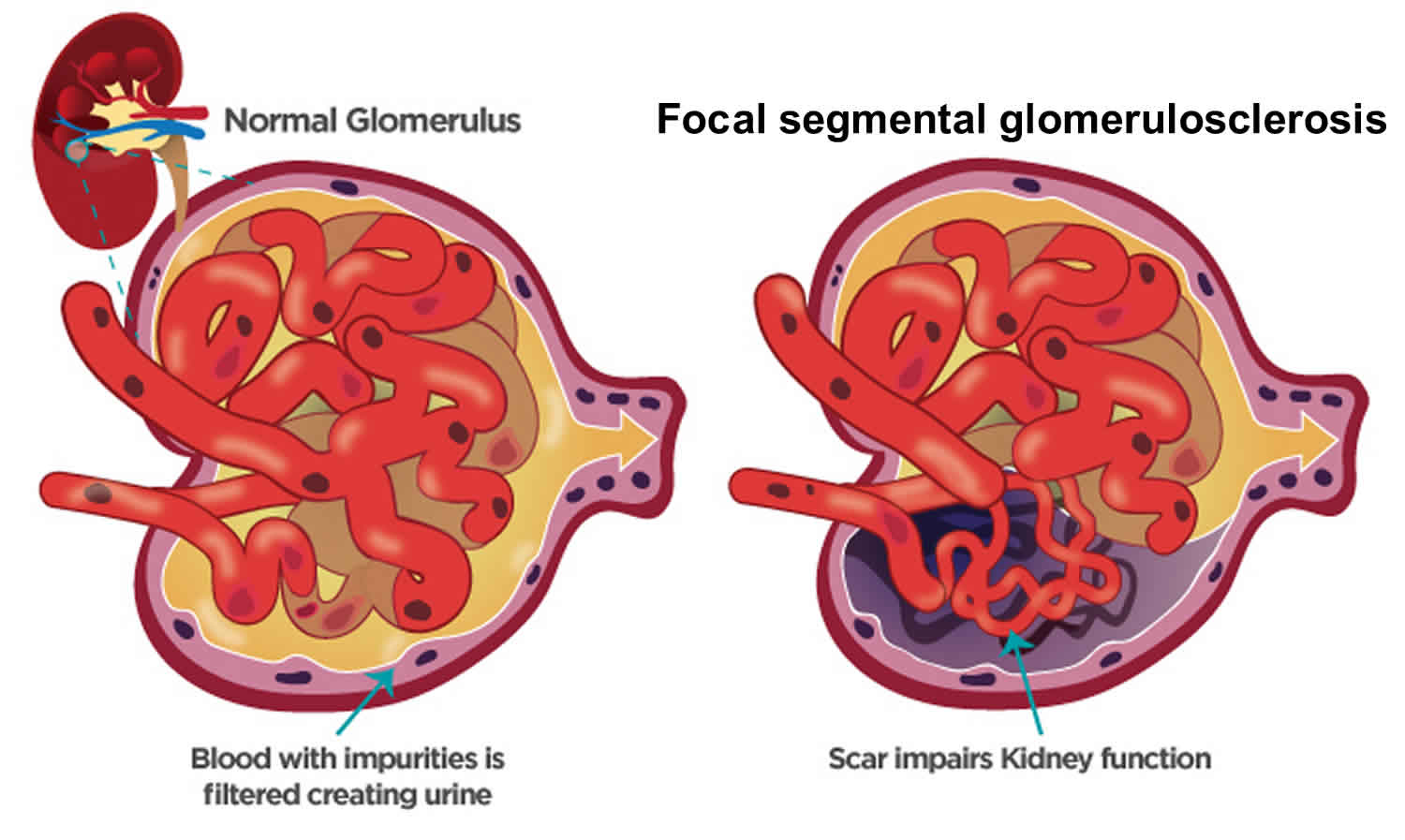
Focal segmental glomerulosclerosis
Glomerulosclerosis is scarring or hardening of the tiny blood vessels called glomerulus (more than one glomerulus are called glomeruli) where your blood is cleaned within your kidneys. Glomerulosclerosis is caused by the activation of glomerular cells to produce scar material. This may be stimulated by molecules called growth factors, which may be made by glomerular cells themselves or may be brought to the glomerulus by the circulating blood that enters the glomerular filter. Damaged glomeruli can allow proteins and sometimes red blood cells to leak into your urine. One of the proteins in your blood is albumin. If too much albumin leaks into your urine, fluid can build up in your body, leading to swelling in your face, hands, feet, or legs. In some cases, glomerular disease can also prevent your kidneys from properly removing waste products, causing wastes to build up in your blood. Focal segmental glomerulosclerosis also called FSGS is one of the most common causes of primary glomerular diseases in adults 1, 2. Focal segmental glomerulosclerosis (FSGS) is scar tissue in the filtering unit of the kidney called the glomerulus 3. The glomeruli serve as filters that help the body get rid of harmful substances. Each kidney has thousands of glomeruli. “Focal” means that some of the glomeruli become scarred. Others remain normal. “Segmental” means that only part of an individual glomerulus is damaged. Focal segmental glomerulosclerosis is a pathologic term, referring to glomerulosclerosis (glomerular scarring, representing an increase in collagens and other proteins) that is found in a focal distribution (initially some glomeruli are affected while most glomeruli are entirely normal) and a segmental distribution (early in disease, the affected glomerular show sclerosis in a segmental pattern while other portions of that glomerulus remains normal). As the disease progresses, this pattern may shift so that some glomeruli are globally (totally) sclerosed and that all glomeruli have at least some sclerosis. Focal segmental glomerulosclerosis causes asymptomatic proteinuria or nephrotic syndrome with or without renal insufficiency. Histologically, focal segmental glomerulosclerosis is characterized by segmental scarring, involving a part of the glomerulus, and affects some but not all glomeruli sampled. Recent research has shed light on the pathogenesis of focal segmental glomerulosclerosis which is podocyte injury and damage, leading to protein loss and subsequent development of focal sclerosing lesions 4. Generally, focal segmental glomerulosclerosis is a progressive form of kidney disease, accounting for 2.3% of end-stage renal disease (ESRD).
Patients with focal segmental glomerulosclerosis (FSGS) may present in different ways. First some present with swelling (edema), either a sudden onset similar to that seen in patients with minimal change disease or slower onset over weeks to months, in their face, hands, feet, legs or ankles. Second, some patients have no symptoms and are found to have no symptoms (asymptomatic) protein in the urine (proteinuria), found on routine urinalysis performed as part of a physical examination. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin.
Focal segmental glomerulosclerosis affects both children and adults. It occurs slightly more often in men and boys. Focal segmental glomerulosclerosis is also more common in African American men. Focal segmental glomerulosclerosis causes up to 40% of cases in adults and 20% in children 5. Focal segmental glomerulosclerosis is the most common cause of idiopathic (or primary) nephrotic syndrome among adults in the US. Though usually idiopathic (unknown cause), focal segmental glomerulosclerosis can occur in association with other factors (secondary focal segmental glomerulosclerosis), including drugs (e.g., heroin, lithium, interferon alfa, pamidronate, cyclosporine, or NSAIDs [causing analgesic nephropathy]), atheroembolic disease affecting the kidneys, obesity, HIV infection (HIV-associated nephropathy), and disorders causing nephron loss (eg, reflux nephropathy, subtotal nephrectomy, renal dysgenesis [eg oligomeganephronia: renal hypoplasia with a decreased number of nephrons]). Familial cases exist.
In focal segmental glomerulosclerosis, because charge as well as size ultrafiltration barriers are defective, protein in the urine (proteinuria) is typically nonselective, affecting high molecular-weight proteins (eg, immunoglobulins [IGs] or antibodies) as well as albumin. Kidneys tend to be small.
A blood test, urine test, and a kidney biopsy will determine if you have FSGS.
- Urine test: A urine test will help find protein and blood in your urine.
- Blood test: A blood test will help find levels of protein, cholesterol, and wastes in your blood.
- Glomerular filtration rate (GFR): A blood test will be done to know how well your kidneys are filtering the wastes from your body.
- Kidney ultrasound. Earlier in the course of illness, kidney ultrasound will reveal normal or enlarged kidneys with increased echogenicity, indicating diffuse intrinsic medical renal disease 6. In advanced kidney failure, kidneys are shrunken and small, suggesting severe interstitial fibrosis and glomerular scarring. In HIV-associated FSGS, ultrasound study generally reveals large echogenic kidneys.
- Kidney biopsy: In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope.
- Genetic testing: A genetic test may be done to see if you were born with genes that caused your kidney disease. This information may help your doctor decide what type of treatment is best for you.
FSGS is a serious condition that can lead to kidney failure, which can only be treated with dialysis or kidney transplant. Treatment options for focal segmental glomerulosclerosis (FSGS) depend on the type you have.
Types of focal segmental glomerulosclerosis (FSGS) include:
- Primary focal segmental glomerulosclerosis. Many people diagnosed with FSGS have no known cause for their condition. This is called primary (idiopathic) FSGS.
- Secondary focal segmental glomerulosclerosis. Several factors, such as infection, drug toxicity, diseases including diabetes or sickle cell disease, obesity, and even other kidney diseases can cause secondary FSGS. Controlling or treating the underlying cause often slows ongoing kidney damage and might lead to improved kidney function over time.
- Genetic focal segmental glomerulosclerosis. This is a rare form of FSGS caused by genetic changes. It is also called familial FSGS. It’s suspected when several members of a family show signs of FSGS. Familial FSGS can also occur when neither parent has the disease but each one carries a copy of an altered gene that can be passed on to the next generation.
- Unknown focal segmental glomerulosclerosis. In some cases, the underlying cause of FSGS cannot be determined despite the evaluation of clinical symptoms and extensive testing.
Usually, treatments for FSGS include:
- Corticosteroids (often called “steroids”)
- Immunosuppressive drugs
- Corticosteroids and immunosuppressive drugs: These medications are used to calm your immune system (your body’s defense system) and stop it from attacking your glomeruli.
- Plasmapheresis. Sometimes your doctor may order plasmapheresis, a special blood filtering process to remove harmful proteins from your blood caused by immune problems. The fluid part of the blood that contains antibodies is removed and replaced with intravenous fluids or donated plasma (that does not contain antibodies). Removing antibodies may reduce inflammation in your kidney tissues.
- Angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs). These are blood pressure medications used to reduce protein loss and control blood pressure.
- Diuretics (water pills). These medications help your body get rid of excess fluid and swelling. These can be used to lower your blood pressure too.
- Diet changes. Some diet changes may be needed, such as reducing salt (sodium) and protein in your food choices to lighten the load of wastes on the kidneys.
Figure 1. Focal segmental glomerulosclerosis
How your kidneys work
You have two kidneys, each about the size of an adult fist, located on either side of the spine just below the rib cage. Although they are small, your kidneys perform many complex and vital functions that keep the rest of the body in balance. Your kidneys remove waste and excess fluid from your blood through filtering units called nephrons. Each nephron contains a filter (glomerulus) that has a network of tiny blood vessels called capillaries. When blood flows into a glomerulus, tiny molecules — water, essential minerals and nutrients, and wastes — pass through the capillary walls. Large molecules, such as proteins and red blood cells, do not. The filtered solution then passes into another part of the nephron called the tubule. The water, nutrients and minerals your body needs are transferred back to the bloodstream. The excess water and waste become urine that flows to the bladder.
Kidney functions:
- Help remove waste and excess fluid
- Filter the blood, keeping some compounds while removing others
- Control the production of red blood cells
- Make vitamins that control growth
- Release hormones that help regulate blood pressure
- Help regulate blood pressure, red blood cells, and the amount of certain nutrients in the body, such as calcium and potassium.
Here’s how kidneys perform their important work:
- Blood enters the kidneys through an artery from the heart
- Blood is cleaned by passing through millions of tiny blood filters
- Waste material passes through the ureter and is stored in the bladder as urine
- Newly cleaned blood returns to the bloodstream by way of veins
- Bladder becomes full and urine passes out of the body through the urethra.
The kidneys perform their life-sustaining job of filtering and returning to the bloodstream about 200 quarts of fluid every 24 hours. Approximately two quarts are eliminated from the body in the form of urine, while the remainder, about 198 quarts, is retained in the body. The urine we excrete has been stored in the bladder for approximately one to eight hours.
Figure 2. How kidneys work
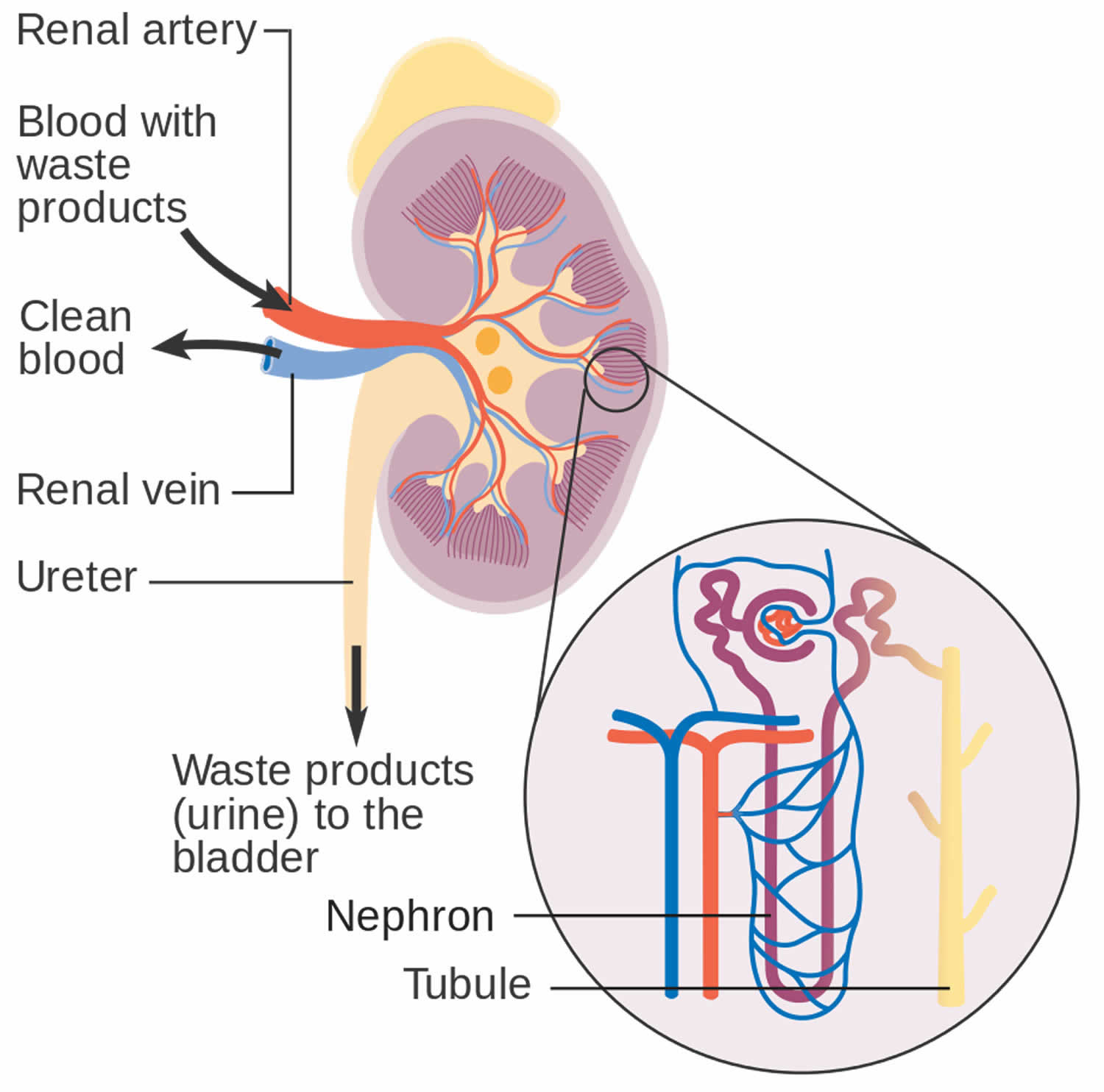
Figure 3. Glomerulus
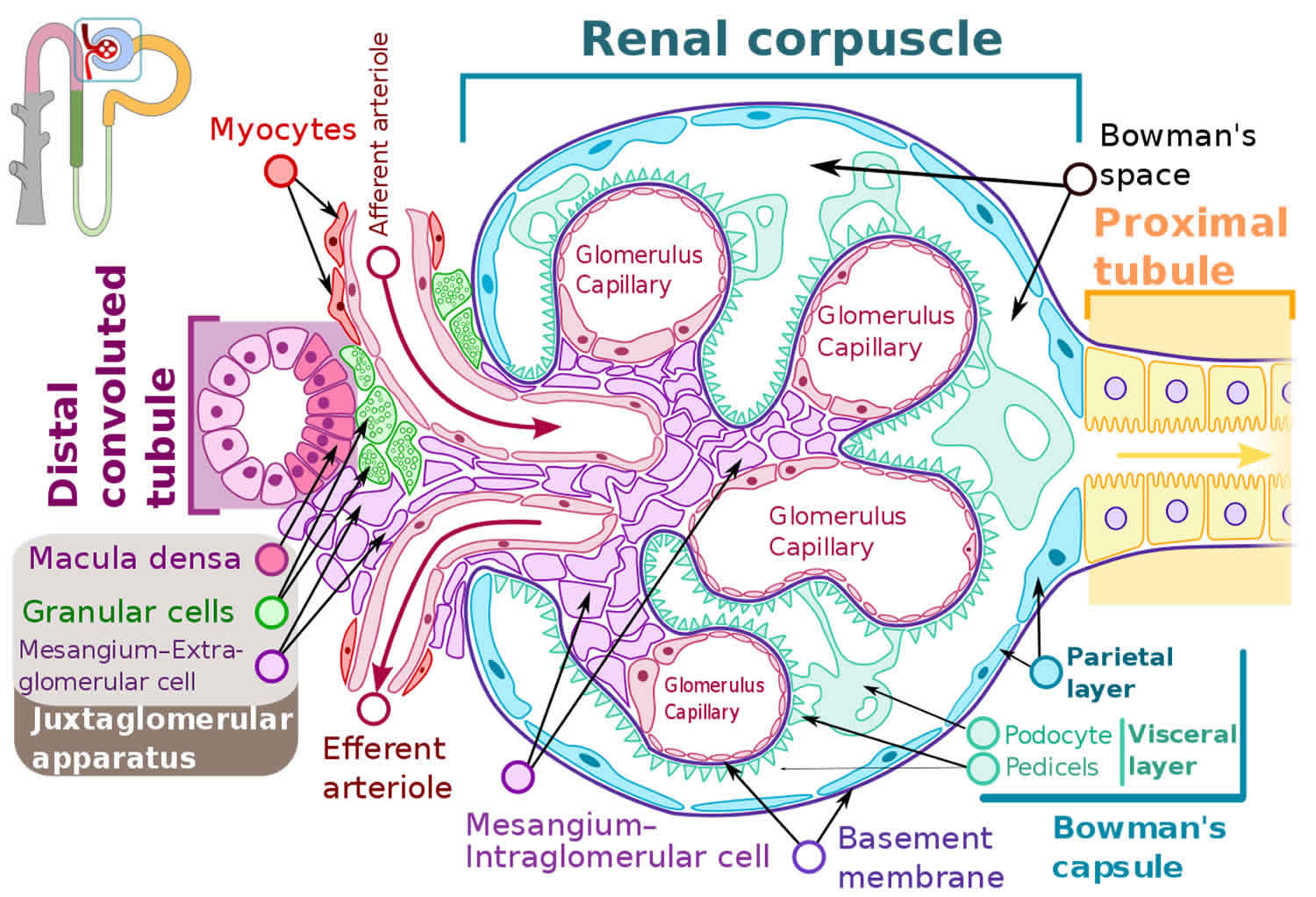
How do glomerular diseases interfere with kidney function?
Glomerular diseases damage the glomeruli, letting protein and sometimes red blood cells leak into the urine. Sometimes a glomerular disease also interferes with the clearance of waste products by the kidney, so they begin to build up in the blood. Furthermore, loss of blood proteins like albumin in the urine can result in a fall in their level in the bloodstream. In normal blood, albumin acts like a sponge, drawing extra fluid from the body into the bloodstream, where it remains until the kidneys remove it. But when albumin leaks into the urine, the blood loses its capacity to absorb extra fluid from the body. Fluid can accumulate outside the circulatory system in the face, hands, feet, or ankles and cause swelling.
What are renal failure and end-stage renal disease?
Renal failure is any acute or chronic loss of kidney function and is the term used when some kidney function remains. Total kidney failure, sometimes called end-stage renal disease (ESRD), indicates permanent loss of kidney function. Depending on the form of glomerular disease, kidney function may be lost in a matter of days or weeks or may deteriorate slowly and gradually over
the course of decades.
Acute renal failure (acute kidney failure)
A few forms of glomerular disease cause very rapid deterioration of kidney function. For example, post-streptococcal glomerulonephritis (PSGN) can cause severe symptoms (hematuria, proteinuria, edema) within 2 to 3 weeks after a sore throat or skin infection develops. The patient may temporarily require dialysis to replace kidney function. This rapid loss of kidney function is called acute renal failure (acute kidney failure). Although acute renal failure (acute kidney failure) can be life-threatening while it lasts, kidney function usually returns after the cause of the kidney failure has been treated. In many patients, acute kidney failure is not associated with any permanent damage. However, some patients may recover from acute renal failure and subsequently develop chronic kidney disease (CKD).
Chronic kidney disease (CKD)
Most forms of glomerular disease develop gradually, often causing no symptoms for many years. Chronic kidney disease (CKD) is the slow, gradual loss of kidney function. Some forms of chronic kidney disease (CKD) can be controlled or slowed down. For example, diabetic nephropathy can be delayed by tightly controlling blood glucose levels and using angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs) to reduce proteinuria and control blood pressure. But chronic kidney disease (CKD) cannot be cured. Partial loss of kidney function means that some portion of the patient’s nephrons have been scarred, and scarred nephrons cannot be repaired. In many cases, CKD leads to total kidney failure.
Total kidney failure
To stay alive, a patient with total kidney failure must go on dialysis, either hemodialysis or peritoneal dialysis or receive a new kidney through kidney transplantation. Patients with chronic kidney disease (CKD) who are approaching total kidney failure should learn as much about their treatment options as possible so they can make an informed decision when the time comes. With the help of dialysis or kidney transplantation, many people continue to lead full, productive lives after reaching total kidney failure.
Will I have kidney failure because of focal segmental glomerulosclerosis (FSGS)?
You should talk with your doctor about your condition because the progression of the disease depends on many factors. FSGS is a chronic disease, because the scarred glomeruli cannot be repaired. Treatment can slow the process of kidney disease. Everyone is different in how they respond to treatment. Over time, some patients with FSGS gradually get worse until they reach kidney failure, If this occurs, they will need a kidney transplant or dialysis to stay alive. Some people respond well to treatment and may live with the disease for many years while being monitored for any signs of change.
Focal segmental glomerulosclerosis causes
Focal segmental glomerulosclerosis (FSGS) is not caused by a single disease. FSGS can have many different causes. The scarring may happen because of an infection, or drug, or a disease that affects the entire body, like diabetes, HIV infection, sickle cell disease or lupus. Focal segmental glomerulosclerosis can also be caused by another glomerular disease that you had before you got FSGS. Focal segmental glomerulosclerosis (FSGS) has different types based on the cause.
Below are the types of focal segmental glomerulosclerosis:
- Primary focal segmental glomerulosclerosis. Many people diagnosed with focal segmental glomerulosclerosis have no known cause for their condition. This is called primary (idiopathic) focal segmental glomerulosclerosis.
- Secondary focal segmental glomerulosclerosis. Several factors, such as infection, drug toxicity, diseases such as diabetes or sickle cell disease, obesity, and even other kidney diseases can cause secondary focal segmental glomerulosclerosis. Controlling or treating the underlying cause often halts ongoing kidney damage and might lead to improved kidney function over time.
- Genetic focal segmental glomerulosclerosis also called familial FSGS. This rare form of focal segmental glomerulosclerosis is caused by genetic mutations. It’s suspected when several members of a family show signs of focal segmental glomerulosclerosis. Familial focal segmental glomerulosclerosis can also occur when neither parent has the disease, but each carries one copy of an abnormal gene that can be passed on to the next generation.
- Unknown focal segmental glomerulosclerosis. In some cases, the underlying cause of FSGS cannot be determined despite the evaluation of clinical symptoms and extensive testing.
It is essential to obtain an extensive history, including birth history (low birth weight/premature birth, congenital renal malformations), family history, medical comorbidities, pre-existing renal disease, exposure to drugs/toxins, recent viral illnesses, and family history to identify secondary causes of FSGS 3.
Focal segmental glomerulosclerosis is broadly categorized into primary (idiopathic) and secondary forms, and such distinction carries both prognostic and therapeutic implications 7. Idiopathic focal segmental glomerulosclerosis is of unknown cause and probably is the most common form. Primary focal segmental glomerulosclerosis has long been thought to be due to the presence of circulating permeability factors/cytokines which causes foot process effacement and proteinuria. These include cardiotrophin-like cytokine factor 1, apoA1b, anti-CD40 Ab and suPAR 8.
Known causes also called acquired focal segmental glomerulosclerosis include:
- Drugs such as heroin, bisphosphonates, anabolic steroids, interferon, lithium, pamidronate and mTOR inhibitors 9
- Infection
- Viral causes include HIV, parvo B19, CMV, EBV, hepatitis C, and Simian virus 40 10.
- Inherited genetic problems
- Diabetes mellitus,
- Hypertension,
- Renal aplasia, hypoplasia or dysplasia,
- Renal artery stenosis,
- Cholesterol emboli 11
- Obesity
- Reflux nephropathy (a condition in which urine flows backward from the bladder to the kidney)
- Sickle cell disease
- Some medicines
- Vascular disease 11
Post-adaptive focal segmental glomerulosclerosis arises after a phase in which the glomeruli are exposed to higher than normal blood flow for a number of years. Increased glomerular blood flow is typical of two settings. First, it occurs when the number of glomeruli is reduced, as happens following bilateral kidney surgery or congenital kidney abnormalities or chronic scarring of the kidney tubules (for example, due to reflux of urine from the bladder back into the kidney). Second, it occurs with obesity, sickle cell anemia and a few other conditions; the reasons for the increase in glomerular blood flow in these settings are unknown.
Occasionally other glomerular diseases, such as IgA nephropathy or membranous nephropathy or lupus nephritis may show focal and segmental glomerular scarring which may be referred by some pathologists as focal segmental glomerulosclerosis. These patients should be treated according to their underlying glomerular disease and the term focal segmental glomerulosclerosis is best not applied in these situations.
Most focal segmental glomerulosclerosis is sporadic (no family history). Some focal segmental glomerulosclerosis is familial (there is more than one affected family member). Genetic mutations may be seen in sporadic focal segmental glomerulosclerosis and familial focal segmental glomerulosclerosis, but is more common in the latter. Several genes encoding slit diaphragm proteins, cell membrane proteins, cytoskeleton proteins, nuclear proteins, mitochondrial proteins, and lysosomal proteins have been identified to be abnormal/mutated leading to loss of integrity of glomerular filtration barrier resulting in focal segmental glomerulosclerosis 12. To date eight genes have been associated with focal segmental glomerulosclerosis. This is an area of intense research and more genes are likely to be identified soon. Several inheritance patterns have been identified and the five most commonly-mutated genes will be discussed below.
- Autosomal recessive inheritance. The inheritance pattern skips generations; parents each carry one copy of the mutant gene and are themselves normal. In children with focal segmental glomerulosclerosis patients who also have a family history of focal segmental glomerulosclerosis, perhaps 35% will have two mutations in podocin. When families are small, podocin mutations can also appear as sporadic focal segmental glomerulosclerosis (no family history); perhaps 20% of children with sporadic focal segmental glomerulosclerosis will have podocin mutations. Reasons for pursuing a diagnosis of podocin mutation include the following: 1) steroid-resistance is typical; 2) the risk of focal segmental glomerulosclerosis recurrence after kidney translantation is lower than with idiopathic focal segmental glomerulosclerosis; 3) family counseling. Podocin mutations almost never cause adult-onset focal segmental glomerulosclerosis
- Autosomal dominant inheritance. This inheritance pattern affects every generation, with one parent and approximately 50% of children affected. Two genes are have been reported (alpha actinin-4 and TRPC6); in both cases focal segmental glomerulosclerosis appears during adulthood.
- Other patterns. WT-1 mutations cause focal segmental glomerulosclerosis appearing in the teenage years. Due to the effects of this mutation on sex development, these individuals may appear to be female but may lack ovaries and a uterus and consequently do not begin menstruation. Mitochondrial DNA mutations may be associated with isolated focal segmental glomerulosclerosis or the focal segmental glomerulosclerosis may be accompanied by other symptoms, include acidosis, stroke-like episodes, muscle weakness, deafness and diabetes.
Individuals of African descent are at increased risk for focal segmental glomerulosclerosis, by a factor of approximately 4-fold compared to those of other races and ethnic groups. It is likely that this has a genetic component but the specific gene or genes have not been identified.
Finally certain medications have been associated with focal segmental glomerulosclerosis.
Focal segmental glomerulosclerosis pathophysiology
The pathogenesis of focal segmental glomerular sclerosis involves a complex interplay of several cell types including podocytes, endothelial cells, and the basement membrane. Podocytes are terminally differentiated cells that provide structural support to the glomerulus and are essential in maintaining an intact glomerular filtration barrier essential to prevent nephrotic range proteinuria. Injury and loss of podocytes result in podocyte hypertrophy of remaining podocytes to cover the glomerular capillary surface resulting in effacement and protein loss 13, 14. Foot process effacement and the proliferation of mesangial, endothelial, and epithelial cells earlier in the course of illness, followed by collapse/shrinkage of glomerular capillaries, all result in scarring (glomerulosclerosis) 15.
The proposed mechanism for podocyte injury includes viral- or toxin-mediated insult and intrarenal hemodynamic alterations, such as high intraglomerular capillary pressure and glomerular hyperperfusion. Many morphologic subsets, such as a collapsing variant (FSGS with mesangial hypercellularity), a cellular variant (endocapillary and extracapillary hypercellularity), and FSGS with tip lesions, are known 16.
Understanding the pathophysiology of FSGS has improved with the discovery that mutations in several proteins responsible for maintaining podocyte structure, function, or both not only result in FSGS but can predict disease characteristics, such as steroid responsiveness 17. For instance, FSGS with mutations in NPHS2 or TRPC6 is challenging to treat with immunosuppressive therapy; however, when such patients undergo kidney transplantation, the disease does not usually recur. APOL1 G1/G2 variants have been associated with a poor renal prognosis and steroid resistance in nephrotic syndrome or FSGS 18.
Proposed circulating factors linked to the development of FSGS include candidate molecules, such as hemopexin, cardiotrophin-like cytokine 1, and vascular endothelial growth factor. One molecule that has been extensively studied is a form of urokinase receptor (suPAR) 19.
Focal segmental glomerulosclerosis histopathology
Histologically, focal segmental glomerular sclerosis (FSGS) is characterized by sclerosis, hyalinosis, adhesions or synechiae formation, resulting in segmental obliteration of glomerular capillaries 3. On electron microscopy, foot process effacement is the predominant finding without significant basement membrane abnormalities. Immunofluorescence shows staining for IgM and C3 in sclerotic areas. Juxtamedullary nephrons are affected first and hence inadequate sampling may miss focal lesions.
Histologically, focal segmental glomerular sclerosis (FSGS) is classified into five variants: perihilar, tip, cellular, collapsing and not otherwise specified (NOS) 20, 21, 22.
Perihilar focal segmental glomerulosclerosis
The sclerosing lesion is located at the vascular pole of the glomerulus. Perihilar focal segmental glomerulosclerosis is commonly seen in adaptive FSGS due to increased pressure in the glomerulus which is in close proximity to the afferent arteriole. Foot process effacement is mild, resulting in subnephrotic proteinuria and relatively normal serum albumin levels 3.
Tip focal segmental glomerulosclerosis
The segmental lesion involves the tubular pole of the glomerulus 3. Tip focal segmental glomerulosclerosis is commonly seen in Caucasians, presenting with diffuse foot process effacement and abrupt onset of nephrotic syndrome. These patients have lower baseline creatinine, have an excellent response to treatment, and the lowest rate of progression 23.
Cellular focal segmental glomerulosclerosis
Cellular focal segmental glomerulosclerosisis the least common variant of focal segmental glomerulosclerosis, characterized by hypercellular glomerulus including endocapillary and glomerular epithelial cell hyperplasia. Cellular focal segmental glomerulosclerosisis presents with diffuse foot process effacement and full-blown nephrotic syndrome 24.
Collapsing focal segmental glomerulosclerosisis
Collapsing focal segmental glomerulosclerosisisis is characterized by hyperplasia and hypertrophy of visceral glomerular epithelial cells leading to the collapse of the glomerular tuft 3. This is commonly seen in viral (parvovirus B19, CMV, HIV) and drug-associated forms of focal segmental glomerulosclerosis (interferon-alpha, interferon-beta, interferon-gamma & pamidronate) and presents with diffuse effacement of foot processes, heavy proteinuria with the lowest rate of remission, and the worst prognosis 25, 26, 27.
Not otherwise specified (NOS) focal segmental glomerulosclerosisisis
NOS focal segmental glomerulosclerosisisis is the most common subtype of focal segmental glomerulosclerosis and does not fit into any other morphological forms of focal segmental glomerulosclerosis 3. NOS focal segmental glomerulosclerosisisis presents with a variable degree of effacement and proteinuria. Histopathology may sometimes resemble nodular sclerosis as in diabetes and other conditions 28.
Focal segmental glomerulosclerosis signs and symptoms
Children with focal segmental glomerular sclerosis (FSGS) typically present with the full-blown nephrotic syndrome (edema, massive proteinuria, hypoalbuminemia, hypercholesterolemia). Adults can have nephrotic or sub-nephrotic proteinuria, hypertension, microscopic hematuria, or present with renal insufficiency. Patients with primary focal segmental glomerulosclerosis often have profound hypoalbuminemia and edema, but these are rare in secondary forms.
Patients with focal segmental glomerulosclerosis may present in different ways. First some present with edema, either a sudden onset similar to that seen in patients with minimal change disease or slower onset over weeks to months. Second, some patients have no symptoms and are found to have asymptomatic (no symptoms) proteinuria, found on routine urinalysis performed as part of a physical examination. Other features of the nephrotic syndrome are often present, including hypercholesterolemia (high serum cholesterol) and low serum albumin.
Generally, edema develops over a few weeks; however, the onset may be abrupt, with sudden weight gain of 15-20 lbs (6.8 to 9 kg) or more. Frequently, a recent upper respiratory tract infection precedes edema.
Pleural effusion and ascites could be present, although pericardial effusions are rare. Gross edema could predispose patients to infections and ulcerations in dependent areas, such as the lower extremities. Abdominal pain may be a sign of peritonitis, a common finding in children. Rarely, xanthomas may be seen in cases of severe hyperlipidemia. In many patients, physical examination is normal except for edema. Severe hypertension is not uncommon, particularly in Black patients with renal impairment 29. Rarely do patients experience severe kidney failure with features of advanced uremia, such as nausea, vomiting, seizures, bleeding, or altered mental status. Patients with FSGS secondary to conditions such as reflux nephropathy, massive obesity, and renal dysplasia/agenesis usually present with non-nephritic proteinuria. These patients may often experience worsening renal function over the course of months to years.
Focal segmental glomerulosclerosis signs and symptoms include:
- Foamy urine caused by high protein levels in your urine (called proteinuria)
- Poor appetite
- Swelling in body parts like your legs, ankles and around your eyes (called edema)
- Weight gain due to extra fluid building in your body
- High fat levels in the blood (high cholesterol)
- Low levels of protein in your blood
Focal segmental glomerulosclerosis can cause nephrotic syndrome.
- Nephrotic syndrome: A set of symptoms that happen together and affect your kidneys. These include:
- Swelling in body parts like your legs, ankles, or around your eyes (edema)
- Large amounts of protein in your urine (proteinuria)
- Loss of protein in your blood
- High levels of fat lipids in your blood (high cholesterol)
- High blood pressure (in some cases)
If the condition is advanced, the symptoms may be like those of kidney failure. People may report fatigue, a poor appetite, headache, itchy skin, shortness of breath and/or nausea.
Focal segmental glomerulosclerosis complications
Focal segmental glomerulosclerosis cmplications may include:
- Chronic kidney failure
- End-stage kidney disease
- Infection
- Malnutrition
- Nephrotic syndrome
Focal segmental glomerulosclerosis diagnosis
Your doctor will perform a physical exam. This exam may show tissue swelling (edema) and high blood pressure. Signs of kidney (renal) failure and excess fluid may develop as the condition gets worse.
Tests may include:
- Kidney biopsy. In this test, a tiny piece of your kidney is removed with a special needle, and looked at under a microscope.
- Kidney function tests (blood and urine)
- Glomerular filtration rate (GFR): A blood test will be done to know how well your kidneys are filtering the wastes from your body.
- 24-hour urine collection for protein quantification
- Urinalysis
- Urine microscopy
- Urine protein
- Hepatitis and HIV serology
- Complement levels
- Serum and urine protein electrophoresis in elderly to rule out paraproteinemias
- Genetic testing: A genetic test may be done to see if you were born with genes that caused your kidney disease. This information may help your doctor decide what type of treatment is best for you.
Ultimately, a kidney biopsy is required to confirm the diagnosis of focal segmental glomerulosclerosis. The characteristic finding in FSGS is segmental solidification of the glomeruli, typically in the perihilar region and occasionally in the peripheral areas, such as the tubular pole 30. In the diseased glomeruli, the accumulation of acellular matrix and hyaline deposits obliterates capillaries in a segmental fashion. Coarsely granular deposits of C3 and IgM are often seen in these areas. Diffuse foot process fusion is seen predominantly in the sclerotic segments, whereas partial effacement is observed overlying normal-appearing lobules. In HIV-associated FSGS, electron microscopy of the kidney shows tubuloreticular inclusions in mesangial and endothelial cells, an indirect indication of viral disease 31.
In patients with focal segmental glomerulosclerosis, urinalysis shows large amounts of protein and casts (hyaline and broad waxy), although red blood cell casts are usually absent. In advanced cases, broad casts may be evident. Serum creatinine (SCr) and creatinine clearance (CrCl) are usually within the reference range in the early stages. Features of nephrotic syndrome (proteinuria >3.5 g/day, serum albumin <30 g/L, with or without edema) may or may not be present.
In idiopathic FSGS, investigations for an underlying cause are usually negative. Such conditions include the following:
- Systemic lupus erythematosus (antinuclear antibody/anti-DNA titers, serum complement C4/C3 levels)
- Hepatitis B or C infection
- Vasculitis (serum protein electrophoresis, antineutrophil cytoplasmic antibody titers)
In patients suspected to have secondary FSGS, the following should be obtained:
- HIV antibody, CD4, and viral load
- Serology for hepatitis B and C
- Parvovirus testing
FSGS in morbidly obese patients is diagnosed by excluding other causes. The common features in obesity-related FSGS include glomerular hyperfiltration and activation of the renin-angiotensin-aldosterone system 32. FSGS may be considered in patients with proteinuria; however, in younger patients with no red blood cell casts and negative serologic studies, the definitive diagnosis is made on a kidney biopsy.
Ultrasonography
Earlier in the course of illness, ultrasonography will reveal normal or enlarged kidneys with increased echogenicity, indicating diffuse intrinsic medical renal disease 6. In advanced kidney failure, kidneys are shrunken and small, suggesting severe interstitial fibrosis and glomerular scarring. In HIV-associated FSGS, ultrasound study generally reveals large echogenic kidneys.
Focal segmental glomerulosclerosis treatment
The type of treatment you get depends on the cause. Everyone is different and your doctor will make a treatment plan that is right for your type of focal segmental glomerulosclerosis. Corticosteroids (daily or every other day) are first line of treatment in children and adults with focal segmental glomerular sclerosis (FSGS) with the best chance of inducing a sustained complete remission; they may also induce a partial remission. While some patients with a complete remission experience relapse, they often undergo a second remission with a second course of therapy. Steroids may also induced a sustained partial remission. Standard courses of steroid therapy include daily or alternate day steroids for period of 2 to 4 months, possibly extended longer with a taper off if the response has been good.
In patients with subnephrotic proteinuria (adaptive focal segmental glomerulosclerosis), a trial of renin-angiotensin system (RAS) inhibition (e.g., angiotensin-converting enzyme inhibitors [ACE inhibitors] and angiotensin 2 receptor blockers [ARBs]), and sodium restriction can be tried. ACE inhibitors and angiotensin 2 receptor blockers are blood pressure medications used to reduce protein loss and control blood pressure. In other secondary forms of focal segmental glomerulosclerosis, removing the offending agent or treating the underlying disorder is recommended. In addition, optimization of blood pressure, treatment of edema with diuretics, statin therapy for hypercholesterolemia and anticoagulation in select patients at risk for thrombosis/embolization are indicated.
Children respond within a few weeks, but adults may take months to respond. Glucocorticoids are associated with a remission rate of approximately 30% compared to about 50% in patients treated with calcineurin inhibitor. Rituximab, mTOR inhibitors, and plasmapheresis have been tried in select patients with varied results.
Corticosteroids
- Oral prednisone: 2 mg/kg/day for 6 weeks followed by 1 mg/kg/day on alternate days for 6 weeks in children and 1mg/kg/day for 3 to 6 months in adults
- High-dose corticosteroid therapy with prednisolone is started at an initial dose of 1 mg/kg/day daily single dose (maximum 80 mg) or alternate day dose of 2 mg/kg/day (maximum 120 mg) for at least four weeks and until complete remission is achieved or a maximum of 16 weeks treatment, whichever is earlier 33, 34. Patients with the potential to remit would likely show some decline in proteinuria before 16 weeks of high-dose steroid therapy. Hence, it is unnecessary to persist with high-dose steroids treatment until 16 weeks if proteinuria persists or worsens. This becomes more important in patients who are experiencing side effects of steroids 35.
- Tapering off steroids should start after at least four weeks of achieving remission with high-dose therapy or after two weeks of the disappearance of proteins, whichever is longer. Prednisolone is reduced by 5 mg every one to two weeks to complete a total duration of six months. If partial remission is achieved within eight to twelve weeks of high-dose steroid therapy, continue until 16 weeks to ensure complete remission. After that, the prednisolone dose is reduced by 5 mg every one to two weeks to complete a total six-month duration of 6 months.
Immunosuppressive drugs
Patients who are resistant or intolerant to steroids are treated with immunosuppressive drugs with calcineurin inhibitors, mycophenolate mofetil, or rituximab 36, 37, 38.
- Calcineurin inhibitors:
- Cyclosporine 3 to 5 mg/kg/day (target trough levels 100 to 175 ng/ml)
- Tacrolimus 0.05 to 0.1 mg/kg/day (target trough levels 5 to 10 ng/ml) 39
- Trough levels should be monitored to prevent drug toxicity. The duration of determining the efficacy of cyclosporin or tacrolimus is at least six months, after which the patient can be labeled calcineurin inhibitor-resistant. Calcineurin inhibitorss should be continued for at least 12 months in patients with partial or complete remission to prevent relapses. The dose of calcineurin inhibitors is to be slowly tapered over 6-12 months as tolerated.
- In patients resistant or intolerant to calcineurin inhibitors, there is a lack of evidence regarding any particular agent. Mycophenolate mofetil, high-dose dexamethasone, rituximab, and adrenocorticotropic hormone (ACTH) have been studied 40. In addition, rituximab, mTOR inhibitors, and plasmapheresis have been tried in select patients with varied results.
- For those who are steroid-responsive but experience one or more relapses when steroids are stopped, other therapies include cyclophosphamide or cyclosporine.
- For those who are steroid-resistant, other therapies include cyclosporine or (less well studied) mycophenolate mofetil.
- Mycophenolate mofetil: 25 to 35 mg/kg/day +/- dexamethasone
- Edema is managed by dietary salt restriction (low sodium diet) and diuretics, either administered orally or intravenously. Diuretics are medications help your body get rid of excess fluid and swelling. These can be used to lower your blood pressure too.
Focal segmental glomerulosclerosis prognosis
Several features predict outcome in focal segmental glomerulosclerosis including, race (Blacks have worse outcomes), degree of proteinuria, presence of renal insufficiency, histological variant (tip variant had the best outcome and collapsing variant had the worst outcome), degree of interstitial fibrosis or tubular atrophy and response to treatment with patients attaining partial or complete remission having better prognosis 3. Also, patients with primary focal segmental glomerulosclerosis did worse when compared to those with adaptive or secondary causes of focal segmental glomerulosclerosis 41, 34.
Focal segmental glomerulosclerosis life expectancy
At ten years after diagnosis, approximately 50% of individuals with focal segmental glomerulosclerosis will developed end-stage kidney disease (ESRD), requiring dialysis or kidney transplant. This statistic includes both individuals who received treatment and did not respond favorably and individuals who came to medical attention too late for effective therapy to be attempted. This prognosis makes focal segmental glomerulosclerosis the most serious primary glomerular disease. The prognosis in those who enter a complete remission is excellent, even when they experience a relapse. The long-term prognosis in those who enter a partial remission is less good, but is substantially better than those who did not respond to therapy or did not take therapy.
Recurrence after kidney transplant
Approximately 25% of patients who have focal segmental glomerulosclerosis in their own kidneys will experience recurrent focal segmental glomerulosclerosis after kidney transplant. When focal segmental glomerulosclerosis recurs, it typically does so within days to weeks of transplant, and essentially always within one year of transplant. The cause of recurrent focal segmental glomerulosclerosis is unknown but is believed to be a circulating protein. Patients at increased risk for recurrent focal segmental glomerulosclerosis include the following: rapid progression (from diagnosis to end-stage kidney disease in less than 3 years, recurrent focal segmental glomerulosclerosis in a prior transplant (risk of recurrent focal segmental glomerulosclerosis is approximately 70%), and white race (weak risk factor). In patients with focal segmental glomerulosclerosis, it is prudent to check the urine weekly by dipstick for protein or every 2 to 4 weeks by measuring a random urine protein/creatinine ratio. The appearance of proteinuria should prompt a transplant kidney biopsy, which would initially show just podocyte foot process effacement and after weeks to months of proteinuria will show focal segmental glomerulosclerosis. If the diagnosis can be made within 2 to 4 weeks of proteinuria onset, therapy may include plasma exchange and possibly cyclophosphamide.
References- Rosenberg AZ, Kopp JB. Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2017 Mar 7. 12 (3):502-517.
- Focal Segmental Glomerulosclerosis (FSGS). https://www.kidney.org/atoz/content/focal
- Guruswamy Sangameswaran KD, Hashmi MF, Baradhi KM. Focal Segmental Glomerulosclerosis. [Updated 2023 Feb 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532272
- Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007 Jun;71(12):1205-14.
- McGrogan A, Franssen CF, de Vries CS. The incidence of primary glomerulonephritis worldwide: a systematic review of the literature. Nephrol. Dial. Transplant. 2011 Feb;26(2):414-30.
- Hogan JJ. A Case of Focal Segmental Glomerulosclerosis. Clin J Am Soc Nephrol. 2021 Aug;16(8):1272-1274. doi: 10.2215/CJN.19591220
- D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am. J. Kidney Dis. 2004 Feb;43(2):368-82
- Königshausen E, Sellin L. Circulating Permeability Factors in Primary Focal Segmental Glomerulosclerosis: A Review of Proposed Candidates. Biomed Res Int. 2016;2016:3765608.
- Petersen CE, Amaral S, Frosch E. Lithium-induced nephrotic syndrome in a prepubertal boy. J Child Adolesc Psychopharmacol. 2008 Apr;18(2):210-3
- Chandra P, Kopp JB. Viruses and collapsing glomerulopathy: a brief critical review. Clin Kidney J. 2013 Feb;6(1):1-5.
- Kriz W, Lemley KV. Mechanical challenges to the glomerular filtration barrier: adaptations and pathway to sclerosis. Pediatr. Nephrol. 2017 Mar;32(3):405-417
- Yu H, Artomov M, Brähler S, Stander MC, Shamsan G, Sampson MG, White JM, Kretzler M, Miner JH, Jain S, Winkler CA, Mitra RD, Kopp JB, Daly MJ, Shaw AS. A role for genetic susceptibility in sporadic focal segmental glomerulosclerosis. J. Clin. Invest. 2016 Apr 01;126(4):1603
- Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol. 2015 Feb;26(2):258-69. doi: 10.1681/ASN.2014030278
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998 Sep;54(3):687-97. doi: 10.1046/j.1523-1755.1998.00044.x
- Barisoni L, Schnaper HW, Kopp JB. Advances in the biology and genetics of the podocytopathies: implications for diagnosis and therapy. Arch Pathol Lab Med. 2009 Feb;133(2):201-16. doi: 10.5858/133.2.201
- Shabaka A, Tato Ribera A, Fernández-Juárez G. Focal Segmental Glomerulosclerosis: State-of-the-Art and Clinical Perspective. Nephron. 2020;144(9):413-427. doi: 10.1159/000508099
- Shankland SJ, Pollak MR. A suPAR circulating factor causes kidney disease. Nat Med. 2011 Aug 4;17(8):926-7. doi: 10.1038/nm.2443
- Gribouval O, Boyer O, Knebelmann B, Karras A, Dantal J, Fourrage C, Alibeu O, Hogan J, Dossier C, Tête MJ, Antignac C, Servais A. APOL1 risk genotype in European steroid-resistant nephrotic syndrome and/or focal segmental glomerulosclerosis patients of different African ancestries. Nephrol Dial Transplant. 2019 Nov 1;34(11):1885-1893. doi: 10.1093/ndt/gfy176
- McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010 Nov;5(11):2115-21. doi: 10.2215/CJN.03800609
- D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004 Feb;43(2):368-82. doi: 10.1053/j.ajkd.2003.10.024
- Stokes MB, D’Agati VD. Morphologic variants of focal segmental glomerulosclerosis and their significance. Adv Chronic Kidney Dis. 2014 Sep;21(5):400-7. doi: 10.1053/j.ackd.2014.02.010
- Thomas DB, Franceschini N, Hogan SL, Ten Holder S, Jennette CE, Falk RJ, Jennette JC. Clinical and pathologic characteristics of focal segmental glomerulosclerosis pathologic variants. Kidney Int. 2006 Mar;69(5):920-6. doi: 10.1038/sj.ki.5000160
- Stokes MB, Markowitz GS, Lin J, Valeri AM, D’Agati VD. Glomerular tip lesion: a distinct entity within the minimal change disease/focal segmental glomerulosclerosis spectrum. Kidney Int. 2004 May;65(5):1690-702. doi: 10.1111/j.1523-1755.2004.00563.x
- Stokes MB, Valeri AM, Markowitz GS, D’Agati VD. Cellular focal segmental glomerulosclerosis: Clinical and pathologic features. Kidney Int. 2006 Nov;70(10):1783-92. doi: 10.1038/sj.ki.5001903
- Moudgil A, Nast CC, Bagga A, Wei L, Nurmamet A, Cohen AH, Jordan SC, Toyoda M. Association of parvovirus B19 infection with idiopathic collapsing glomerulopathy. Kidney Int. 2001 Jun;59(6):2126-33. doi: 10.1046/j.1523-1755.2001.00727.x
- Tomlinson L, Boriskin Y, McPhee I, Holwill S, Rice P. Acute cytomegalovirus infection complicated by collapsing glomerulopathy. Nephrol Dial Transplant. 2003 Jan;18(1):187-9. doi: 10.1093/ndt/18.1.187
- Markowitz GS, Nasr SH, Stokes MB, D’Agati VD. Treatment with IFN-{alpha}, -{beta}, or -{gamma} is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010 Apr;5(4):607-15. doi: 10.2215/CJN.07311009. Epub 2010 Mar 4. Erratum in: Clin J Am Soc Nephrol. 2010 Jul;5(7):1353.
- Baradhi KM, Gary Abuelo J, Stillman IE. The Case: diabetic nephropathy in a nondiabetic smoker? Kidney Int. 2012 Nov;82(10):1141-2. doi: 10.1038/ki.2012.333
- Freedman BI, Hicks PJ, Bostrom MA, Cunningham ME, Liu Y, Divers J, Kopp JB, Winkler CA, Nelson GW, Langefeld CD, Bowden DW. Polymorphisms in the non-muscle myosin heavy chain 9 gene (MYH9) are strongly associated with end-stage renal disease historically attributed to hypertension in African Americans. Kidney Int. 2009 Apr;75(7):736-45. doi: 10.1038/ki.2008.701
- Chen YM, Liapis H. Focal segmental glomerulosclerosis: molecular genetics and targeted therapies. BMC Nephrol. 2015 Jul 9;16:101. doi: 10.1186/s12882-015-0090-9
- Winston JA, Burns GC, Klotman PE. The human immunodeficiency virus (HIV) epidemic and HIV-associated nephropathy. Semin Nephrol. 1998 Jul;18(4):373-7.
- Rüster C, Wolf G. The role of the renin-angiotensin-aldosterone system in obesity-related renal diseases. Semin Nephrol. 2013 Jan;33(1):44-53. doi: 10.1016/j.semnephrol.2012.12.002
- Nagai R, Cattran DC, Pei Y. Steroid therapy and prognosis of focal segmental glomerulosclerosis in the elderly. Clin Nephrol. 1994 Jul;42(1):18-21.
- Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran DC; Toronto Glomerulonephritis Registry Group. Focal and segmental glomerulosclerosis: definition and relevance of a partial remission. J Am Soc Nephrol. 2005 Apr;16(4):1061-8. doi: 10.1681/ASN.2004070593
- Costello R, Patel R, Humphreys J, McBeth J, Dixon WG. Patient perceptions of glucocorticoid side effects: a cross-sectional survey of users in an online health community. BMJ Open. 2017 Apr 3;7(4):e014603. doi: 10.1136/bmjopen-2016-014603
- Banfi G, Moriggi M, Sabadini E, Fellin G, D’Amico G, Ponticelli C. The impact of prolonged immunosuppression on the outcome of idiopathic focal-segmental glomerulosclerosis with nephrotic syndrome in adults. A collaborative retrospective study. Clin Nephrol. 1991 Aug;36(2):53-9.
- Cattran DC, Appel GB, Hebert LA, Hunsicker LG, Pohl MA, Hoy WE, Maxwell DR, Kunis CL. A randomized trial of cyclosporine in patients with steroid-resistant focal segmental glomerulosclerosis. North America Nephrotic Syndrome Study Group. Kidney Int. 1999 Dec;56(6):2220-6. doi: 10.1046/j.1523-1755.1999.00778.x
- Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, Li J, Mattiazzi A, Ciancio G, Chen L, Zilleruelo G, Abitbol C, Chandar J, Seeherunvong W, Ricordi C, Ikehata M, Rastaldi MP, Reiser J, Burke GW 3rd. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. 2011 Jun 1;3(85):85ra46. doi: 10.1126/scitranslmed.3002231
- Ramachandran R, Kumar V, Rathi M, Nada R, Jha V, Gupta KL, Sakhuja V, Kohli HS. Tacrolimus therapy in adult-onset steroid-resistant nephrotic syndrome due to a focal segmental glomerulosclerosis single-center experience. Nephrol Dial Transplant. 2014 Oct;29(10):1918-24. doi: 10.1093/ndt/gfu097
- Canetta PA, Radhakrishnan J. Impact of the National Institutes of Health Focal Segmental Glomerulosclerosis (NIH FSGS) clinical trial on the treatment of steroid-resistant FSGS. Nephrol Dial Transplant. 2013 Mar;28(3):527-34. doi: 10.1093/ndt/gfs563
- Chun MJ, Korbet SM, Schwartz MM, Lewis EJ. Focal segmental glomerulosclerosis in nephrotic adults: presentation, prognosis, and response to therapy of the histologic variants. J Am Soc Nephrol. 2004 Aug;15(8):2169-77. doi: 10.1097/01.ASN.0000135051.62500.97


{kind=link}