the only gene associated with dermatosparaxis Ehlers-Danlos syndrome. Dermatosparaxis Ehlers-Danlos syndrome is inherited in the autosomal recessive pattern.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Kyphoscoliotic Ehlers-Danlos syndrome (kEDS)
Major criteria are:
- Congenital muscle hypotonia;
- Congenital or early onset kyphoscoliosis (progressive or non-progressive); and
- Generalized joint hypermobilitywith dislocations/subluxations (shoulders, hips and knees in particular).
There are ten minor criteria, as well as gene-specific minor criteria (four for PLOD1 and four for FKBP14). Minimal criteria suggestive for kyphoscoliotic Ehlers-Danlos syndrome are 1 and 2 of the major criteria—congenital muscle hypotonia and congential/early onset kyphoscoliosis—plus either: major criterion 3, or three minor criteria (either general of gene-specific).
A final diagnosis requires confirmation by testing. The majority of patients with kyphoscoliotic Ehlers-Danlos syndrome harbor biallelic mutations in PLOD1; recently, biallelic mutations have been identified in FKBP14 in patients displaying a phenotype that clinically largely overlaps with kyphoscoliotic Ehlers-Danlos syndrome-PLOD1. Laboratory confirmation should start with a urine test using high-performance liquid chromatography (to evaluate the ratio of lysyl-pyridinoline to hydroxylysyl-pyridinoline crosslinks; a normal ratio is ~0.2, whereas kyphoscoliotic Ehlers-Danlos syndrome-PLOD1 range is 2-9). This method is fast and cost-effective and it can also be used to determine the pathogenic status of a variant of uncertain significance. Molecular analysis can follow if the urine test is normal. Whereas absence of an abnormal urinary LP/HP ratio excludes the diagnosis of kyphoscoliotic Ehlers-Danlos syndrome-PLOD1, absence of the confirmatory genetic findings does not exclude the diagnosis of kyphoscoliotic Ehlers-Danlos syndrome, as other yet-to-be-discovered genes may be associated with this phenotype; however, alternative diagnoses should be considered in the absence of PLOD1 or FKBP14 mutations. Kyphoscoliotic Ehlers-Danlos syndrome is inherited in the autosomal recessive pattern.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Brittle Cornea Syndrome
Major criteria are:
- Thin cornea, with or without rupture (central corneal thickness often <400 µm);
- Early onset progressive keratoconus;
- Early onset progressive keratoglobus; and
- Blue sclerae.
There are fourteen minor criteria. Minimal criteria required to suggest brittle cornea syndrome are the first major criterion, plus either: at least one other major criterion; or three minor criteria.
A final diagnosis requires confirmation through molecular testing. Brittle Cornea Syndrome is caused by biallelic mutations in either ZNF469 or PRDM5. At least one family with a clinical brittle cornea syndrome phenotype did not harbor mutations in these genes, suggesting that at least one other gene might be associated with brittle cornea syndrome. Brittle cornea syndrome is inherited in the autosomal recessive pattern.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Spondylodysplastic Ehlers-Danlos syndrome (spEDS)
Major criteria are:
- Short stature (progressive in childhood);
- Muscle hypotonia (ranging from severe congenital, to mild later-onset); and
- Bowing of limbs.
There are five general minor criteria, plus gene-specific criteria for B4GALT7, B3GALT6, and SLC39A13. Minimal criteria required to suggest a diagnosis for spondylodysplastic Ehlers-Danlos syndrome are the first and second major criteria, plus characteristic radiographic abnormalities and at least three minor criteria (either general or gene-specific). Spondylodysplastic Ehlers-Danlos syndrome is inherited in the autosomal recessive pattern.
Final diagnosis requires confirmation through molecular testing.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Musculocontractural Ehlers-Danlos Syndrome (mcEDS)
Major criteria are:
- Congenital multiple contractures, characteristically adduction-flexion contractures and/or talipes equinovarus (clubfoot);
- Characteristic craniofacial features, which are evident at birth or in early infancy; and
- Characteristic cutaneous features including skin hyperextensibility, easy bruisability, skin fragility with atrophic scars, increased palmar wrinkling.
There are fifteen minor criteria. The minimum criteria required to suggest musculocontractural Ehlers-Danlos Syndrome are: at birth or in early childhood, major criteria 1 and 2; in adolescence and adulthood, major criteria 1 and 3.
A final diagnosis requires confirmation through molecular testing. Musculocontractural Ehlers-Danlos Syndrome is caused by biallelic mutations in CHST14. A few mutations have been identified in the DSE gene in patients with a similar phenotype. Musculocontractural Ehlers-Danlos Syndrome is inherited in the autosomal recessive pattern.
For more information, please see The Ehlers-Danlos Syndromes, Rare Types 2.
Myopathic Ehlers-Danlos syndrome (mEDS)
Major criteria are:
- Congenital muscle hypotonia, and/or muscle atrophy, that improves with age;
- Proximal joint contractures (knee, hip and elbow); and
- Hypermobility of distal joints.
There are four minor criteria. The minimal criteria required to suggest a diagnosis of myopathic Ehlers-Danlos syndrome are the first major criterion plus either: one other major criterion, or three minor criteria.
A final diagnosis requires molecular testing; myopathic Ehlers-Danlos syndrome is caused by heterozygous or biallelic mutations in COL12A1, and the clinical phenotype highly overlaps with collagen type VI-related myopathies. It is currently unknown whether other, yet to be discovered genes, are associated with this phenotype. In case no COL12A1 mutations are identified alternative diagnoses, especially collagen VI-related Ullrich Congenital Muscular Dystrophy and Bethlem Myopathy, should be considered. Myopathic Ehlers-Danlos syndrome is inherited in either the autosomal dominant or the autosomal recessive pattern.
Periodontal Ehlers-Danlos syndrome (pEDS)
Major criteria are:
- Severe and intractable periodontitis of early onset (childhood or adolescence);
- Lack of attached gingiva;
- Pretibial plaques; and
- Family history of a first-degree relative who meets clinical criteria.
There are eight minor criteria. The minimal criteria required to suggest periodontal Ehlers-Danlos syndrome are the first criterion or the second criterion, plus at least two other major criteria and one minor criterion.
A final diagnosis requires molecular testing; periodontal Ehlers-Danlos syndrome is caused by heterozygous gain-of-function mutations in C1R or C1S. At present it cannot be stated whether absence of a C1R or C1S mutations excludes the diagnosis because the experience with the molecular diagnosis is limited. Periodontal Ehlers-Danlos syndrome (pEDS) is inherited in the autosomal recessive pattern.
Genetic classification structure of Ehlers-Danlos syndrome
There is an additional genetic classification structure of the Ehlers-Danlos syndrome into groups according to similarities in the way the responsible genes affect the body.
- Group A: Disorders of collagen primary structure and collagen processing, comprised of:
- Classical Ehlers-Danlos syndrome,
- Vascularl Ehlers-Danlos syndrome,
- Arthrochalasiaa Ehlers-Danlos syndrome,
- Dermatosparaxis Ehlers-Danlos syndrome, and
- Cardiac-valvular Ehlers-Danlos syndrome.
- Group B: Disorders of collagen folding and collagen crosslinking, comprised of:
- Kyphoscoliotic Ehlers-Danlos syndrome-PLOD1 and
- Kyphoscoliotic Ehlers-Danlos syndrome-FKB14.
- Group C: Disorders of structure and function of the myomatrix, comprised of:
- Classical-like Ehlers-Danlos syndrome and
- Myopathic Ehlers-Danlos syndrome.
- Group D: Disorders of glycosaminoglycan biosynthesis, comprised of :
- Spondylodysplastic Ehlers-Danlos syndrome-B4GALT7,
- Spondylodysplastic Ehlers-Danlos syndrome-b3GALT6,
- Musculocontractural Ehlers-Danlos syndrome-CHST14, and
- Musculocontractural Ehlers-Danlos syndrome-DSE.
- Group E: Defects in complement pathway, comprised of Periodontal Ehlers-Danlos syndrome.
- Group F: Disorders of intracellular processes, comprised of Spondylodysplastic Ehlers-Danlos syndrome-SLC39A13 and Brittle Cornea syndrome.
- Group G: Unresolved forms of Ehlers-Danlos syndrome, comprised of Hypermobile Ehlers-Danlos syndrome (previously known as Ehlers Danlos syndrome type 3).
Conditions no longer included in the Ehlers-Danlos syndrome spectrum are occipital horn syndrome, fibronectin-deficient (EDS X), familial articular hypermobility (EDS XI), X-linked Ehlers-Danlos syndrome with muscle hematoma (EDS V), and filamin A related Ehlers-Danlos syndrome with periventricular nodular heterotopia.
Ehlers-Danlos Syndrome prognosis
The prognosis depends on the type of Ehlers-Danlos syndrome and the individual. Life expectancy can be shortened for those with the Vascular Ehlers-Danlos syndrome due to the possibility of organ and vessel rupture and risk of heart and lung complications. For other types of Ehlers-Danlos syndrome life expectancy and quality of life is normal, though caution is advised with some activities. Complications include scarring, slow wound healing and joint dislocations. There can be a wide or narrow range of severity within a family, but each person’s case of Ehlers-Danlos syndrome will be unique. Pregnancy may pose a risk in some patients; this needs to be discussed in patients planning to start a family. While there is no cure for the Ehlers-Danlos syndromes, there is treatment for symptoms, and there are preventative measures that are helpful for most.
Ehlers-Danlos Syndrome complications
Complications depend on the types of signs and symptoms you have. For example, overly flexible joints can result in joint dislocations and early-onset arthritis. Fragile skin may develop prominent scarring.
People who have Ehlers-Danlos syndrome, vascular type, are at risk of often fatal ruptures of major blood vessels. Some organs, such as the uterus and intestines, also may rupture. Pregnancy can increase these risks.
Ehlers Danlos syndrome causes
Different types of Ehlers-Danlos syndrome are associated with a variety of genetic causes, some of which are inherited and passed on from parent to child. If you have the most common varieties of Ehlers-Danlos syndrome, there’s a 50 percent chance that you’ll pass on the gene to each of your children. But in some Ehlers-Danlos syndrome cases it occurs by chance without a family history of the condition.
The two main ways Ehlers-Danlos syndrome is inherited are:
- autosomal dominant inheritance (hypermobile, classical and vascular Ehlers-Danlos syndrome) – the faulty gene that causes Ehlers-Danlos syndrome is passed on by one parent and there’s a 50% risk of each of their children developing the condition
- autosomal recessive inheritance (kyphoscoliotic Ehlers-Danlos syndrome) – the faulty gene is inherited from both parents and there’s a 25% risk of each of their children developing the condition
A person with Ehlers-Danlos syndrome can only pass on the same type of Ehlers-Danlos syndrome to their children. For example, the children of someone with hypermobile Ehlers-Danlos syndrome can’t inherit vascular Ehlers-Danlos syndrome.
The severity of the condition can vary within the same family.
Genetic Changes
Mutations in at least 19 genes have been found to cause the Ehlers-Danlos syndromes. Mutations in the COL5A1 or COL5A2 gene, or rarely in the COL1A1 gene, can cause the classical type. Mutations in the TNXB gene cause the classical-like type and have been reported in a very small percentage of cases of the hypermobile type (although in most people with this type, the cause is unknown). The cardiac-valvular type and some cases of the arthrochalasia type are caused by COL1A2 gene mutations; mutations in the COL1A1 gene have also been found in people with the arthrochalasia type. Most cases of the vascular type result from mutations in the COL3A1 gene, although rarely this type is caused by certain COL1A1 gene mutations. The dermatosparaxis type is caused by mutations in the ADAMTS2 gene. PLOD1 or FKBP14 gene mutations result in the kyphoscoliotic type. Other rare forms of Ehlers-Danlos syndrome result from mutations in other genes.
Some of the genes associated with the Ehlers-Danlos syndromes, including COL1A1, COL1A2, COL3A1, COL5A1, and COL5A2, provide instructions for making pieces of several different types of collagen. These pieces assemble to form mature collagen molecules that give structure and strength to connective tissues throughout the body. Other genes, including ADAMTS2, FKBP14, PLOD1, and TNXB, provide instructions for making proteins that process, fold, or interact with collagen. Mutations in any of these genes disrupt the production or processing of collagen, preventing these molecules from being assembled properly. These changes weaken connective tissues in the skin, bones, and other parts of the body, resulting in the characteristic features of the Ehlers-Danlos syndromes.
Some genes associated with recently described types of Ehlers-Danlos syndrome have functions that appear to be unrelated to collagen. For many of these genes, it is not clear how mutations lead to hypermobility, elastic skin, and other features of these conditions.
Inheritance Pattern
The inheritance pattern of the Ehlers-Danlos syndromes varies by type. The classical, vascular, arthrochalasia, and periodontal forms of the disorder, and likely the hypermobile type, have an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new (de novo) gene mutations and occur in people with no history of the disorder in their family.
The classical-like, cardiac-valvular, dermatosparaxis, kyphoscoliotic, spondylodysplastic, and musculocontractural types of Ehlers-Danlos syndrome, as well as brittle cornea syndrome, are inherited in an autosomal recessive pattern. In autosomal recessive inheritance, two copies of a gene in each cell are altered. Most often, the parents of an individual with an autosomal recessive disorder are carriers of one copy of the altered gene but do not show signs and symptoms of the disorder.
The myopathic type of Ehlers-Danlos syndrome can have either an autosomal dominant or autosomal recessive pattern of inheritance.
Figure 5. Autosomal dominant inheritance pattern
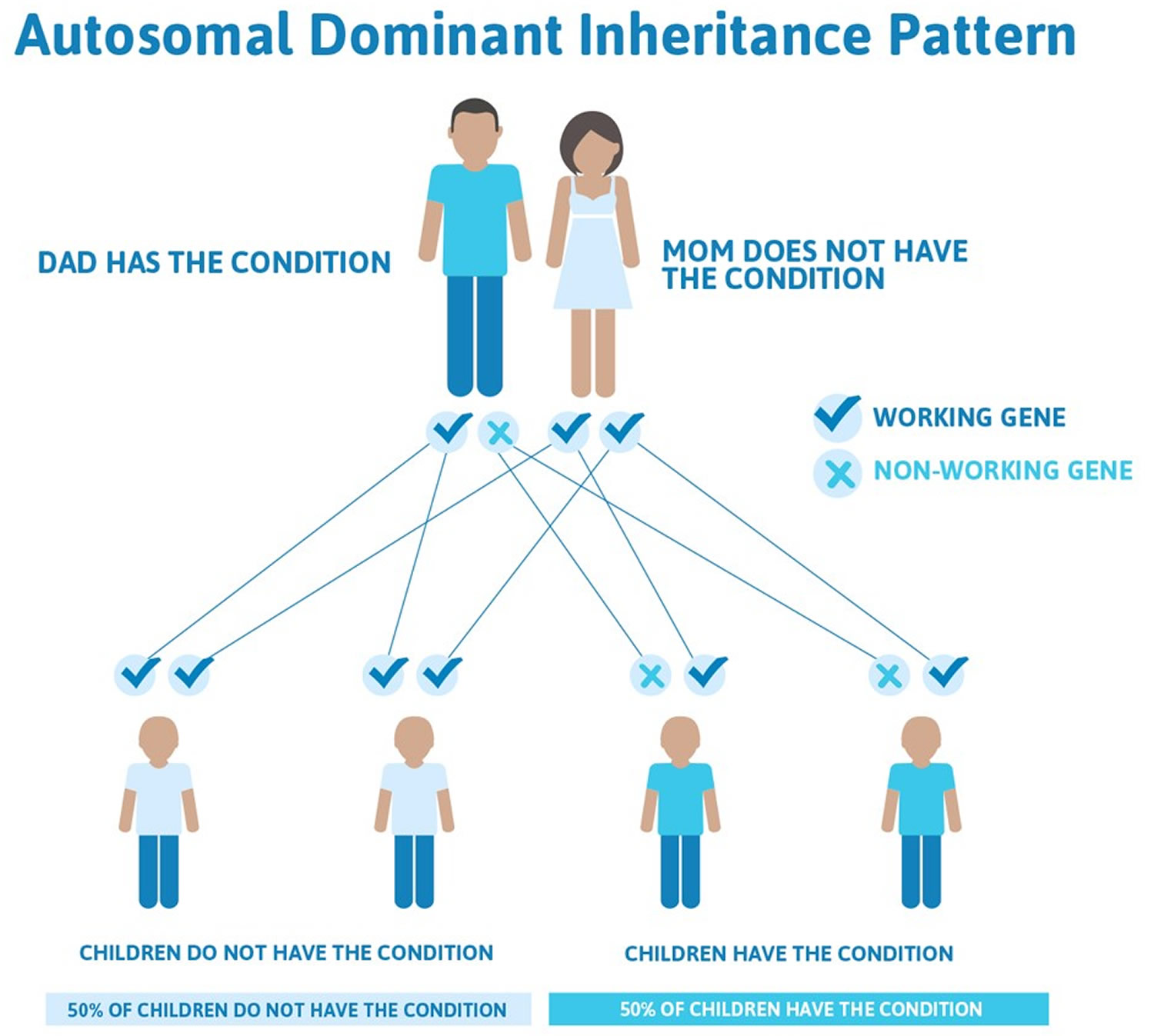
Figure 6. Autosomal recessive inheritance pattern
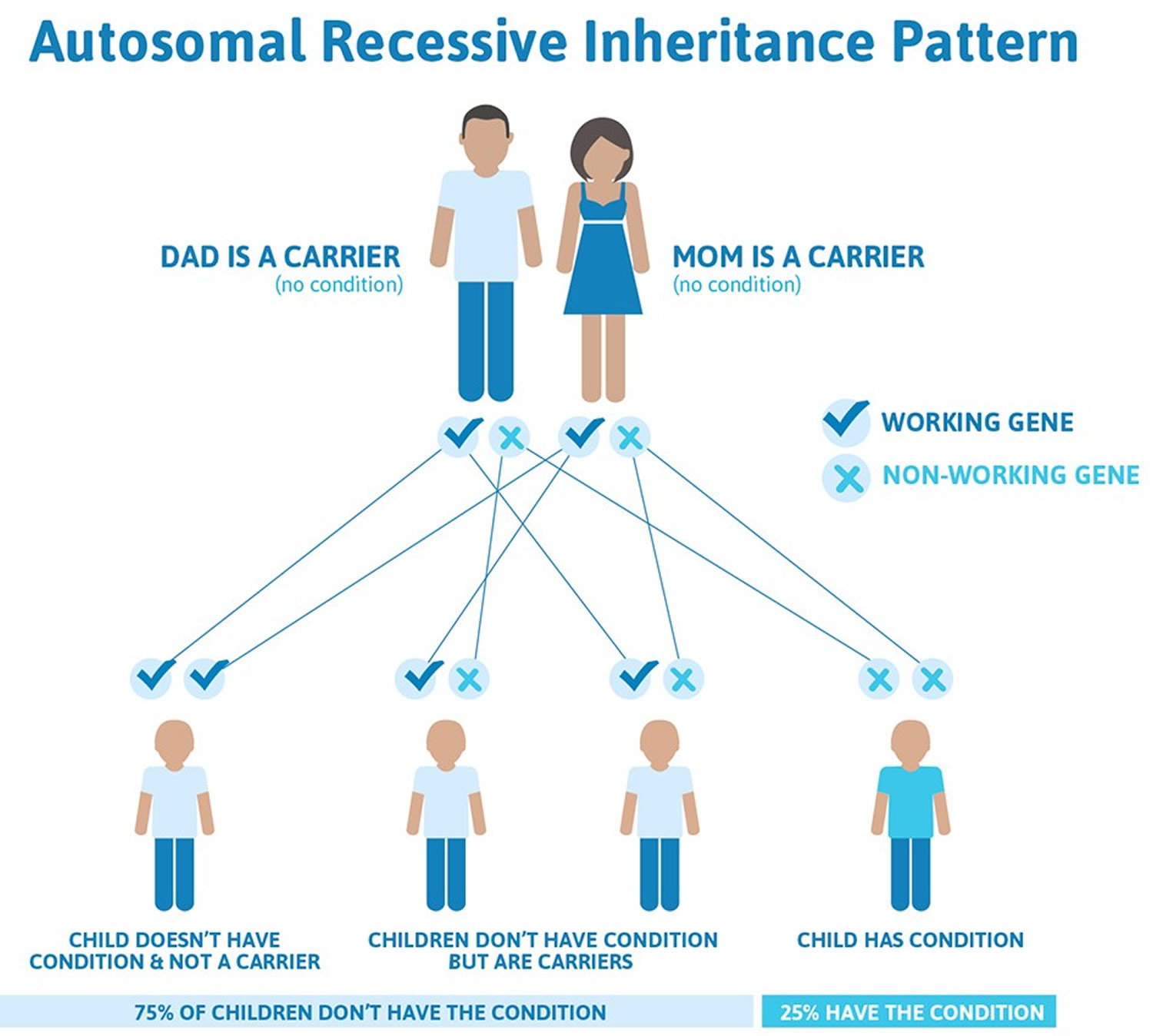
Ehlers-Danlos syndrome prevention
If you have a personal or family history of Ehlers-Danlos syndrome and you’re thinking about starting a family, you may benefit from talking to a genetic counselor — a health care professional trained to assess the risk of inherited disorders. Genetic counseling can help you understand the inheritance pattern of the type of Ehlers-Danlos syndrome that affects you and the risks it poses for your children.
Ehlers Danlos syndrome symptoms
Clinical manifestations of an Ehlers-Danlos syndrome are most often joint and skin related and may include:
Joints
Joint hypermobility; loose/unstable joints which are prone to frequent dislocations and/or subluxations; joint pain; hyperextensible joints (they move beyond the joint’s normal range); early onset of osteoarthritis.
Skin
Soft velvety-like skin; variable skin hyper-extensibility; fragile skin that tears or bruises easily (bruising may be severe); severe scarring; slow and poor wound healing; development of molluscoid pseudo tumors (fleshy lesions associated with scars over pressure areas).
Miscellaneous/Less Common
Chronic, early onset, debilitating musculoskeletal pain (usually associated with the Hypermobility Type); arterial/intestinal/uterine fragility or rupture (usually associated with the Vascular Type); scoliosis at birth and scleral fragility (associated with the Kyphoscoliosis Type); poor muscle tone (associated with the Arthrochalasia Type); mitral valve prolapse; and gum disease.
Each type of Ehlers-Danlos syndrome is defined as a distinct problem in connective tissue. Connective tissue is what the body uses to provide strength and elasticity; normal connective tissue holds strong proteins that allow tissue to be stretched but not beyond its limit, and then safely return that tissue to normal. Connective tissue is found throughout the body, and Ehlers-Danlos syndromes are structural problems. An analogy: If one builds a house with faulty materials, say half the necessary wood or with soft aluminum nails, it is certain there will be problems. Some problems are more likely to show up than others, but because those materials were used everywhere and are not necessarily visible, one can be surprised by where a problem shows up or how serious it is.
It is much the same thing with an Ehlers-Danlos Syndrome and connective tissue.
The connective tissue a person with Ehlers-Danlos syndrome is built with is not structured the way it should be. With a badly-constructed or processed connective tissue, some or all of the tissue in the Ehlers-Danlos syndrome-affected body can be pulled beyond normal limits which causes damage. Connective tissue can be found almost anywhere, in skin, muscles, tendons and ligaments, blood vessels, organs, gums, eyes, and so on.
The problems resulting from one’s body being built out of a protein that behaves unreliably can be widespread and in a wide range of severity. It shows up in places that seem unrelated until the underlying connection to an Ehlers-Danlos syndrome is recognized.
Hypermobile Ehlers-Danlos syndrome
Hypermobile Ehlers-Danlos syndrome (hEDS) is often thought to be the same as or very similar to another condition called joint hypermobility syndrome.
People with hypermobile Ehlers-Danlos syndrome may have:
- Joint hypermobility. Because the connective tissue that holds joints together is looser, your joints can move far past the normal range of motion. Joint pain and dislocations are common.
- Stretchy skin. Weakened connective tissue allows your skin to stretch much more than usual. You may be able to pull a pinch of skin up away from your flesh, but it will snap right back into place when you let go. Your skin might also feel exceptionally soft and velvety.
- Fragile skin. Damaged skin often doesn’t heal well. For example, the stitches used to close a wound often will tear out and leave a gaping scar. These scars may look thin and crinkly.
- Loose, unstable joints that dislocate easily
- Joint pain and clicking joints
- Extreme tiredness (fatigue)
- Skin that bruises easily
- Digestive problems, such as heartburn and constipation
- Dizziness and an increased heart rate after standing up
- Problems with internal organs, such as mitral valve prolapse or organ prolapse
- Problems with bladder control (stress incontinence)
Currently, there are no tests to confirm whether someone has hypermobile Ehlers-Danlos syndrome. The diagnosis is made based on a person’s medical history and a physical examination.
Classical Ehlers-Danlos syndrome
Classical Ehlers-Danlos syndrome (cEDS) is less common than hypermobile Ehlers-Danlos syndrome and tends to affect the skin more.
People with classical Ehlers-Danlos syndrome may have:
- joint hypermobility
- loose, unstable joints that dislocate easily
- stretchy skin
- fragile skin that can split easily – especially over the forehead, knees, shins and elbows
- smooth, velvety skin that bruises easily
- wounds that are slow to heal and leave wide scars
- hernias and organ prolapse
Vascular type Ehlers-Danlos syndrome,
People who have vascular Ehlers-Danlos syndrome (vEDS) – considered to be the most serious, often share distinctive facial features of a thin nose, thin upper lip, small earlobes and prominent eyes. They also have thin, translucent skin that bruises very easily. In fair-skinned people, the underlying blood vessels are very visible through the skin.
Ehlers-Danlos syndrome, vascular type, can weaken your heart’s largest artery (aorta), as well as the arteries to other regions of your body. A rupture of any of these larger blood vessels can be fatal. The vascular type can also weaken the walls of the uterus or large intestines — which also may rupture.
People with vascular Ehlers-Danlos syndrome (vEDS) may have:
- skin that bruises very easily
- thin skin with visible small blood vessels, particularly on the upper chest and legs
- fragile blood vessels that can bulge or tear, resulting in serious internal bleeding
- a risk of organ problems, such as the bowel tearing, the womb tearing (in late pregnancy) and partial collapse of the lung
- hypermobile fingers and toes, unusual facial features, (such as a thin nose and lips, large eyes and small earlobes), varicose veins and delayed wound healing
Kyphoscoliotic Ehlers-Danlos syndrome
Kyphoscoliotic Ehlers-Danlos syndrome (kEDS) is rare.
People with kyphoscoliotic Ehlers-Danlos syndrome may have:
- curvature of the spine – this starts in early childhood and often gets worse in the teenage years
- joint hypermobility
- loose, unstable joints that dislocate easily
- weak muscle tone from childhood (hypotonia) – this may cause a delay in sitting and walking, or difficulty walking if symptoms get worse
- fragile eyes that can easily be damaged
- soft, velvety skin that is stretchy, bruises easily and scars
Ehlers Danlos syndrome diagnosis
If you think you might have one of the Ehlers-Danlos syndromes or hypermobility spectrum disorders (HSD), and particularly if someone in your immediate family has been diagnosed, ask your doctor if a diagnosis fits your symptoms. If they choose to, any doctor who can diagnosis a disease is able to diagnose Ehlers-Danlos syndrome/hypermobility spectrum disorders (HSD); but most likely you’ll be given a referral to a geneticist, because Ehlers-Danlos syndrome are genetic disorders and geneticists are most adept at distinguishing between those diseases, as well as in doing any testing necessary to differentiate Ehlers-Danlos syndrome/hypermobility spectrum disorders from the more than 200 other heritable connective tissue disorders.
A diagnosis is important because, although Ehlers-Danlos syndrome/hypermobility spectrum disorders (HSD) are not curable, they are treatable. Knowing the type of Ehlers-Danlos syndrome/hypermobility spectrum disorders gives you and your medical team some idea of where problems might come from and why they’re happening. When eventually there is a cure, you’ll know to use it. And as more of us are diagnosed, Ehlers-Danlos syndrome/hypermobility spectrum disorders gain the attention all of us need, increasing the likelihood of expanded research that might lead to finding that cure.
Your path to an Ehlers-Danlos syndrome/hypermobility spectrum disorders (HSD) diagnosis starts with an examination. There may be physical testing: using the Beighton Scale to assess how mobile your joints are, a search for abnormal scarring and testing your skin to determine what it feels like and how much it stretches, as well as any additional tests your particular doctor feels are needed. There’s likely to be a look into your medical history to look for conditions and problems associated with Ehlers-Danlos syndrome/hypermobility spectrum disorders, and a discussion of your family to help determine if an Ehlers-Danlos syndrome/hypermobility spectrum disorders was inherited.
Diagnosis of an Ehlers-Danlos syndrome subtype comes by finding the one that most matches your symptoms. There are clinical criteria that help guide diagnosis; your signs and symptoms will be matched up to the major and minor criteria to identify the subtype that is the most complete fit. There is substantial symptom overlap between the Ehlers-Danlos syndrome subtypes and the other connective tissue disorders including hypermobility spectrum disorders, as well as a lot of variability between them. So a definitive diagnosis for all the Ehlers-Danlos syndrome subtypes—except for hypermobile Ehlers-Danlos syndrome (hEDS)—also calls for confirmation by testing to identify the responsible variant for the gene affected in each subtype. These molecular testing results also provide the basis for genetic counseling for our families, guidance on treatment options for ourselves, and help in reaching research goals.
The genetic basis for hypermobile Ehlers-Danlos syndrome is still unknown, so an hypermobile Ehlers-Danlos syndrome (or hypermobility spectrum disorders) diagnosis rests on the criteria and what your doctor finds during your examination. The hypermobile Ehlers-Danlos syndrome criteria also established serious consideration of joint hypermobility with all related symptoms and conditions, with hypermobile Ehlers-Danlos syndrome at one end of the spectrum. hypermobility spectrum disorders can be no less consequential than hypermobile Ehlers-Danlos syndrome (hEDS), either to your health or concern for treatment.
Ehlers Danlos syndrome treatment
There is no cure for Ehlers-Danlos syndrome, but treatment can help you manage your symptoms and prevent further complications.
Medications
Your doctor may prescribe drugs to help you control:
- Pain. Over-the-counter pain relievers — such as acetaminophen (Tylenol, others) ibuprofen (Advil, Motrin IB, others) and naproxen sodium (Aleve) — are the mainstay of treatment. Stronger medications are only prescribed for acute injuries.
- Blood pressure. Because blood vessels are more fragile in some types of Ehlers-Danlos syndrome, your doctor may want to reduce the stress on the vessels by keeping your blood pressure low.
Physical therapy
Joints with weak connective tissue are more likely to dislocate. Exercises to strengthen the muscles and stabilize joints are the primary treatment for Ehlers-Danlos syndrome. Your physical therapist might also recommend specific braces to help prevent joint dislocations.
Surgical and other procedures
Surgery may be recommended to repair joints damaged by repeated dislocations. However, your skin and the connective tissue of the affected joint may not heal properly after the surgery.
Surgery may be necessary to repair ruptured blood vessels or organs in people with Ehlers-Danlos syndrome, vascular type.
Home remedies
If you have Ehlers-Danlos syndrome, it’s important to prevent injuries. Here are a few things you can do to safeguard yourself.
- Choose sports wisely. Walking, swimming, tai chi, recreational biking, using an elliptical machine or a stationary bike are all good choices. Avoid contact sports, weightlifting and other activities that increase your risk of injury. Minimize stress on your hips, knees and ankles.
- Rest your jaw. To protect your jaw joint, avoid chewing gum, hard rolls and ice. Take breaks during dental work to close your mouth.
- Avoid certain musical instruments. To prevent a collapsed lung, avoid playing reeded wind or brass instruments. A violin, viola or piano would be safer options and would take advantage of the increased flexibility of your hands.
Coping and support
Coping with a lifelong illness is challenging. Depending on the severity of your symptoms, you may face challenges at home, at work and in your relationships with others. Here are some suggestions that may help you cope:
- Increase your knowledge. Knowing more about Ehlers-Danlos syndrome can help you take control of your condition. Find a doctor who’s experienced in the management of this disorder.
- Tell others. Explain your condition to family members, friends and your employer. Ask your employer about any accommodations that you feel will make you a more productive worker.
- Build a support system. Cultivate relationships with family and friends who are positive and caring. It also may help to talk to a counselor or clergy member. Many people with Ehlers-Danlos syndrome can benefit from seeing a psychologist or social worker.
Support groups, either online or in person, help people share common experiences and potential solutions to problems.
Helping your child cope
If you are a parent of a child with Ehlers-Danlos syndrome, consider these suggestions to help your child:
- Maintain normalcy. Treat your child like other children. Ask others — grandparents, aunts, uncles, teachers — to do the same.
- Be open. Allow your child to express his or her feelings about having Ehlers-Danlos syndrome, even if it means being angry at times. Make sure your child’s teachers and other caregivers know about your child’s condition. Review with them appropriate caregiving skills, particularly in the event of a fall or injury.
- Promote safe activity. Encourage your child to participate in physical activities with appropriate boundaries. Discourage contact sports while encouraging non-weight-bearing activities, such as swimming. Your child’s doctor or physical therapist also may have recommendations.
- EDS Types. https://www.ehlers-danlos.com/eds-types/#chart[↩]
- The Ehlers–Danlos Syndromes, Rare Types. American Journal of Medical Genetics Part C (Seminars in Medical Genetics) 175C:70–115 (2017). https://www.ehlers-danlos.com/pdf/2017-FINAL-AJMG-PDFs/Brady_et_al-2017-American_Journal_of_Medical_Genetics_Part_C-_Seminars_in_Medical_Genetics.pdf[↩][↩][↩][↩][↩][↩][↩][↩]
- Diagnosis, natural history, and management in vascular Ehlers–Danlos syndrome. American Journal of Medical Genetics Part C (Seminars in Medical Genetics) 175C:40-47 (2017). https://www.ehlers-danlos.com/pdf/2017-FINAL-AJMG-PDFs/Byers_et_al-2017-American_Journal_of_Medical_Genetics_Part_C-_Seminars_in_Medical_Genetics.pdf[↩]




 and fragile.){kind=link}