Kearns Sayre syndrome
Kearns-Sayre syndrome is a rare neuromuscular disorder defined by the triad of onset before age 20 years, pigmentary retinopathy (a “salt-and-pepper” pigmentation in the retina that can affect vision, but often leaves it intact), and progressive external ophthalmoplegia 1. Kearns-Sayre syndrome is the result of abnormalities in the DNA of mitochondria (mtDNA) – small rod-like structures found in every cell of the body that produce the energy that drives cellular functions. The mitochondrial diseases correlate with specific DNA mutations that cause problems with many of the organs and tissues in the body.
The prevalence of Kearns-Sayre syndrome is approximately 1 to 3 per 100,000 individuals 2.
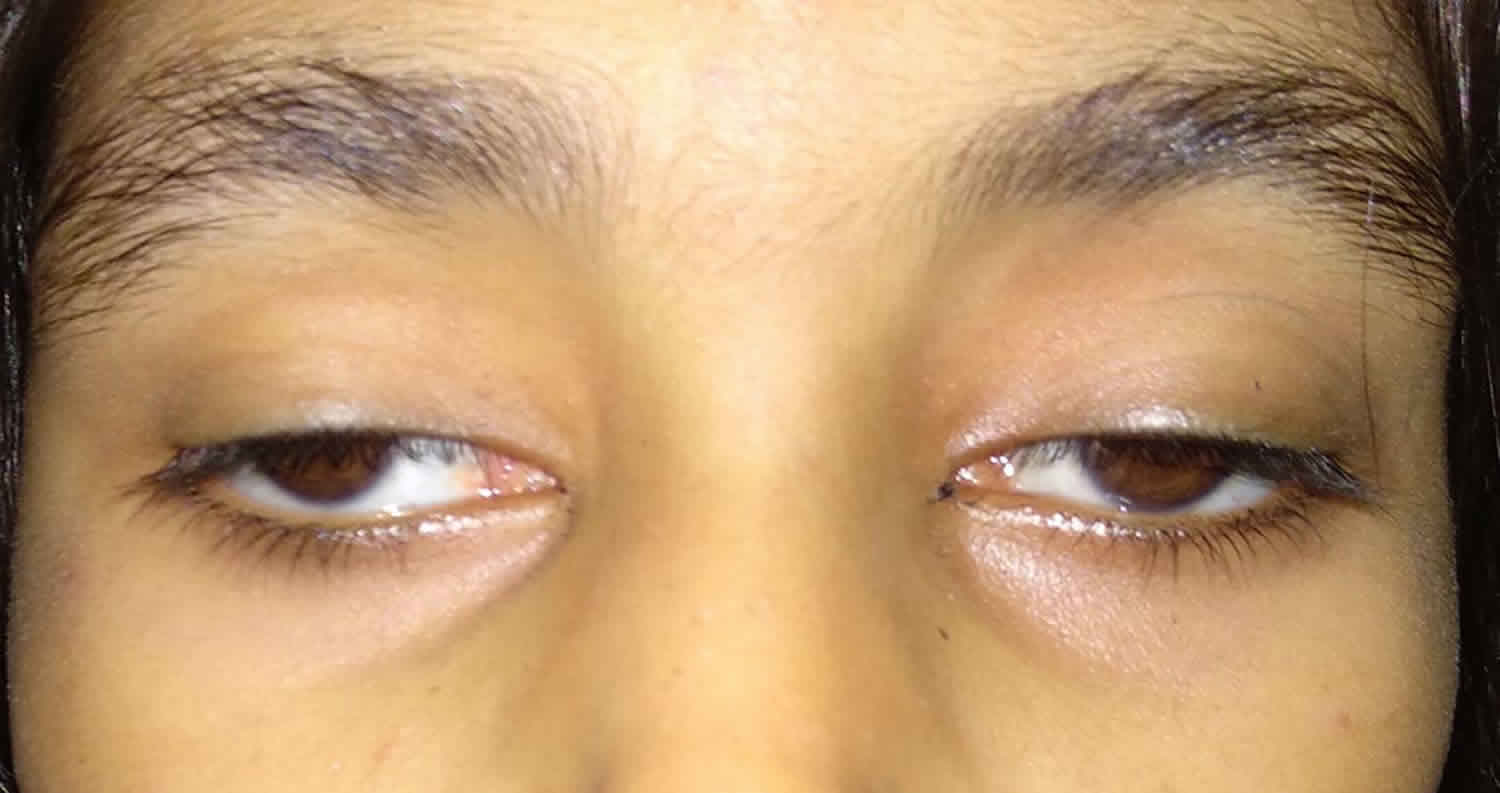
Kearns-Sayre syndrome is characterized by progressive limitation of eye movements until there is complete immobility (progressive external ophthalmoplegia), accompanied by eyelid droop (ptosis). Kearns-Sayre syndrome is also associated with abnormal accumulation of pigmented material on the membrane lining the eyes called pigmentary retinopathy, which results from breakdown (degeneration) of the light-sensing tissue at the back of the eye (the retina) that gives it a speckled and streaked appearance. The pigmentary retinopathy may cause loss of vision. Kearns-Sayre syndrome may be associated with pigmentary retinopathy similar to that seen in patients with retinitis pigmentosa 3. Additional symptoms may include mild skeletal muscle weakness in the limbs, heart block (a cardiac conduction defect), short stature, hearing loss (deafness), abnormally high levels of protein in the cerebrospinal fluid (CSF) with protein concentration greater than 100 mg/dL, an inability to coordinate voluntary movements (ataxia), impaired cognitive function (dementia), kidney problems and diabetes mellitus. Seizures are infrequent. Affected individuals often have short stature. Several endocrine disorders can be associated with Kearns-Sayre syndrome, including delayed sexual maturation, hypothyroidism, and growth hormone deficiency 1.
When the muscle cells of affected individuals are stained and viewed under a microscope, these cells usually appear abnormal. The abnormal muscle cells contain an excess of structures called mitochondria and are known as ragged-red fibers.
A related condition called ophthalmoplegia-plus may be diagnosed if an individual has many of the signs and symptoms of Kearns-Sayre syndrome but not all the criteria are met.
There is currently no effective way to treat mitochondria abnormalities in Kearns-Sayre syndrome. Treatment is generally symptomatic and supportive 4. Management of Kearns-Sayre syndrome involves multiple specialties depending on the organs involved. The most essential is a regular and long-term follow-up with cardiologists. Conduction problems of heart impulse like heart block may be treated with a pacemaker. Other consultations may include audiology, ophthalmology, endocrinology, neurology, and neuropsychiatry. Hearing aids may be required. There is typically no treatment for limitation in eye movement. Endocrinology abnormalities can be treated with drugs.
Kearns-Sayre syndrome causes
Kearns-Sayre syndrome is a condition caused by defects in mitochondria, which are structures within cells that use oxygen to convert the energy from food into a form cells can use. This process is called oxidative phosphorylation. Although most DNA is packaged in chromosomes within the nucleus (nuclear DNA), mitochondria also have a small amount of their own DNA, called mitochondrial DNA (mtDNA). This type of DNA contains many genes essential for normal mitochondrial function. People with Kearns-Sayre syndrome have a single, large deletion of mtDNA, ranging from 1,000 to 10,000 DNA building blocks (nucleotides). The cause of the deletion in affected individuals is unknown.
The mtDNA deletions that cause Kearns-Sayre syndrome result in the loss of genes important for mitochondrial protein formation and oxidative phosphorylation. The most common deletion removes 4,997 nucleotides, which includes twelve mitochondrial genes. Deletions of mtDNA result in impairment of oxidative phosphorylation and a decrease in cellular energy production. Regardless of which genes are deleted, all steps of oxidative phosphorylation are affected. Researchers have not determined how these deletions lead to the specific signs and symptoms of Kearns-Sayre syndrome, although the features of the condition are probably related to a lack of cellular energy. It has been suggested that eyes are commonly affected by mitochondrial defects because they are especially dependent on mitochondria for energy.
Inheritance pattern
Kearns-Sayre syndrome is generally not inherited but arises from mutations in the body’s cells that occur after conception. This alteration is called a somatic mutation and is present only in certain cells.
Rarely, Kearns-Sayre syndrome is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
The children of males with Kearns-Sayre syndrome are not at risk 1.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Kearns-Sayre syndrome symptoms
Kearns-Sayre syndrome is characterized by progressive external ophthalmoplegia (progressive paralysis of certain eye muscles) and pigmentary retinopathy (a “salt-and-pepper” pigmentation in the retina that can affect vision, but often leaves it intact). These symptoms typically develop before 20 years of age 1. At least one of the following must also be present: cardiac conduction block, cerebrospinal fluid protein concentration greater than 100 mg/dL, or cerebellar ataxia 1.
Additional symptoms may include mild skeletal muscle weakness, short stature, hearing loss, impaired cognitive function, and diabetes mellitus. Seizures are infrequent. Several endocrine disorders can be associated with Kearns-Sayre syndrome, including delayed sexual maturation, hypothyroidism, and growth hormone deficiency 1.
In most cases, the first physical characteristic of this disorder is retardation of growth. In addition, drooping of the upper eyelid (ptosis) due to weakness of one of the muscles of the eyelid (levator palpebrae superioris) is also seen early during infancy. Other muscles involved in coordinating eye movements may be affected next, growing progressively weaker and eventually resulting in paralysis of certain eye movements (progressive external ophthalmoplegia). Eventually, muscle weakness may extend to other portions of the face, throat (pharynx), neck, and/or shoulders. Muscle weakness in such areas may hinder talking and/or swallowing (dysphagia). As the disease progresses, the upper arms and legs may be affected, resulting in progressive impairment of coordinated movement (ataxia) and/or a staggered or halting gait (titubation).
Most individuals with Kearns-Sayre syndrome will also have visual difficulties due to the abnormal accumulation of colored (pigmented) material on the delicate membrane that lines the eyes (atypical retinitis pigmentosa) and progressive degeneration of certain portions of the eye (pigmentary degeneration of the retina). This degenerative process may eventually affect the optic nerve (optic atrophy), the layers of membranes behind the retina (choroid), and/or the tough, white outer covering of the eyeball (sclera). In some cases, affected individuals may also experience night blindness; rapid, involuntary eye movements (nystagmus); and a decrease in the sharpness of vision (visual acuity). In rare cases, abnormal clouding of the front portion of the eyeball (cornea) may also contribute to nystagmus and decreased visual acuity. About 40 percent of people with Kearns-Sayre syndrome experience profound visual problems.
The third primary finding in people with Kearns-Sayre syndrome is an interference with the transfer of nerve impulses (conduction) that control the activity of heart muscles (heart block). The severity of such conduction abnormalities may vary among affected individuals.
The normal heart has four chambers. The two upper chambers, known as atria, are separated from each other by a fibrous partition known as the atrial septum. The two lower chambers are known as ventricles and are separated from each other by the ventricular septum. Valves connect the atria (left and right) to their respective ventricles. In the mild form of heart block, the two upper chambers of the heart (atria) beat normally, but the contractions of the two lower chambers (ventricles) slightly lag behind. In the more severe forms, only a half to a quarter of the atrial beats are conducted to the ventricles. In complete heart block, the atria and ventricles beat separately. In some cases, heart block may lead to blackouts (syncope), breathlessness, and/or irregular heartbeats (arrhythmias).
Individuals with Kearns-Sayre syndrome may also exhibit a variety of other physical characteristics and symptoms. The number and severity of these symptoms may vary greatly from patient to patient; in some people, individuals may exhibit a partial or incomplete form of the disorder. The additional physical characteristics and symptoms associated with Kearns-Sayre syndrome may include developmental delays; short stature (dwarfism); diminished muscle tone (hypotonia); hearing loss, eventually leading to deafness; cognitive impairment; progressive memory loss and deterioration of intellectual abilities (dementia), and/or abnormalities affecting various of parts of the brain (e.g., white and gray matter, brain stem, and/or cerebellum).
In some affected individuals, Kearns-Sayre syndrome may also be associated with several disorders involving the function of structures and organs that secrete hormones into the blood system (multiple endocrine dysfunction). The most common of these disorders occurring in association with Kearns-Sayre syndrome include hypoparathyroidism, diabetes mellitus, and/or primary failure of the ovaries or testes (gonads). These disorders may result in short stature, a delay in reaching puberty, excessive fatigue, and/or muscle cramps. The relationship between Kearns-Sayre syndrome and endocrine abnormalities is not fully understood.
On rare occasions, Kearns-Sayre syndrome may also be associated with other disorders or conditions including absence of certain reflexes, kidney (renal) abnormalities, and/or peripheral neuropathy. peripheral neuropathy is a rare disorder that may affect one or several nerves of the body, causing pain and weakness. Peripheral neuropathy may affect sensory, motor, reflex, or blood vessel function.
Kearns-Sayre syndrome diagnosis
The diagnosis of Kearns-Sayre syndrome may be suspected when the three primary characteristics associated with this disorder occur in association with one another. These include paralysis of certain eye muscles (progressive external ophthalmoplegia), abnormal coloration of the delicate membrane lining the eyes (atypical retinitis pigmentosa) and other changes in the structures of the eye (pigmentary degeneration of the retina), and disease affecting the heart (cardiomyopathy), especially conduction disorders (e.g., heart block). Diagnosis of Kearns-Sayre syndrome may be confirmed by a thorough clinical evaluation and a variety of specialized tests.
Such specialized tests may include an electrocardiogram to detect the presence and evaluate the severity of heart block, blood and spinal fluid lactic acid levels, a muscle biopsy to demonstrate the presence of characteristic abnormalities in muscle tissue (ragged-red fibers), and/or a spinal tap to determine whether there are elevated levels of certain cerebrospinal fluid (CSF) proteins. The muscle biopsy can determine the presence of deleted mtDNA, which is generally not detected in the blood sample. In some cases of Kearns-Sayre syndrome, the levels of other substances (i.e., serum creatine kinase, blood lactate, gamma globulin, and/or pyruvate) may be elevated in the blood.
Microscopic examination of biopsy tissue samples under an electron microscope may reveal large numbers of abnormal mitochondria in skeletal and eye muscle tissue. In some cases, a CT scan or tomography may be used to identify abnormal accumulation of calcium in and/or lesions affecting certain areas of the brain.
Kearns-Sayre syndrome treatment
Treatment for Kearns-Sayre syndrome is generally symptomatic and supportive. Management options include placement of cardiac pacemakers in individuals with cardiac conduction blocks, eyelid slings for severe ptosis, cochlear implants and hearing aids for neurosensory hearing loss, hormone replacement for endocrinopathies, dilation of the upper esophageal sphincter to alleviate cricopharyngeal achalasia, folinic acid supplementation in individuals with Kearns-Sayre syndrome with low cerebral spinal fluid folic acid, administration of coenzyme Q10 and L-carnitine, physical and occupational therapy, and treatment of depression. The U.S. Food and Drug Administration (FDA) has granted Orphan Drug designation to coenzyme Q10 (UbiQGel) for the treatment of Kearns-Sayre syndrome and other mitochondrial cytopathies. L-Carnitine (a specialized amino acid) and vitamin therapy including coenzyme Q10, riboflavin, and vitamins C and K are being tested as possible treatments for mitochondrial disorders. These options may be used alone or in conjunction with one another for the treatment of mitochondrial disorders. More studies are needed to determine the long-term safety and effectiveness of these potential treatments for mitochondrial disorders such as Kearns-Sayre syndrome.
Ibedenone, Thiamine, Vitamin E (antioxidant), and dichloroacetate may be also used but their effects have not yet been scientifically validated.
Antioxidants may ameliorate damage from reactive oxygen species; percutaneous endoscopic gastrostomy may improve nutritional intake and prevent aspiration pneumonia in individuals with severe dysphagia. Surveillance includes electrocardiogram (ECG) and echocardiogram every six to 12 months and yearly audiometry and endocrinologic evaluation 1.
Separate treatment options for associated disorders (e.g., diabetes mellitus or hypoparathyroidism) may be necessary. In some cases, treatment may include hormone replacement therapies. Other treatments will depend on the specific conditions present.
Genetic counseling may be of benefit for affected individuals and their families. A team approach for infants with this disorder may also be of benefit and may include special social support and other medical services including physical and occupational therapies. Other treatment is symptomatic and supportive.
Kearns-Sayre syndrome prognosis
Kearns-Sayre syndrome is a slowly progressive disorder. The prognosis for individuals with Kearns-Sayre syndrome varies depending on the severity and the number of organs involved. Early diagnosis and periodic electrocardiogram (ECG) are important since heart block can cause death in 20 percent of patients. Early pacemaker implantation can be of great benefit and offer a longer life expectancy in many patients.
References- Goldstein A, Falk MJ. Mitochondrial DNA Deletion Syndromes. 2003 Dec 17 [Updated 2019 Jan 31]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1203
- Kearns-Sayre syndrome. https://ghr.nlm.nih.gov/condition/kearns-sayre-syndrome
- Padhy SK, Kumar V, Mandal S. Pigmentary retinopathy in Kearns-Sayre syndrome. Case Reports 2018;2018:bcr-2018-227394.
- Kearns-Sayre Syndrome Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Kearns-Sayre-Syndrome-Information-Page





{kind=link}