Laron syndrome
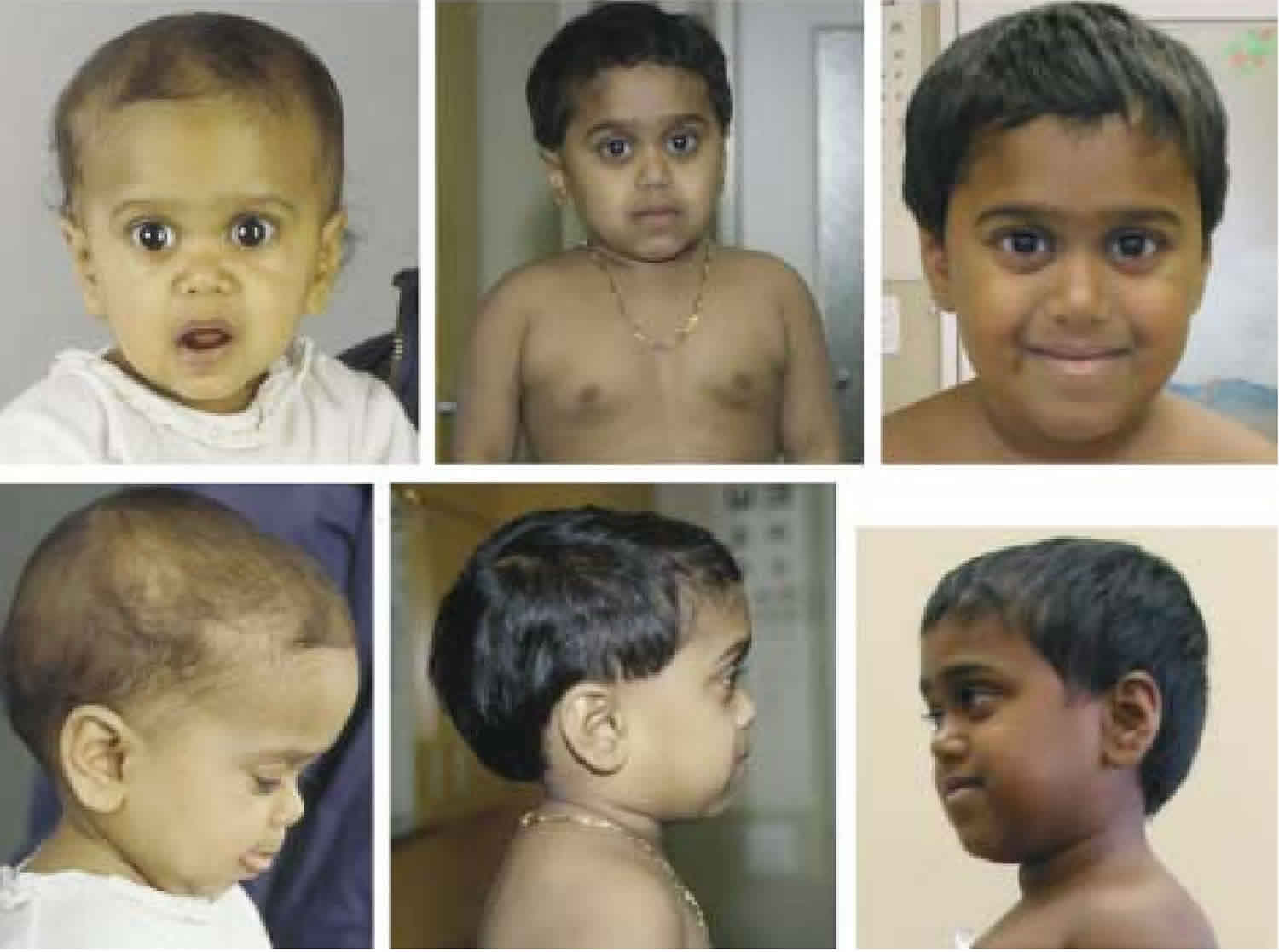
Laron syndrome also called growth hormone insensitivity syndrome, growth hormone receptor deficiency or Laron dwarfism, is a rare form of short stature that results from the body’s inability to use growth hormone, a substance produced by the brain’s pituitary gland that helps promote growth 1. Affected individuals are close to normal size at birth, but they experience slow growth from early childhood that results in very short stature. If Laron syndrome is not treated, adult males typically reach a maximum height of about 4.5 feet; adult females may be just over 4 feet tall.
Other features of untreated Laron syndrome include reduced muscle strength and endurance, low blood sugar levels (hypoglycemia) in infancy, small genitals and delayed puberty, hair that is thin and fragile, and dental abnormalities. Many affected individuals have a distinctive facial appearance, including a protruding forehead, a sunken bridge of the nose (saddle nose), and a blue tint to the whites of the eyes (blue sclerae). Affected individuals have short limbs compared to the size of their torso, as well as small hands and feet. Adults with this condition tend to develop obesity. However, the signs and symptoms of Laron syndrome vary, even among affected members of the same family.
Studies suggest that people with Laron syndrome have a significantly reduced risk of cancer and type 2 diabetes. Affected individuals appear to develop these common diseases much less frequently than their unaffected relatives, despite having obesity (a risk factor for both cancer and type 2 diabetes). However, people with Laron syndrome do not seem to have an increased lifespan compared with their unaffected relatives.
Laron syndrome is a rare disorder. About 350 people have been diagnosed with the condition worldwide. The largest single group of affected individuals (about 100 people) lives in an area of southern Ecuador.
There is currently no cure for Laron syndrome. Treatment is primarily focused on improving growth 2. The only specific treatment available for this condition is subcutaneous injections of insulin-like growth factor 1 (a growth-promoting hormone), often called IGF-1. IGF-1 stimulates linear growth (height) and also improves brain growth and metabolic abnormalities caused by long-term IGF-1 deficiency. It has also been shown to raise blood glucose levels, reduce cholesterol, and increase muscle growth. IGF-1 and GH levels should be closely monitored in people undergoing this treatment because overdosage of IGF-1 causes a variety of health problems.
Laron syndrome cause
Laron syndrome is caused by mutations in the GHR gene on chromosome 5p13-p12. The GHR gene provides instructions for making a protein called the growth hormone receptor. The growth hormone receptor is present on the outer membrane of cells throughout the body, particularly liver cells. As its name suggests, the growth hormone receptor attaches (binds) to growth hormone; the two proteins fit together like a key in a lock. When growth hormone is bound to its receptor, it triggers signaling that stimulates the growth and division of cells. This signaling also leads to the production, primarily by liver cells, of another important growth-promoting hormone called insulin-like growth factor 1 (IGF-1).
Growth hormone and IGF-1 have a wide variety of effects on the growth and function of many parts of the body. For example, these hormones stimulate the growth and division of cells called chondrocytes, which play a critical role in producing new bone tissue. Growth hormone and IGF-1 also influence metabolism, including how the body uses and stores carbohydrates, proteins, and fats from food.
Mutations in the GHR gene impair the receptor’s ability to bind to growth hormone or to trigger signaling within cells. As a result, even when growth hormone is available, cells are unable to respond by producing IGF-1 and stimulating growth and division. The cells’ inability to react to growth hormone, which is described as growth hormone insensitivity, disrupts the normal growth and function of many different tissues. Short stature results when growth hormone cannot adequately stimulate the growth of bones. Changes in metabolism caused by insensitivity to growth hormone and the resulting shortage of IGF-1 cause many of the other features of the condition, including obesity.
Researchers are working to determine how mutations in the GHR gene may protect people with Laron syndrome from developing cancer and type 2 diabetes. Studies suggest that insensitivity to growth hormone may help prevent the uncontrolled growth and division of cells that can lead to the development of cancerous tumors. Growth hormone insensitivity also appears to alter how the body responds to insulin, which is a hormone that regulates blood sugar levels. Resistance to the effects of insulin is a major risk factor for type 2 diabetes. People with Laron syndrome have the opposite situation, an increased sensitivity to insulin, which likely helps explain their reduced risk of this common disease.
Laron syndrome inheritance pattern
Most cases of Laron syndrome are inherited in an autosomal recessive pattern, which means both copies of the GHR gene in each cell have mutations (see Figure 1). The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
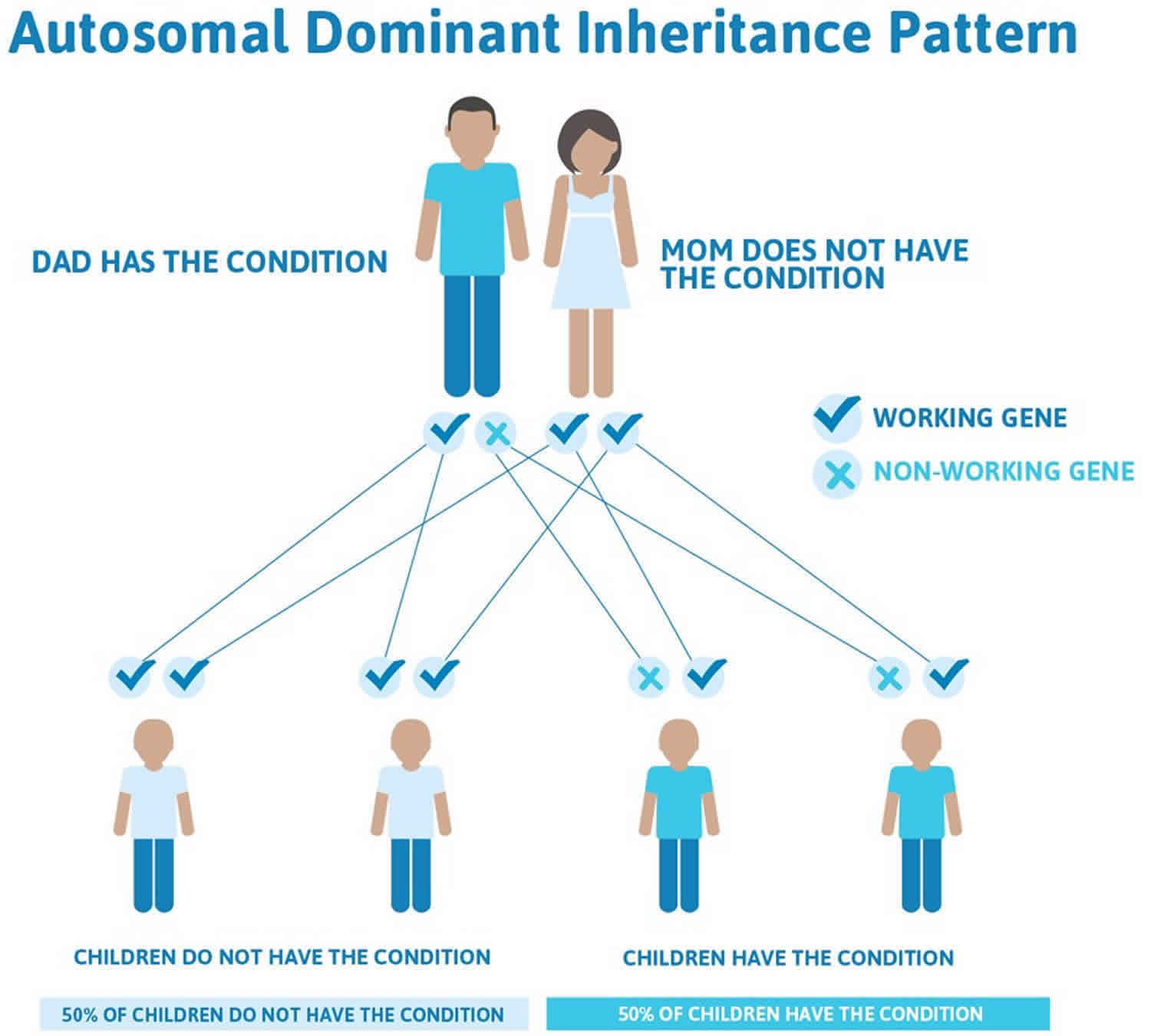
Much less commonly, Laron syndrome has an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder (see Figure 2). In most of these cases, an affected person has one parent with the condition.
Figure 1. Laron syndrome autosomal recessive inheritance pattern

Figure 2. Laron syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Laron syndrome symptoms
Laron syndrome is a rare condition in which the body is unable to use growth hormone. The primary symptom is short stature. Although affected people are generally close to average size at birth, they experience slow growth from early childhood. If left untreated, adult males with Laron syndrome typically reach a maximum height of about 4.5 feet and adult females may be just over 4 feet tall 3.
Other signs and symptoms associated with Laron syndrome vary but may include 3:
- Reduced muscle strength and endurance
- Hypoglycemia in infancy
- Delayed puberty
- Small genitals
- Thin, fragile hair
- Dental abnormalities
- Short limbs (arms and legs)
- Obesity
- Distinctive facial features (protruding forehead, a sunken bridge of the nose, and blue sclerae)
People affected by Laron syndrome appear to have a reduced risk of cancer and type 2 diabetes 1.
Laron syndrome diagnosis
Laron syndrome diagnosis is based on clinical and biological findings. Hormonal tests reveal normal or increased level of growth hormone (GH) and low IGF-1 levels, which fail to rise after exogenous growth hormone administration. Growth hormone binding protein levels (GHBP, structurally identical to the extracellular domain of growth hormone receptor) levels are low in cases with mutations in the extracellular domain of the GHR and normal in cases with mutations in the intracellular domain. Genetic tests should be performed to make a precise etiological diagnosis.
Laron syndrome treatment
There is currently no cure for Laron syndrome. Treatment is primarily focused on improving growth 2. The only specific treatment available for Laron syndrome is subcutaneous injections of insulin-like growth factor 1 (IGF-1) a growth-promoting hormone. IGF-1 stimulates linear growth (height) and also improves brain growth and metabolic abnormalities caused by long-term IGF-1 deficiency. It has also been shown to raise blood glucose levels, reduce cholesterol, and increase muscle growth 4. IGF-1 and growth hormone levels should be closely monitored in people undergoing this treatment because overdosage of IGF-I causes a variety of health problems 5.
The medication(s) listed below have been approved by the U.S. Food and Drug Administration (FDA) as orphan products for treatment of Laron syndrome.
- Mecasermin (Brand name: Increlex®) – Manufactured by Tercica, Inc. FDA-approved indication: Long-term treatment of growth failure in children with severe primary IGF-1 deficiency (Primary IGFD) or with growth hormone (GH) gene deletion who have developed neutralizing antibodies to growth hormone.
- Mecasermin rinfabate (Brand name: Iplex®) – Manufactured by Insmed, Inc. FDA-approved indication: Treatment of growth failure in children with severe primary IGF-1 deficiency (Primary IGFD) or with growth hormone (GH) gene deletion who have developed neutralizing antibodies to growth hormone.
Recombinant human insulin-like growth factor 1 (IGF-1) therapy provides limited benefit in improving height. In an observational study containing 28 patients with Laron syndrome, the results of treatment with 120 mg/kg/day IGF-1 for a mean duration of 5 years increased height from -6.1 standard deviation (SD) to -5.1 SD 6. In the first year of treatment there was a marked increase in height velocity from 2.8 to 8.7 cm but height velocity markedly decreased after the first year of treatment. In a separate report of 21 individuals with growth hormone insensitivity, 5 of whom had Laron syndrome, there was an increase in height from baseline of +1.9 SD with treatment of 120 mcg/kg/day IGF-1 for a mean of 10.5 years 7. The treatment effect is markedly lower than that of growth hormone in children with severe congenital growth hormone deficiency 3. While growth hormone therapy stimulates both hepatic and local IGF-1 production, subcutaneous injections of IGF-1 do not simulate this local IGF-1 production. In addition, growth hormone therapy normalises not only IGF-1 levels but levels of IGF binding protein 3 (IGFBP-3) and acid labile subunit whereas in growth hormone insensitive subjects treated with IGF-1 there is no increase in IGFBP-3 or acid-labile subunit concentrations. Thus, it would be expected that the injected IGF-1 would have a much lower half life than endogenous IGF-1. A combined therapy of IGF-1 with IGFBP-3 disappointingly was less effective in improving height 8.
Laron syndrome prognosis
The long-term outlook (prognosis) for people with Laron syndrome is generally good. Laron syndrome does not appear to affect lifespan and is associated with a reduced risk of cancer and type 2 diabetes.
References- Laron syndrome. https://ghr.nlm.nih.gov/condition/laron-syndrome
- Laron syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=633
- Murray PG, Clayton PE. Disorders of Growth Hormone in Childhood. [Updated 2016 Nov 16]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK278971
- Laron Z. Growth hormone insensitivity (Laron syndrome). Rev Endocr Metab Disord. December 2002; 3(4):347-355.
- Laron Z. Insulin-like growth factor-I treatment of children with Laron syndrome (primary growth hormone insensitivity). Pediatr Endocrinol Rev. March 2008; 5(3):766-771.
- Chernausek SD, Backeljauw PF, Frane J, Kuntze J, Underwood LE, Group GHISC. Long-term treatment with recombinant insulin-like growth factor (IGF)-I in children with severe IGF-I deficiency due to growth hormone insensitivity. The Journal of clinical endocrinology and metabolism 2007;92:902-10.
- Backeljauw PF, Kuntze J, Frane J, Calikoglu AS, Chernausek SD. Adult and near-adult height in patients with severe insulin-like growth factor-I deficiency after long-term therapy with recombinant human insulin-like growth factor-I. Hormone research in paediatrics 2013;80:47-56.
- Tonella P, Fluck CE, Mullis PE. Insulin-like growth factor-I treatment in primary growth hormone insensitivity: effect of recombinant human IGF-I (rhIGF-I) and rhIGF-I/rhIGF-binding protein-3 complex. Hormone research in paediatrics 2010;73:140-7.





{kind=link}