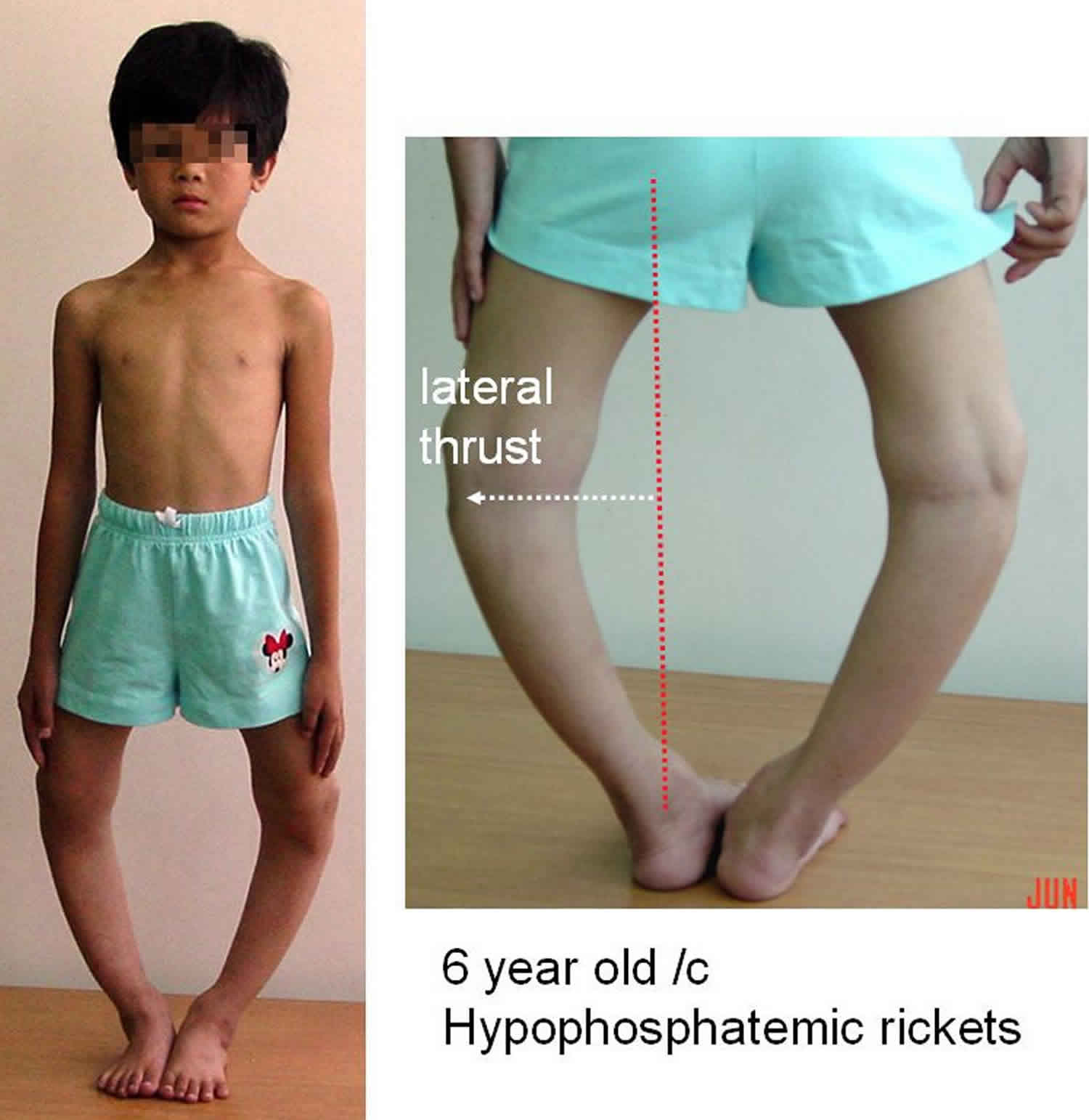
Hypophosphatemic rickets
Hypophosphatemic rickets also known as vitamin D-resistant rickets, hereditary hypophosphatemic rickets or familial hypophosphatemic rickets, is an inherited disorder related to low levels of phosphate in the blood (hypophosphatemia). Phosphate is a mineral that is essential for the normal formation of bones and teeth.
In most cases, the signs and symptoms of hereditary hypophosphatemic rickets begin in early childhood. The features of the disorder vary widely, even among affected members of the same family. Mildly affected individuals may have hypophosphatemia without other signs and symptoms. More severely affected children experience slow growth and are shorter than their peers. They develop bone abnormalities that can interfere with movement and cause bone pain. The most noticeable of these abnormalities are bowed legs or knock knees. These abnormalities become apparent with weight-bearing activities such as walking. If untreated, they tend to worsen with time.
Other signs and symptoms of hereditary hypophosphatemic rickets can include premature fusion of the skull bones (craniosynostosis) and dental abnormalities. The disorder may also cause abnormal bone growth where ligaments and tendons attach to joints (enthesopathy). In adults, hypophosphatemia is characterized by a softening of the bones known as osteomalacia.
Researchers have described several forms of hereditary hypophosphatemic rickets, which are distinguished by their pattern of inheritance and genetic cause. The most common form of the disorder is known as X-linked hypophosphatemic rickets. It has an X-linked dominant pattern of inheritance. X-linked recessive, autosomal dominant, and autosomal recessive forms of the disorder are much rarer.
X-linked hypophosphatemic rickets affects both males and females. In some families it has been anecdotally observed that females may have less severe features of the disease than males. However, such a great variation in degree of severity exists overall, that it is not clear that this is always the case.
Another rare type of the disorder is known as hereditary hypophosphatemic rickets with hypercalciuria. In addition to hypophosphatemia, this condition is characterized by the excretion of high levels of calcium in the urine (hypercalciuria).
X-linked hypophosphatemic rickets is the most common form of rickets that runs in families. It affects about 1 in 20,000 newborns 1. Each of the other forms of hereditary hypophosphatemic rickets has been identified in only a few families.
Diagnosis is by serum phosphate, alkaline phosphatase (ALP), and 1,25-dihydroxyvitamin D3 (cholecalciferol) levels. Treatment is oral phosphate plus calcitriol; burosumab is given for X-linked hypophosphatemic rickets.
If a person with X-linked hypophosphatemic rickets increases their vitamin D intake, will their calcium levels rise?
Yes. Taking the activated form of vitamin D, called calcitriol, is a standard part of X-linked hypophosphatemic rickets treatment in children and is considered for adults with X-linked hypophosphatemic rickets in certain situations (e.g., those with skeletal pain, recurring bone fractures, upcoming orthopedic surgery, or osteomalacia with high alkaline phosphatase). Calcitriol increases calcium levels by promoting calcium absorption in the intestines, and calcium retention in kidneys 2. This is done carefully because over treatment with calcitrol can lead to too much calcium which can harm the kidneys and other body tissues 3.
Can males with X-linked hypophosphatemic rickets pass the condition to their children?
Yes. Males with X-linked hypophosphatemic rickets will always pass the condition to their daughters, but never pass the condition to their sons 1.
X-linked hypophasphatemic rickets (XLH) is most often inherited in an X-linked dominant fashion. X-linked hypophosphatemic rickets occurs when a person inherits one copy of a PHEX gene mutation. PHEX is located on the X chromosome. X chromosomes are one of the two sex chromosomes. Females have two X chromosomes. Males have a X and a Y chromosome 1.
Females with a PHEX mutation on one copy of their X chromosomes have X-linked hypophosphatemic rickets. Women with X-linked hypophosphatemic rickets have a 1 in 2 or 50% chance with each pregnancy of passing X-linked hypophosphatemic rickets to their offspring, regardless of the child’s gender 1.
Males who inherit a PHEX mutation on their X chromosome also have X-linked hypophosphatemic rickets. A male will always pass his Y chromosome to his male offspring (who will be unaffected), and his X chromosome with the PHEX mutation to his daughters (who will be affected) 1.
Because I have hypophosphatemic rickets, are my children at risk to have the same condition?
Hypophosphatemic rickets is a genetic condition. This puts your children at risk to inherit the gene mutation that causes the disease. The chance that your child would inherit the condition depends on the inheritance pattern in your family. Hypophosphatemic rickets is most commonly inherited in an X-linked dominant pattern, but some families have been found to have an X-linked recessive, autosomal dominant and autosomal recessive inheritance patterns 1. To learn about which inheritance pattern is in your family and the specific risks to your future children, you should consult with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Can hypophosphatemic rickets lead to frequent dental abscesses?
The presence of severe dental complications among people with Hypophosphatemic Rickets is now well recognized and is an important part of symptom management 4. Spontaneous dental abscesses are frequently encountered in hypophosphatemic rickets 5. Both primary and permanent teeth may be affected 6. These abscesses occur in the absence of a history of trauma or dental decay and result from hypomineralization of the dentine and enlargement of the pulp 7.
How might the dental features of hypophosphatemic rickets be managed?
The challenge for the dentist is to prevent and treat these lesions 5. The use of calcitriol and phosphate helps to insure good dentin development and mineralization, and may prevent clinical anomalies such as the dental necrosis classically associated with the disease. Topical application of fluoride to the teeth and the use of fissure sealants may also be useful in preventing dental disease. Starting treatment during early childhood and good adherence to the therapy are necessary to observe beneficial effects 8.
If dental features are already present, the use of non-surgical root canal and partial or full crowns may be utilized 8.
How are adults with hypophosphatemic rickets treated?
The goal of treatment of adults with hypophosphatemic rickets is to manage bone pain and in some cases aid in the healing of bone fracture. Adults with hypoposphatemic rickets that are not experiencing symptoms may not benefit from therapy, but could experience therapy related complication (e.g., nephrocalcinosis and hyperparathyroidism). Treatment for symptomatic adults involves daily divided doses of Calcitriol and phosphate. Adults with hypophospatemic rickets undergoing treatment should carefully follow their prescribed dosing schedule. All adults with hypophosphatemic rickts should be regularly monitored for serum phosphorus, calcium, creatinine, and parathyroid hormone (PTH) 9.
How are high calcium levels in adults with hypophosphatemic rickets treated?
High levels of calcium in adults with hypophosphatemic rickets may be due to an excessive calcitriol dose or inadequate dosing of phosphate 9. Treatment of high calcium levels in adults may involve adjusting calcitrol or phosphate dosing levels/schedule 9. You should to talk with your doctor regarding your high calcium levels and treatment options.
Hypophosphatemic rickets causes
Hereditary hypophosphatemic rickets can result from mutations in several genes. Mutations in the PHEX gene, which are responsible for X-linked hypophosphatemic rickets, occur most frequently. Mutations in other genes cause the less common forms of the condition. Rarer, sporadic, acquired hypophosphatemic rickets cases are sometimes associated with benign (non-cancerous) mesenchymal tumors that decrease resorption of phosphate 10.
Hereditary hypophosphatemic rickets is characterized by a phosphate imbalance in the body. Among its many functions, phosphate plays a critical role in the formation and growth of bones in childhood and helps maintain bone strength in adults. Phosphate levels are controlled in large part by the kidneys. The kidneys normally excrete excess phosphate in urine, and they reabsorb this mineral into the bloodstream when more is needed. However, in people with hereditary hypophosphatemic rickets, the kidneys cannot reabsorb phosphate effectively and too much of this mineral is excreted from the body in urine. As a result, not enough phosphate is available in the bloodstream to participate in normal bone development and maintenance.
The genes associated with hereditary hypophosphatemic rickets are involved in maintaining the proper balance of phosphate. Many of these genes, including the PHEX gene, directly or indirectly regulate a protein called fibroblast growth factor 23 (produced from the FGF23 gene). This protein normally inhibits the kidneys’ ability to reabsorb phosphate into the bloodstream. Gene mutations increase the production or reduce the breakdown of fibroblast growth factor 23 (FGF23). The resulting overactivity of this protein reduces phosphate reabsorption by the kidneys, leading to hypophosphatemia and the related features of hereditary hypophosphatemic rickets.
In familial hypophosphatemia, symptoms occur, at least in part, because of an impaired ability of the kidneys to retain phosphate. If the blood levels of phosphate become abnormally low, bone mineralization becomes impaired, thereby weakening the bones and leading to osteomalacia and bowed bones.
A second renal abnormality in x linked hypophosphatemic rickets and autosomal dominant hypophosphatemia rickets is evident: impaired activation of vitamin D. Active vitamin D formation is required for the body to maintain a normal handling of calcium, another important mineral important to bones. Both of these abnormalities of kidney function that of phosphate conservation and of vitamin D activation, are mediated by the high levels of circulating FGF23.
Although much has been learned about the cause of hypophosphatemic rickets, a great deal more remains undiscovered.
Table 1. Hypophosphatemic rickets causes
| Gene | Transmission | Reference | Main features | |
|---|---|---|---|---|
| Hypophosphatemic rickets sharing elevated FGF23 circulating levels and inappropriately low or normal 1,25-diOH-vitamin D | ||||
| X-linked hypophosphatemic rickets | PHEX | X-linked | 11 | Hypophosphatemic rickets with similar phenotype in males and females |
| Autosomal dominant hypophosphatemic rickets | FGF23 | Autosomal dominant | 12 | Hypophosphatemic rickets |
| Autosomal recessive hypophosphatemic rickets | ||||
| DMP1 | Autosomal recessive | 13 | Hypophosphatemic rickets | |
| Autosomal recessive hypophosphatemic rickets type 2 | ENPP1 | Autosomal recessive | 14 | Hypophosphatemic rickets associated with arterial calcifications of infancy (GACI syndrome) |
| Autosomal recessive hypophosphatemic rickets type 3 | FAM20c | Autosomal recessive | 15 | Hypophosphatemia associated with osteosclerosis of the bone rather than rickets, dysmorphy, and cerebral calcifications; severe dental phenotype |
| Osteoglophonic dysplasia | FGFR1 | 16 | Hypophosphatemic rickets associated with frequent craniosynostosis, dysmorphy, and dwarfism | |
| Hypophosphatemic rickets associated with congenital sporadic disorders due to heterozygous post-zygotic mutations in genes activating signaling pathways and elevated FGF23 | ||||
| McCune–Albright syndrome | 17 | Characterized by the triad precocious puberty, cafe-au-lait spots and fibrous dysplasia; hypophosphatemic rickets is rare, and secondary to increased FGF23 production by the fibrous dysplasia | ||
| Mosaic cutaneous disorders include nevus sebaceous and Schimmelpenning syndrome | KRAS and NRAS | 18 | Hypophosphatemic rickets associated with bone lesions and extended cutaneous congenital lesions | |
| Hypophosphatemic rickets associated with mesenchymatous tumors secreting FGF23 | ||||
| Tumor-induced osteomalacia | 19 | Acquired and often severe hypophosphatemia and phosphate wasting. Hypocalcemia may be present as the consequence of suppressed 1,25-dihydroxyvitamin D production | ||
| Hypophosphatemic rickets sharing appropriately suppressed FGF23 and elevated 1,25-diOH-vitamin D; defects in renal phosphate transporters | ||||
| Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) | SLC34A3 | Autosomal recessive | 20 | Hypophosphatemic rickets with nephrocalcinosis and kidney stones |
| Diseases affecting the renal distal tubule | ||||
| Lowe syndrome, Dent syndrome (CLCN5 gene), Toni-Debré-Fanconi | ||||
Hereditary hypophosphatemic rickets inheritance pattern
Hereditary hypophosphatemic rickets can have several patterns of inheritance. When the condition results from mutations in the PHEX gene (resulting in a variant type of PHEX protein), it is inherited in an X-linked dominant pattern. The PHEX gene is located on the X chromosome, which is one of the two sex chromosomes. In females (who have two X chromosomes), a mutation in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
The PHEX protein is a member of an enzyme family of proteins, but it is not precisely clear what the cellular function of PHEX is. The bone cells that express PHEX also secrete an important hormone called FGF23, which is produced in increased amounts when the PHEX protein loses its function, as occurs in X-linked hypophosphatemic rickets. This factor has been shown to act on the kidney to result in excessive urinary excretion of phosphate. The mechanism by which the elevated FGF23 levels occur in the setting of PHEX dysfunction is also not understood.
Similarly, autosomal dominant hypophosphatemia rickets may be caused by specific mutations of the FGF23 (fibroblast growth factor 23) gene located on chromosome 12. These changes result in a variant type of FGF23 protein that persists for longer than normal periods of time in the body, and can result in elevated FGF23 blood levels.
Less commonly, hereditary hypophosphatemic rickets can have an X-linked recessive pattern of inheritance. This form of the condition is often called Dent disease. Like the PHEX gene, the gene associated with Dent disease is located on the X chromosome. In males, one altered copy of the gene in each cell is sufficient to cause the condition. In females, a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females.
In a few families, hereditary hypophosphatemic rickets has had an autosomal dominant inheritance pattern, which means one copy of an altered gene in each cell is sufficient to cause the disorder. The rare condition hereditary hypophosphatemic rickets with hypercalciuria has an autosomal recessive pattern of inheritance, which means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. However, some parents of children with hereditary hypophosphatemic rickets with hypercalciuria have experienced hypercalcuria and kidney stones.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Hypophosphatemic rickets symptoms
Signs and symptoms of familial hypophosphatemic rickets vary greatly, and are usually first noticed at about eighteen months of age. Children often present with progressive bow or knock-knee deformities, delayed walking, waddling gait, and/or short stature. Bone pain and/or joint pain often develops when the child is actively engaged in physical activities. Adults may complain of osteomalacia-related pain, propensity to fracture, arthritis, or pain attributable to excess mineralization of tendons at the site of muscular attachments.
Infants may have an abnormally tall, narrow head (dolichocephaly), a relative enlargement of the front-to-back dimension (scaphocephaly), or abnormally early fusion of the skull bones (craniosynostosis). Toddlers may have an abnormal “waddling” walk (gait) due to abnormally bowed legs (genu varus). In some patients, the knees are bent inwards such that they are too close together (knock knees or genu valgum). Hip deformities in which the thighbone angles towards the center of the body (coxa vara) may occur. Affected individuals often reach a shorter adult height than would otherwise be expected. In older adults, narrowing of the spine (spinal stenosis), and abnormal side-to-side curvature of the spine (scoliosis) may occur.
Symptoms such as weakness and intermittent muscle cramps may also occur, although this is not a usual finding in childhood. Cases of hereditary hypophosphatemic rickets may range from mild to severe. Some individuals may have no noticeable symptoms while others may be marked by pain and/or stiffness of the back, hips, and shoulders possibly limiting mobility. In later adulthood, calcification of tendons and ligaments and the development of bone spurs or bony protrusions can further limit mobility and cause pain.
Dental problems such as decay and spontaneous dental abscesses or late eruption of teeth may develop in individuals with hereditary hypophosphatemic rickets. Less frequently, affected individuals develop enamel defects and an increased frequency of cavities (caries). In some affected individuals, hearing impairment due to malformation of the inner ears (sensorineural hearing loss) may also be present.
Hypophosphatemic rickets diagnosis
Medical history
The earliest clinical sign of hypophosphatemic rickets is usually a somewhat slowed growth rate in the first year of life. The next clinical sign is the patient’s reluctance to bear weight when beginning to stand or walk. Oddly, affected individuals do not have seizures and other systemic signs related to muscle function or oxidative metabolism.
To the degree that heterozygous females are affected, the patient’s maternal family history is likely to include short stature and rickets. Short stature in men is also expected. Older children may have a history of late dentition or multiple dental abscesses.
Physical examination
Affected newborns have normal weight, but infants may show growth retardation. Intellectual development is unaffected. Widened joint spaces and flaring at the knees may become apparent in children by their first birthday, particularly in boys. When a child begins to stand and walk, bowing of the weight-bearing long bones quickly becomes clinically evident. Dentition may be absent or delayed in very young children; older children may experience multiple dental abscesses.
Laboratory studies
Begin clinical laboratory evaluation of rickets with assessment of serum calcium, phosphate, and alkaline phosphatase levels. In hypophosphatemic rickets, calcium levels may be within or slightly below the reference range; alkaline phosphatase levels will be significantly above the reference range.
Carefully evaluate serum phosphate levels in the first year of life, because the concentration reference range for infants (5.0-7.5 mg/dL) is high compared with that for adults (2.7-4.5 mg/dL). Hypophosphatemia can easily be missed in a baby.
Serum parathyroid hormone levels are within the reference range or slightly elevated, while calcitriol (1,25-dihydroxyvitamin D) levels are low or within the lower reference range. Most importantly, urinary loss of phosphate is above the reference range.
Renal tubular phosphate reabsorption
The renal tubular reabsorption of phosphate (TRP) is calculated with the following formula:
- 1 – [Phosphate Clearance (CPi) / Creatinine Clearance (Ccr)] X 100
The following formula calculates Phosphate Clearance (CPi):
- [Urine Phosphate (mg/dL) X Volume (mL/min)] / Plasma Phosphate (mg/dL)
By substituting creatinine values for phosphate in the same formula, Creatinine Clearance (Ccr) can also be calculated. A single early morning urine sample can be used, because Phosphate Clearance (CPi) divided by Creatinine Clearance (Ccr) causes units of urine volume to cancel each other.
- The tubular reabsorption of phosphate in X-linked hypophosphatemia is 60%; normal tubular reabsorption of phosphate exceeds 90% at the same reduced plasma phosphate concentration.
Imaging studies
In all cases of rickets, the study of choice is radiography of the wrists, knees, ankles, and long bones. However, no pathognomonic sign on radiographs distinguishes hypophosphatemic rickets from any other etiology.
On the other hand, a study by Lempicki et al indicated that in hypophosphatemic rickets, magnetic resonance imaging (MRI) reveals characteristics of the knee—including maximum physial widening and transverse degree of widening—that correlate with alkaline phosphatase levels 21
Hypophosphatemic rickets treatment
The conventional treatment for X-linked hypophosphatemic rickets since the 1982 has included use of oral phosphate salts and activated forms of vitamin D, such as calcitriol, given in a multiple daily dosing regimen. Symptomatic and supportive measures are important as well. The usual medication regimen must be carefully monitored to prevent excess blood or urinary calcium levels 22. The approach does not completely cure the disorder. The vitamin D compounds help with phosphate balance and also assist with preventing the complications of too much secretion of parathyroid hormone (PTH). Phosphate enhances the bone healing, but also does not completely cure the disease.
Treatment of affected individuals with this combination of vitamin D and phosphate may result in several side effects, including calcium deposits in the kidneys (nephrocalcinosis), excess levels of calcium in the blood (hypercalcemia), and excess levels of calcium in the urine (hypercalciuria).
In children receiving treatment, periodic renal ultrasonography studies are important to monitor for the development of nephrocalcinosis. Originally thought to be a complication of the disease, this complication is now recognized as an iatrogenic result of therapy 2. Monitoring the ratio of calcium to creatinine in the urine is also important. A ratio of more than 0.25:1 requires reduction of the vitamin D dosage to avoid nephrocalcinosis. Consult a nephrologist for help treating any patient with possible kidney involvement.
Most recently, in 2018, burosumab (Crysvita), a monoclonal IgG1 antibody that inhibits FGF23 activity, was approved by the FDA to treat adults and children ages 1 year and older with X-linked hypophosphatemia 23. For children, burosumab is given by subcutaneous injection every 2 weeks, whereas adults are dosed every 4 weeks.
Covering teeth with sealants has been suggested as a preventive measure for the spontaneous abscesses associated with familial hypophosphatemic rickets.
Attempts have been made in clinical trials to address patient stature by adding growth hormone (GH) to the usual treatment protocol to stimulate growth plates in the long bones. At least one study reported a mild degree of disproportionate truncal growth, which requires further evaluation 24. Although growth hormone therapy has been effective in promoting short-term growth, its high cost discourages widespread use.
A randomized, controlled, open-label study by Meyerhoff et al 25 suggested that growth hormone (GH) therapy in short prepubertal children with X-linked hypophosphatemic rickets does not effectively increase adult height. In terms of exceeding baseline values, standard deviation scores for adult height, leg length, and arm length were not significant in patients who underwent 3 years of growth hormone treatment (although the standard deviation scores for sitting height was). Body disproportion was not found to be exaggerated by growth hormone.
Genetic counseling is recommended for affected individuals and their families.
Surgical care
Osteotomy to realign extremely distorted leg curvatures may be necessary for children whose diagnosis was delayed or whose initial treatment was inadequate. Skull deformity may require treatment for synostosis 26. Spontaneous abscesses often require periodic dental procedures.
A retrospective study by Gizard et al 27 suggested that in patients with X-linked hypophosphatemic rickets, corrective surgery for leg bowing performed before puberty carries an increased risk that the limb deformity will recur. In the study, patients underwent osteotomy and bone alignment (with the exception of three transient hemiepiphysiodesis procedures). Fourteen out of 49 patients (29%) experienced recurrence, with the number of additional surgeries required being highest in individuals who underwent their first surgery before age 11 years and lowest in those whose first surgery was performed after age 15 years 27.
Hypophosphatemic rickets prognosis
Apart from the short stature of most affected adults, the prognosis for a normal lifespan and normal health is good. If the condition is not treated (especially while children are growing), skeletal deformities may be permanent.
The chief aspects of morbidity in X-linked hypophosphatemic rickets are the metabolic processes linked to phosphate. Clinically, the most obvious of these aspects is the effect on bone formation and growth that causes very severe rickets, especially in affected males. The early development of rickets indicates alterations in the orderly processes of bone growth and remodeling that cause bone deformation. Abnormal dentine formation causes late dentition and spontaneous abscess formation.
In a study of 59 adult patients with X-linked hypophosphatemic rickets, Chesher et al 28 found that 42% required at least one osteotomy, while dental disease, nephrocalcinosis, and hearing impairment were found in 63%, 42%, and 14% of the patients, respectively.
Hypercalcemia, nephrocalcinosis, and hypertension
Acute hypercalcemia (with resulting irritability, confusion, and potential seizures) can occur during treatment. Nephrocalcinosis, the long-term result of overaggressive therapy, may be more damaging. Aside from hypercalcemia from vitamin D supplementations, the phosphate supplementations need to be approached with caution. A long-term study in X-linked hypophosphatemic rickets patients treated with combination therapy demonstrated a distinct relationship between mean phosphate dose and the grade of nephrocalcinosis; if the phosphate supplementations are kept to below 60 mg/kg body weight per day, the risk of nephrocalcinosis is minimized 29. Although ultrasonography reveals that 47% of properly treated patients show evidence of nephrocalcinosis, this condition in X-linked hypophosphatemic rickets apparently does not usually progress to renal failure.
Hypertension has been reported in older children under treatment as a consequence of persistent hyperparathyroidism 30. Nephrocalcinosis did not need to be present for hypertension to occur. Consequently, patients under treatment should be carefully monitored for laboratory signs of hyperparathyroidism.
References- Hereditary hypophosphatemic rickets. https://ghr.nlm.nih.gov/condition/hereditary-hypophosphatemic-rickets
- Hypophosphatemic Rickets Treatment & Management. https://emedicine.medscape.com/article/922305-treatment
- Ruppe MD. X-Linked Hypophosphatemia. 2012 Feb 9 [Updated 2017 Apr 13]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK83985
- Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3(1):R13-R30. Published 2014 Mar 14. doi:10.1530/EC-13-0103 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959730
- Douyere D, Joseph C, Gaucher C, Chaussain C, Courson F. Familial hypophosphatemic vitamin D-resistant rickets–prevention of spontaneous dental abscesses on primary teeth: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107(4):525-530.
- Batra P, Tejani Z, Mars M. X-linked hypophosphatemia: dental and histologic findings. J Can Dent Assoc. 2006;72(1):69-72.
- Baroncelli GI, Angiolini M, Ninni E, Galli V, Saggese R, Giuca MR. Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosphatemic rickets. Eur J Paediatr Dent. 2006;7(2):61-66.
- Sabandal MM, Robotta P, Bürklein S, Schäfer E. Review of the dental implications of X-linked hypophosphataemic rickets (XLHR). Clin Oral Investig. 2015;19(4):759-768. doi:10.1007/s00784-015-1425-4
- Hereditary hypophosphatemic rickets and tumor-induced osteomalacia. https://www.uptodate.com/contents/hereditary-hypophosphatemic-rickets-and-tumor-induced-osteomalacia
- Hypophosphatemic Rickets. https://www.merckmanuals.com/home/children-s-health-issues/congenital-kidney-tubular-disorders/hypophosphatemic-rickets
- Francis F, Hennig S, Korn B, Reinhardt R, de Jong P, Poustka A, Lehrach H, Rowe PSN, Goulding JN, Summerfield T, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nature Genetics. 1995;11:130–136. doi: 10.1038/ng1095-130.
- Consortium A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nature Genetics. 2000;26:345–348. doi: 10.1038/81664
- Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nature Genetics. 2006;38:1248–1250. doi: 10.1038/ng1868
- Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, Manor E, Buriakovsky S, Hadad Y, Goding J, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. American Journal of Human Genetics. 2010;86:273–278. doi: 10.1016/j.ajhg.2010.01.010
- Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, Bjerknes R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. Journal of Bone and Mineral Research. 2013;28:1378–1385. doi: 10.1002/jbmr.1850
- White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. American Journal of Human Genetics. 2005;76:361–367. doi: 10.1086/427956
- Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. Journal of Clinical Investigation. 2003;112:683–692. doi: 10.1172/JCI18399
- Lim YH, Ovejero D, Sugarman JS, Deklotz CM, Maruri A, Eichenfield LF, Kelley PK, Juppner H, Gottschalk M, Tifft CJ, et al. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Human Molecular Genetics. 2014;23:397–407. doi: 10.1093/hmg/ddt429
- Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. PNAS. 2001;98:6500–6505. doi: 10.1073/pnas.101545198
- Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. American Journal of Human Genetics. 2006;78:179–192. doi: 10.1086/499409
- Lempicki M, Rothenbuhler A, Merzoug V, et al. Magnetic Resonance Imaging Features as Surrogate Markers of X-Linked Hypophosphatemic Rickets Activity. Horm Res Paediatr. 2017 Apr 3. 87 (4):244-53.
- Nielsen LH, Rahbek ET, Beck-Nielsen SS, Christesen HT. Treatment of hypophosphataemic rickets in children remains a challenge. Dan Med J. 2014 Jul. 61(7):A4874.
- FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia. https://www.fda.gov/news-events/press-announcements/fda-approves-first-therapy-rare-inherited-form-rickets-x-linked-hypophosphatemia
- Haffner D, Nissel R, Wuhl E, Mehls O. Effects of growth hormone treatment on body proportions and final height among small children with X-linked hypophosphatemic rickets. Pediatrics. 2004 Jun. 113(6):e593-6.
- Meyerhoff N, Haffner D, Staude H, et al. Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol. 2018 Mar. 33 (3):447-56.
- Jaszczuk P, Rogers GF, Guzman R, Proctor MR. X-linked hypophosphatemic rickets and sagittal craniosynostosis: three patients requiring operative cranial expansion: case series and literature review. Childs Nerv Syst. 2016 May. 32 (5):887-91.
- Gizard A, Rothenbuhler A, Pejin Z, et al. Outcomes of orthopedic surgery in a cohort of 49 patients with X-linked hypophosphatemic rickets (XLHR). Endocr Connect. 2017 Nov. 6 (8):566-73.
- Chesher D, Oddy M, Darbar U, et al. Outcome of adult patients with X-linked hypophosphatemia caused by PHEX gene mutations. J Inherit Metab Dis. 2018 Sep. 41 (5):865-876.
- Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med. 1991 Dec 26. 325(26):1843-8.
- Alon US, Monzavi R, Lilien M, et al. Hypertension in hypophosphatemic rickets–role of secondary hyperparathyroidism. Pediatr Nephrol. 2003 Feb. 18(2):155-8.




{kind=link}