Fatal insomnia
Fatal insomnia is a rare prion disease that interferes with sleep and leads to deterioration of mental function and loss of coordination 1. Prion diseases are caused by the accumulation of misfolded prion proteins in the brain. Two other prion diseases, Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker syndrome, may also occur as a result of variations of the prion-related protein (PRPN) gene, although some prion diseases occur in the absence of a genetic variation. Generally, prion disorders are characterized by long incubation periods and short clinical duration, which means the abnormal prions may be build up for many years (long incubation period), but once symptoms begin the disorder rapidly worsens. Death occurs within a few months to a few years.
Fatal insomnia has two forms:
- Fatal familial insomnia (FFI): Fatal familial insomnia is an inherited form and almost all cases of fatal familial insomnia result from mutations in the cellular prion protein (PrP) gene or prion-related protein (PRPN) gene that are inherited in an autosomal dominant manner 2. There are a very small number of reported sporadic cases of fatal familial insomnia 3. Average age at onset is 40 year (ranging from the late 20s to the early 70s). The first symptoms of fatal familial insomnia usually begin in mid-life and may include increasing difficulty falling asleep and maintaining sleep, progressive insomnia, weight loss, lack of appetite, too high or too low body temperature, rapidly progressive dementia (cognitive decline), ataxia, and psychiatric symptoms. Sympathetic hyperactivity (eg, hypertension, tachycardia, hyperthermia, sweating) may occur later. There is currently no effective treatment for fatal familial insomnia, but research for a treatment and cure is ongoing 4. Life expectancy is 7 to 73 months. Death usually occurs within 12-18 months of the first symptoms 5.
- Sporadic fatal insomnia (SFI): Sporadic fatal insomnia occurs spontaneously, without a genetic mutation. Average age at onset is slightly older and life expectancy is slightly longer than in fatal familial insomnia. Early symptoms include cognitive decline and ataxia. Sleep abnormalities are not commonly reported but can usually be observed during a sleep study.
Fatal familial insomnia and sporadic fatal insomnia differ from other prion diseases because they affect predominantly one area of the brain, the thalamus, which influences sleep.
In fatal familial insomnia, symptoms may begin in a person’s late 20s to the early 70s (average is 40 years). Death usually occurs 7 to 73 months after symptoms begin. The sporadic form begins slightly later, and life expectancy is slightly longer.
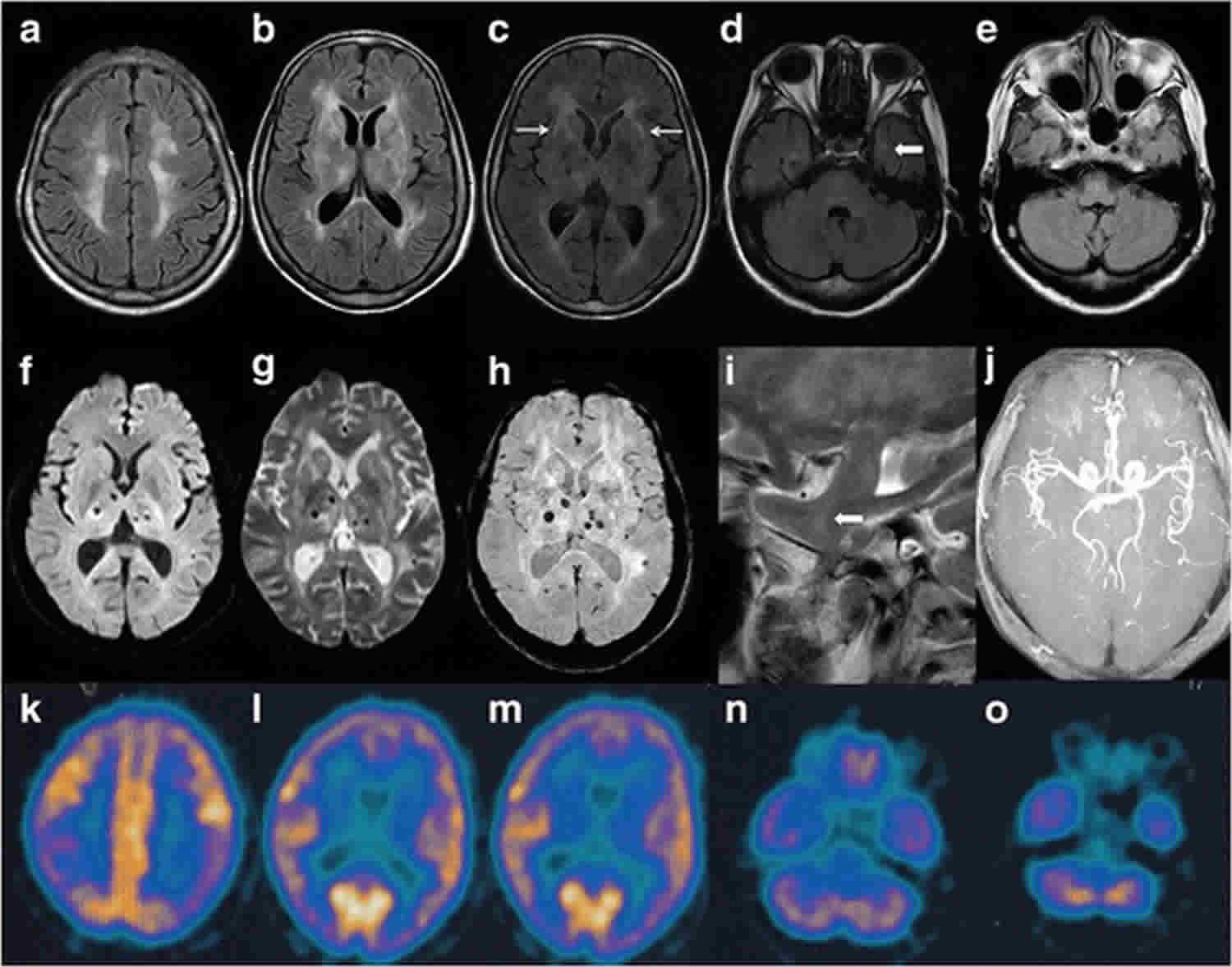
Fatal insomnia should be considered as a rare possibility when patients have rapidly progressive cognitive impairment accompanied by behavioral or mood changes, ataxia, and sleep disturbances. Suspicion of fatal familial insomnia or sporadic fatal insomnia should prompt a sleep study by polysomnography. Genetic testing can confirm the diagnosis of the familial form. MRI and measurement of 14-3-3 protein and tau in CSF are not useful, but polysomnography and PET (which shows thalamic hypometabolism) can confirm the diagnosis.
Fatal familial insomnia is an extremely rare disorder. The exact incidence and prevalence of the disorder is unknown. The sporadic form of fatal familial insomnia, known as sporadic fatal insomnia (SFI), is extremely rare and has only been described in the medical literature in about two dozen people. Collectively, prion disorders affect about 1 in 1,000,000 million people in the general population per year. Genetic prion diseases are believed to make up about 15% of all individuals with prion diseases. Because rare diseases often go undiagnosed or misdiagnosed, it is difficult to determine their true frequency in the general population. Fatal familial insomnia affects men and women in equal numbers. The average age of onset is 45-50 years old, although the disorder has been described occurring in individuals in their teens and as late as their 70s. Fatal familial insomnia has been described in populations around the world.
There is only supportive treatment for fatal insomnia.
Fatal familial insomnia
Fatal familial insomnia (FFI) is a rare inherited prion disease or a transmissible spongiform encephalopathy that mainly affects the thalamus 6. The thalamus is the part of the brain that controls the sleep-wake cycle, but is also known as the “relay center” of the brain because it helps the different parts of the brain communicate with each other 7. Like all prion diseases, fatal familial insomnia is a progressive neurodegenerative disease, which means over time there are fewer neurons (nerve cells). Loss of neurons in the thalamus, as well as other mechanisms not yet fully understood, cause the symptoms of fatal familial insomnia 8.
Fatal familial insomnia (FFI) is characterized by an inability to sleep (insomnia) that may be initially mild, but progressively worsens, leading to significant physical and mental deterioration. Affected individuals may also develop dysfunction of the autonomic nervous system, the part of the nervous system that controls involuntary or automatic body processes, which are things that happen without a person thinking about them, such as body temperature regulation, sweating, breathing or regulating the heart rate. Specific symptoms observed depend on the part of the autonomic nervous system that is affected by the disease. In all instances, fatal familial insomnia is caused by an abnormal variant in the prion-related protein (PRPN) gene, although sometimes, the disorder occurs randomly, without a variant PRPN gene (sporadic fatal insomnia). The PRNP gene regulates the production of the human prion protein. Alterations in this gene lead to the generation of abnormally-shaped (misfolded) prion protein, also known simply as a “prion”, which is toxic to the body. In fatal familial insomnia, the abnormal prions build up primarily within the thalamus of the brain. This leads to the progressive loss of nerve cells (neurons) and the various symptoms associated with this disorder. There is no cure, but investigators are researching ways to best treat and manage fatal familial insomnia.
Fatal familial insomnia causes
Fatal familial insomnia (FFI) is a very rare form of genetic prion disease. In almost every case it is caused by a very specific mutation in the PRNP gene. This mutation causes the prion protein (PrP) that is made from PRNP gene to be a different shape (fold incorrectly). Since the protein has a different shape, it cannot work correctly 6. The exact function of prion protein (PrP) in the body is not fully understood. However, because of the variant gene, the prion protein (PrP) that is produced develops an abnormal 3-dimensional shape that is described simply as “misfolded”. The misfolded prion protein (PrP) is toxic to the body, especially cells of the nervous system. In fatal familial insomnia, misfolded prion protein (PrP) is primarily found in the thalamus, which is a structure deep within the brain that helps to regulate many functions of the body including sleep, appetite, and body temperature. As the misfolded prion protein (PrP) builds up in the thalamus, it results in a progressive destruction of nerve cells (neurons), which leads to the symptoms of the disorder. The damage to brain tissue may appear as sponge-like holes or gaps when examined under a microscope, which is why prion diseases like fatal familial insomnia are called transmissible spongiform encephalopathies.
The term “prion” was coined to designate a “proteinaceous infectious agent” to explain the transmissible nature of prion diseases. Extensive research has shown that a prion is essentially the misfolded prion protein (PrP). However, it is important to know that fatal familial insomnia is not contagious in the traditional sense because the only way to transmit prion disease to a healthy individual is through direct exposure to disease-affected brain tissue, perhaps by ingestion or injection. If a person without an underlying genetic defect develops a prion disease, they are said to have an ‘acquired’ form. For example, variant Creutzfeldt-Jakob disease occurred in the United Kingdom when people ate prion-contaminated beef. A lesser known example is kuru. Kuru is a virtually extinct prion disease that occurred in the Fore people of Papua New Guinea. The disease spread throughout this population because of the villagers’ practice of eating the brains of deceased kuru-affected tribesmen (ritualistic cannibalism).
In rare instances, the change (variation) in the PRPN gene in individuals with fatal familial insomnia occurs spontaneously, without a family history of the disease. This is called a new or de novo variant. The gene variation has occurred at the time of the formation of the egg or sperm for that child only, and no other family member will be affected. The disorder is usually not inherited from or “carried” by a healthy parent. However, the person who has this de novo variant could pass on the variant gene to their offspring in an autosomal dominant manner. As of 2016, there have only been 24 reported cases of sporadic fatal familial insomnia 3. Sporadic fatal familial insomnia occurs when some of a person’s normal PrP (prion protein) spontaneously changes into the abnormal shape which causes fatal familial insomnia, and then somehow changes the shape of prion protein (PrP) in other neurons in a chain reaction 9.
The abnormally shaped PrP (prion protein) causes changes in the thalamus including the progressive loss of neurons (nerve cells) 8. The thalamus relays messages between different parts of the brain. It manages our sleep/wake cycle; the flow of visual, auditory, and motor information; our sense of balance; how we experience pain; aspects of learning, memory, speech and understanding language; and even emotional experiences, expression, and our personalities. Losing neurons in the thalamus causes many of the symptoms of fatal familial insomnia because the thalamus can no longer do all of its jobs well 10.
Although the main target of fatal familial insomnia is the thalamus, other parts of the brain are affected as well including the inferior olives. The inferior olives are part of the medulla oblongata and are important for coordinating our movements (motor control). Losing neurons in the inferior olives can make it harder for a person to control their movements as seen in later stages of fatal familial insomnia. Medical researchers are still working to understand how the abnormally folded PrP causes the progressive changes in the thalamus and other affected brain areas 6.
Fatal familial insomnia symptoms
The characteristic symptom in fatal familial insomnia is progressive insomnia. Insomnia often begins during middle age, but it can occur earlier or later in life. Insomnia may first be mild, but it then become progressively worse until an affected individual gets very little sleep. Insomnia usually begins suddenly and can rapidly worsen over the next few months. When sleep is achieved, vivid dreams may occur. The lack of sleep leads to physical and mental deterioration and the disease ultimately progresses to coma and death.
Although insomnia is usually the first symptom, some individuals may present with progressive dementia, in which there are worsening problems with thought, cognition, memory, language, and behavior. Initially, the signs may be subtle and include unintended weight loss, forgetfulness, inattentiveness, problems concentrating, or speech problems. Episodes of confusion or hallucinations can eventually occur.
Some affected individuals may experience double vision (diplopia) or abnormal, jerky eye movements (nystagmus). There may be problems with swallowing (dysphagia) or slurred speech (dysarthria). Some individuals eventually have trouble coordinating voluntary movements (ataxia). Abnormal movements including tremors or twitchy, jerking muscle spasms (myoclonus), or Parkinson’s-like symptoms may also develop.
Additional symptoms involving dysfunction of the autonomic nervous system often develop. Specific symptoms can vary from one person to another based on the specific part of the autonomic nervous system affected. Common symptoms can include fever, rapid heart rate (tachycardia), high blood pressure (hypertension), increased sweating (hyperhidrosis), increased production of tears, constipation, variations in body temperature, and sexual dysfunction including erectile dysfunction. Anxiety and depression are common findings as well.
Fatal familial insomnia staging
Fatal familial insomnia has been described to have 4 stages 11:
- Stage 1: The first stage of the disease is identified by the subacute onset of insomnia which worsens over a period of few months and causes psychiatric symptoms such as phobia, paranoia and panic attacks. During this time, patients may report lucid dreaming.
- Stage 2: The next 5-month period, psychiatric symptoms worsen along with worsening insomnia and patients experience hallucinations. Autonomic dysfunction in the form of sympathetic hyperactivity is seen.
- Stage 3: This short stage of around 3 months is typically dominated by total insomnia and complete disruptions of the sleep-wake cycle.
- Stage 4: The final stage of the disease can last for 6 months or more and is defined by rapid cognitive decline and dementia. Patients experience an inability to voluntarily move or speak which can be followed by coma and eventual death.
Fatal familial insomnia diagnosis
The diagnosis of fatal familial insomnia (FFI) is first suggested by rapidly progressive cognitive impairment (dementia) along with behavior or mood changes, ataxia and sleep disturbances. Further diagnosis will include a sleep study and possibly a PET scan to confirm thalamic hypometabolism (meaning the thalamus in the brain is less active than it should be) 2. Polysomnography, also called a sleep study, may be performed on affected individuals to demonstrate a reduced amount of time sleeping and difficulties transitioning through the various sleep stages.
Lab tests
- Initial workup should include complete blood count, erythrocyte sedimentation rate, serum chemistry, liver function tests, and ammonia levels along with blood cultures for suspected bacterial infections.
- Additionally, investigating reversible causes of cognitive decline should include thyroid function tests, vitamin B-12, and folate levels along with testing for neurosyphilis is warranted. HIV testing is important in patients with known risk factors.
Imaging studies
Positron emission tomography or PET scan is an advanced imaging technique that can be useful in diagnosing fatal familial insomnia. During a PET scan, three-dimensional images are produced that reflect the brain’s metabolic activity and can show reduced activity within the thalamus (thalamic hypometabolism), as a characteristic feature. The recommended PET scan is the fluorodeoxyglucose positron emission tomography (FDG-PET) 5.
Other advanced imaging techniques include computerized tomography (CT) scanning and magnetic resonance imaging (MRI). CT scanning is not useful in the diagnosis of Fatal familial insomnia or prion disease, while the MRI can show some abnormalities in the scan that may support prion disease, although its application to diagnose Fatal familial insomnia is not well characterized. However, MRI and CT may be helpful in ruling out other conditions that may mimic Fatal familial insomnia or prion disease. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of certain tissue structures. An MRI uses a magnetic field and radio waves to produce cross-sectional images of particular organs and body tissues.
Cerebrospinal fluid (CSF) Studies
Physicians may run tests to detect the presence of the 14-3-3 protein. This is a normal protein that is produced when nerve cells die. Sometimes, in individuals with fatal familial insomnia, levels of this protein increase substantially in the cerebrospinal fluid (CSF). CSF is the colorless fluid that surrounds the brain and spinal cord and provides protection and support. Elevated levels of 14-3-3 in the CSF does not always occur, and normal levels of this protein does not rule out Fatal familial insomnia. The tau protein is also often elevated in the CSF of prion disease, although because of the rarity of Fatal familial insomnia, the usefulness of testing for tau in Fatal familial insomnia is not fully understood. More recently, a test called RTQuIC (real-time quaking induced conversion), that helps to detect low levels of prions in CSF, is now being used to assist in the diagnosis of prion disease and may be useful for Fatal familial insomnia, but there is currently not enough data on that.
Molecular Genetic Testing
Suspected patients should undergo genetic testing in the form of targeted analysis for the pathogenic variant of PRNP or full gene sequencing.
Genetic testing can confirm the diagnosis, but in the United States is only available if the person meets one of the following three criteria:
- Family history of fatal familial insomnia
- Abnormal sleep study or PET scan (consistent with strong suspicion of fatal familial insomnia)
- Diagnosis of fatal familial insomnia (usually through a combination of sleep study results and PET scan results)
Carrier testing for at-risk relatives and prenatal testing are possible for families with a confirmed diagnosis of fatal familial insomnia 5.
How is the sleep study of a person with fatal familial insomnia different from another person with insomnia?
The sleep study EEG of a person who has insomnia but not fatal familial insomnia shows mainly delta wave activity (high-amplitude slow waves of sleep stage 3 or deep sleep) and episodes of “micro-sleep”. There are also brief bursts of K-complexes (large waves in sleep stage 2 or light sleep) and sleep spindles (small bursts of activity in sleep stage 2). Insomnia with this pattern is associated with perceiving something that isn’t there (disturbed perception), moments of not being aware of self or surroundings (lapses of consciousness), and quickly changing and often extreme behavior or emotions (erratic behavior) 12.
In people with Fatal familial insomnia, sleep study EEGs contain a mixture of rhythms that are neither typical of wake nor of light sleep, and may be called “subwakefulness.” There is a clear absence of sleep spindles and K-complexes. Total sleep time is shorter and the sleep study shows only slow activity or desynchronization without rapid eye movements (REM) 12.
Fatal familial insomnia treatment
There is currently no cure for fatal familial insomnia (FFI) or treatment that can slow the disease progression. The management goal is to ease symptoms and keep the person with fatal familial insomnia as comfortable as possible 9. However research is ongoing and a number of potential treatments are being developed 8.
Different treatment modalities mentioned in the literature are as follows 13:
- Discontinuation of medications that may exacerbate confusion, memory and/or insomnia is important.
- Fatal familial insomnia patients show poor response to sedatives. Tinuper P et al. 14 described in lack of effect of barbiturates or benzodiazepines on EEG in fatal familial insomnia patients.
- Problems with swallowing may warrant placement of a feeding tube.
- Reder AT et al. 15 investigated gamma-hydroxybutyrate (GHB), and found its administration to induce short-wave sleep in a patient with fatal familial insomnia.
- Several treatment options using compounds such as pentosane polysulphate, quinacrine, amphotericin B have been studied with inconclusive results.
- An Italian study using doxycycline, as a treatment option, over a period of 10 years is ongoing.
As of 2016, a number of treatments have had some success in slowing disease progression in animal models, including pentosan polysulfate, quinacrine (mepacrine), and amphotericin B. Sadly, the results have been less clear in clinical trials in humans 4. In Italy, a clinical trial trying to prevent symptoms in people known to have the genetic changes for fatal familial insomnia but have not yet developed symptoms is underway using doxyclycline 16. Most promisingly, several forms of immunotherapy have reported success. The three main research areas focus on antibody vaccines, dendritic cell vaccines, and adoptive transfer of physiological prion protein-specific CD4+ T-lymphocytes. More research is being done to study how well these treatments work (effectiveness) and if the treatments are safe, but medical researchers believe that these or similar immunotherapies may offer hope for those with fatal familial insomnia in the future 3.
Fatal familial insomnia prognosis
Presently, after symptoms of fatal familial insomnia (FFI) begin, disease course can last from 7 to 36 months, with an average duration of 18 months leading to eventual death. Fatal familial insomnia usually causes death within 12 to 18 months, with a range of a few months to several years 5. Patients with homozygous (Met-Met) mutation have a shorter mean survival time compared to heterozygous (Met-Val) patients 17. As research continues however, it is hoped a treatment or even a cure will be developed that will dramatically change the outlook for people who have fatal familial insomnia 18.
Sporadic fatal insomnia
Sporadic fatal insomnia (SFI) is a rapid progressive neurodegenerative disease characterised by gradual to perpetual insomnia, followed by dysautonomia, coma and death 19. The cause of sporadic fatal insomnia was recently mapped to a mutation in a protein, the prion, found in the human brain 20. It is the unfolding of the prion that leads to the generation of toxic oligomers that destroy brain tissue and function. Recent studies have confirmed that a methionine mutation at codon 129 of the human Prion is characteristic of sporadic fatal insomnia 20. Current treatment slows down the progression of the disease, but no cure has been found, yet 20.
Sporadic fatal insomnia treatment
Because symptomatic treatment with vitamins B6, B12, iron and Folic Acid has offered some improvement in the wellbeing of patients affected by sporadic fatal insomnia, vitamin supplementation remains a helpful nutritional aspect in the management of the illness 20. These vitamins are found mainly in fish, fruits and vegetables. Dairy products and other aliments also contain them to a lower extent, however dairy products are still important due to their tryptophan content, which is also beneficial. This strategy has led to prolonged symptomatic relief and to sleep restoration effects, up to 5–7 hours nightly sleep for several consecutive days, within months of therapy 21.
Melatonin supplements could also be introduced in the early stages of the disease since plasma melatonin is drastically decreased. Melatonin is a natural endogenous molecule, therefore side effects will be less frequent. Modified release tablets are already commercially available, and in countries like the UK they are prescribed for the treatment of short-term insomnia at a dosage of 2 mg once daily in adults over 55 years old. Adjusting the dosage to induce sleep might also be helpful 20.
More recent studies have been focused on Doxycycline, a common tetracycline antibiotic for systemic use which has been proven to be effective in disrupting amyloid fibril formation. The function is not that of inhibiting the prion protein, but of early prophylaxis. The efficacy of Doxycycline (DOXY®) is possibly associated with its ability to cross the blood brain barrier and it has been frequently reported that its usage brings increased survival in patients 22.
Further modern treatments involve the use of murine monoclonal antibodies targeting the human prion protein. These are currently in clinical trial, but preclinical reports have described their success in extending survival on in vivo rat models 23.
Concomitantly, the neuroleptic drugs Diazepam, used as anxiolytic, and Midazolam, used for sedation, seem to give short-term relief in people suffering from both sporadic fatal insomnia and Creutzfeldt-Jackob Disease (CJD), another prion disorder. Midazolam is usually administered at later stages of the disease 24. The effectiveness of both Diazepam and Midazolam may be connected to the fact that the FI-prion displays high affinity for Gabaergic receptors; Midazolam at high doses has also proven to cause side effects similar to the symptoms shown in fatal familial insomnia. Likewise, Amphotericin B, an antifungal systemic drug has proven to be effective both in vitro and in vivo 25. Even if the antifungal agent has been recently abandoned, it could serve as a template for more efficient antifungal drug design that targets the thalamus and crosses the blood brain barrier. By contrast, dopamine receptor binders have been favored in the treatment of other transmissible spongiform encephalopathies.
Lastly, the class of antipsychotic drugs phenothiazines, widely used in the treatment of insomnia, seems to be successful in short-term therapy. Many patients have confirmed that after days of sleeplessness, Promethazine worked for twenty-four hours. This class of compounds is particularly important to consider, because of their ability to bind and stabilize prion proteins.
Fatal insomnia symptoms
In fatal familial insomnia, early symptoms include minor difficulties falling and staying asleep and occasional muscle twitching, spasms, and stiffness. During sleep, people may move a lot and kick. Eventually, they cannot sleep at all. Later, mental function deteriorates and coordination is lost (called ataxia). The heart rate may become rapid, blood pressure may increase, and people may sweat profusely.
In the sporadic fatal insomnia, early symptoms include a rapid decline in mental function and loss of coordination. People with this form may not report sleep problems, but sleep studies can detect abnormalities.
Fatal insomnia diagnosis
- A doctor’s evaluation
- Polysomnography and positron emission tomography (PET) scan
- For the familial fatal insomnia, genetic testing
Doctors consider fatal insomnia as a rare possible diagnosis when people have typical symptoms, such as rapidly deteriorating mental function, loss of coordination, and/or sleep problems. The following are done to confirm the diagnosis:
- Polysomnography, which can detect abnormalities in sleep patterns
- Positron emission tomography (PET), which can detect certain abnormalities in the thalamus
The diagnosis of fatal familial insomnia is confirmed by genetic testing.
Fatal insomnia treatment
No treatment is available for fatal insomnia. Treatment of fatal insomnia focuses on relieving symptoms and making the person as comfortable as possible. Measures to help people sleep have been tried, but the benefits were only temporary.
References- Fatal Insomnia. https://www.msdmanuals.com/en-au/home/brain,-spinal-cord,-and-nerve-disorders/prion-diseases/fatal-insomnia
- Mastrianni JA. Genetic Prion Diseases. 2003 Mar 27 [Updated 2014 Jan 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1229
- Saa P, Harris DA and Cervenakova L. Mechanisms of prion-induced neurodegeneration. Expert Reveiws in Molecular Medicine. April 8 2016; 18(e5):1-18. https://www.ncbi.nlm.nih.gov/pubmed/27055367
- Burchell JT and Panegyres PK. Prion diseases: immunotargets and therapy. ImmunoTargets and Therapy. June 16 2016; 5:57-68. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4970640
- Geschwind MD. Prion Diseases. Continuum Journal. December 2015; 21(6):1612-1638. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4879966
- Llorens F, Thüne K, Schmitz M, Ansoleaga B, Frau-Méndez MA, Cramm M, Tahir W, Gotzmann N, Berjaoui S, Carmona M, Silva CJ, Fernandez-Vega I, José Zarranz J, Zerr I, and Ferrer I. Identification of new molecular alterations in fatal familial insomnia. Hum Mol Genet. April 7 2016; [Epub ahead of print]:http://www.ncbi.nlm.nih.gov/pubmed/27056979
- When Stroke Affects the Thalamus. http://strokeconnection.strokeassociation.org/Spring-2015/When-Stroke-Affects-the-Thalamus
- Frau-Méndez MA, Fernández-Vega I, Ansoleaga B, Blanco Tech R, Carmona Tech M, Antonio Del Rio J, Zerr I, Llorens F, José Zarranz J, and Ferrer I. Fatal familial insomnia: mitochondrial and protein synthesis machinery decline in the mediodorsal thalamus. Brain Pathol. June 24 2016; [Epub ahead of print]:https://www.ncbi.nlm.nih.gov/pubmed/27338255
- Transmissible Spongiform Encephalopathies Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Transmissible-Spongiform-Encephalopathies-Information-Page
- Caswell J. When Stroke Affects the Thalamus. Stroke Connection Mag. Spring 2015; http://strokeconnection.strokeassociation.org/Spring-2015/When-Stroke-Affects-the-Thalamus
- Lindsley CW. Genetic and Rare Disease of the CNS. Part I: Fatal Familial Insomnia (FFI). ACS Chem Neurosci. 2017 Dec 20;8(12):2570-2572.
- Schenkein J, Montagna P. Self management of fatal familial insomnia. Part 1: what is FFI?. MedGenMed. 2006;8(3):65. Published 2006 Sep 14. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1781306
- Khan Z, Bollu PC. Fatal Familial Insomnia. [Updated 2019 Jun 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482208
- Tinuper P, Montagna P, Medori R, Cortelli P, Zucconi M, Baruzzi A, Lugaresi E. The thalamus participates in the regulation of the sleep-waking cycle. A clinico-pathological study in fatal familial thalamic degeneration. Electroencephalogr Clin Neurophysiol. 1989 Aug;73(2):117-23.
- Reder AT, Mednick AS, Brown P, Spire JP, Van Cauter E, Wollmann RL, Cervenàkovà L, Goldfarb LG, Garay A, Ovsiew F. Clinical and genetic studies of fatal familial insomnia. Neurology. 1995 Jun;45(6):1068-75.
- Forloni G, Tettamanti M, Lucca U, Albanese Y, Quaglio E, Chiesa R, Erbetta A, Villani F, Redaelli V, Tagliavini F, Artuso V, and Roiter I. Preventive study in subjects at risk of fatal familial insomnia: Innovative approach to rare diseases. Prion. March-April 2015; 9(2):75-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4601344
- Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003 Mar;2(3):167-76.
- Bichell RE. A Couple’s Quest To Stop A Rare Disease Before It Takes One Of Them. National Public Radio (NPR). June 19, 2017; http://www.npr.org/sections/health-shots/2017/06/19/527795512/a-couples-quest-to-stop-a-rare-disease-before-it-takes-one-of-them
- Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A, Tinuper P, Zucconi M, Gambetti P. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. New England J of Med. 1986;315:997–1003. doi:10.1056/NEJM198610163151605
- Tabaee Damavandi P, Dove MT, Pickersgill RW. A review of drug therapy for sporadic fatal insomnia. Prion. 2017;11(5):293–299. doi:10.1080/19336896.2017.1368937 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5639864
- Schenkein J, Montagna P. Self-management of fatal familial insomnia. Part 2: case report. MedGenMed. 2006;8(3):66.
- Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, Prusiner SB. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5(11):e1000673. doi:10.1371/journal.ppat.1000673
- Noack A, Noack S, Hoffmann A, Maalouf K, Buettner M, Couraud PO, Romero IA, Weksler B, Alms D, Römermann K, et al. Drug-induced trafficking of p-glycoprotein in human brain capillary endothelial cells as demonstrated by exposure to mitomycin C. PLoS One. 2014;9(2):e88154. doi:10.1371/journal.pone.0088154
- Klyubin I, Nicoll AJ, Khalili-Shirazi A, Farmer M, Canning S, Mably A, Linehan J, Brown A, Wakeling M, Brandner S, et al. Peripheral administration of a humanized anti-PrP antibody blocks alzheimer’s disease αβ synaptotoxicity. J Neurosci. 2014;34(18):6140–45. doi:10.1523/JNEUROSCI.3526-13.2014
- Wieser HG, Schwarz U, Blättler T, Bernoulli C, Sitzler M, Stoeck K, Glatzel M. Serial EEG findings in sporadic and iatrogenic creutzfeldt-jakob disease. Clin Neurophysiol. 2004;115(11):2467–78. doi:10.1016/j.clinph.2004.05.032





{kind=link}