What is neuroplasticity
Neuroplasticity also called neuronal plasticity, can be broadly defined as the ability of the nervous system to respond to intrinsic or extrinsic stimuli by reorganizing its structure, function and connections; can be described at many levels, from molecular to cellular to systems to behavior; and can occur during development, in response to the environment, in support of learning, in response to disease, or in relation to therapy 1. Neuroplasticity (neuronal plasticity) is a process through which external and internal environment of an individual gradually becomes represented in neuronal structure and function during development and through learning 2. One of the most outstanding properties of the nervous system is its ability to modify its structure and function in response to experience, thus allowing individual “self-tuning” to particular environmental drivers 3. The phenomenon of neural plasticity is known to underlie the learning, consolidation and refinement of both adaptive and maladaptive behaviors 4. At the synaptic level, activity-dependent modifications of the strength or efficacy of synaptic transmission shape the response properties of neural circuits.
The extrinsic and intrinsic environment becomes represented in the structure and function of neuronal networks during development through neuronal plasticity 5. In adulthood, this representation is continuously optimized and modulated through neuroplasticity and learning 6. Neuroplasticity occurs at several levels, from neurogenesis (the formation of newborn neurons in proliferative areas) to the adjustment of synaptic weights, and modulates both the structure and function of neuronal networks (Figure 1). Changes in function of the nervous system are essentially always based on a structural change at some level (neuronal, synaptic, protein, or genomic structure), and it is therefore difficult to make clear distinctions between functional and structural plasticity.
Neuroplasticity can be conceptualized as two distinct processes. First, structural variability is generated through the overproduction of immature neuronal structures, which occurs at many levels from new-born neurons to the outgrowth of filopodia (Figure 1). Second, selective stabilization among the overproduced structures retains those that best represent the internal or external environment 7. Structural variability may be stochastic, although it may also be genetically tuned, but selection is an active process driven by neuronal activity that reflects both extrinsic and intrinsic stimuli. Activity-dependent selection guides the stabilization of functionally relevant neurons and connections by utilizing genes involved in survival and synaptogenesis, or it eliminates weakly or incoherently active structures through the use of genes that underlie apoptosis and pruning 8. The elimination of weakly active neurons and connections is critical for optimizing the signal-to-noise ratio in neuronal networks. Neuroplasticity can be compared to auditions for a Broadway show: if many candidates audition, the production team can select an optimal performer for each role (and send those not suitable back home), but if only a few people show up, almost all have to be utilized regardless of their talent.
Figure 1. Proposed model for the levels of neuronal plasticity in antidepressant activity
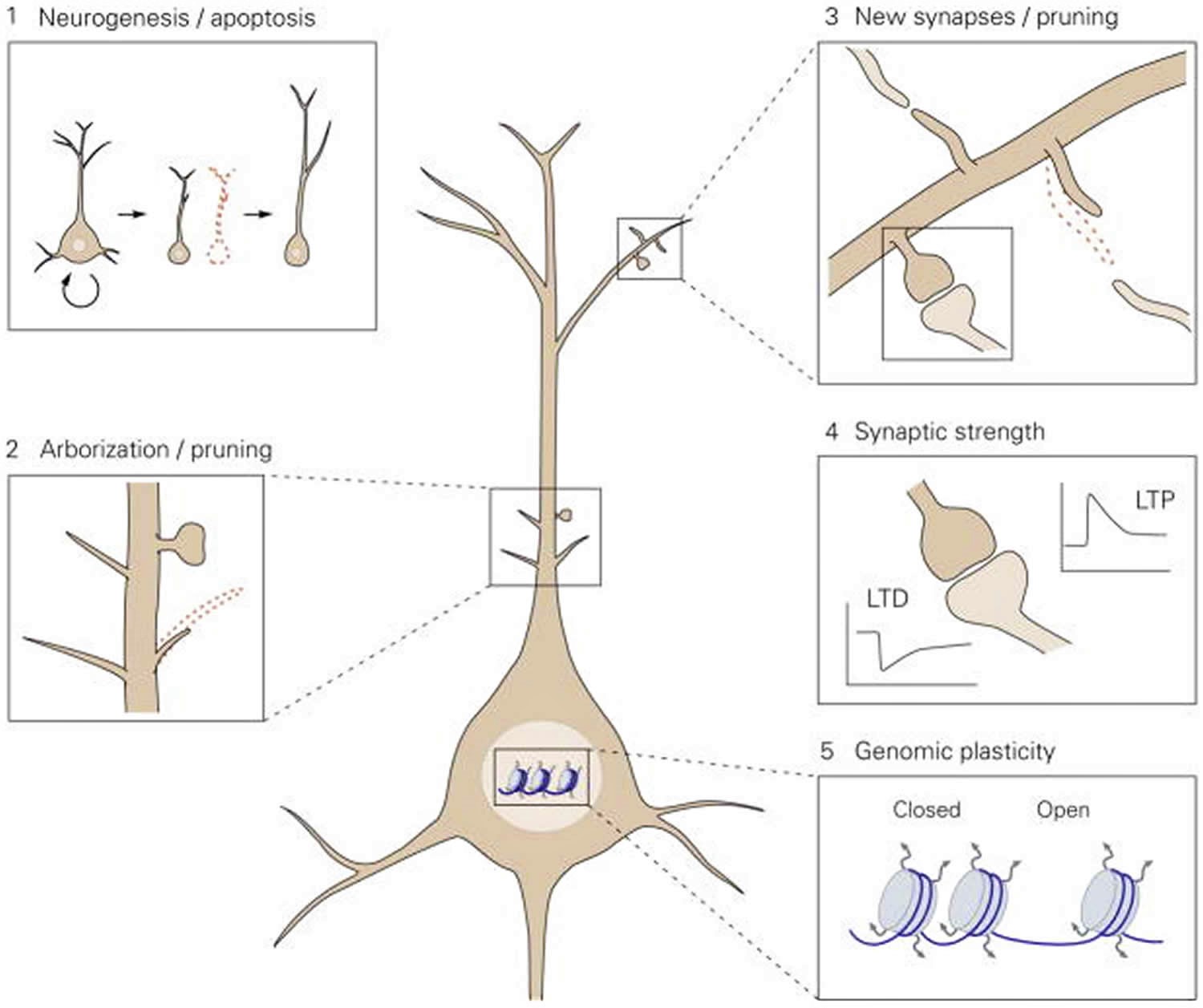
Footnote: Neuroplasticity acts at different structural levels and bidirectionally to influence variability and selection within neuronal networks. Increased formation of structures at any of these levels by, for example, environmental enrichment (EE) or chronic antidepressant treatment, generates variability and promotes competition between similar structures for stabilization, thereby enhancing adaptability, even if the total number of structures is not increased due to the simultaneous increase in structural elimination. 1. Neurogenesis and selective apoptosis. Increased precursor proliferation in the dentate gyrus, induced by antidepressants and EE, leads to the increased survival of newborn neurons that successfully mediate activity within the hippocampal circuitry. Neurons that fail to functionally integrate into the hippocampal circuitry are eliminated through apoptosis. 2. Arborization and pruning of axonal and dendritic branches. The increased dynamics of nascent branches promotes the stabilization of branches containing synapses that successfully represent environmental conditions, whereas arbors without active synapses remain short-lived and are pruned. 3. Synaptogenesis and synaptic elimination. Immature “trial synapses” between two neurons are initiated by filopodial extension from pre- or postsynaptic sites. Synapses that are successfully activated during the trial period are preferentially selected for stabilization, whereas contacts that fail to mediate activity collapse and are eliminated. 4. Plastic regulation of synaptic strength. Information transfer through active synapses is potentiated through the process of long-term potentiation (LTP), whereas inactive or inappropriately active synapses are suppressed through long-term depression (LTD). 5. Environmental activity regulates the transcription and translation of effector genes involved in neuronal plasticity through transcriptional control and epigenetic mechanisms, such as remodeling of chromatin structure from a closed to an open state.
[Source 5 ]Figure 2. Adult neurogenesis brain regions
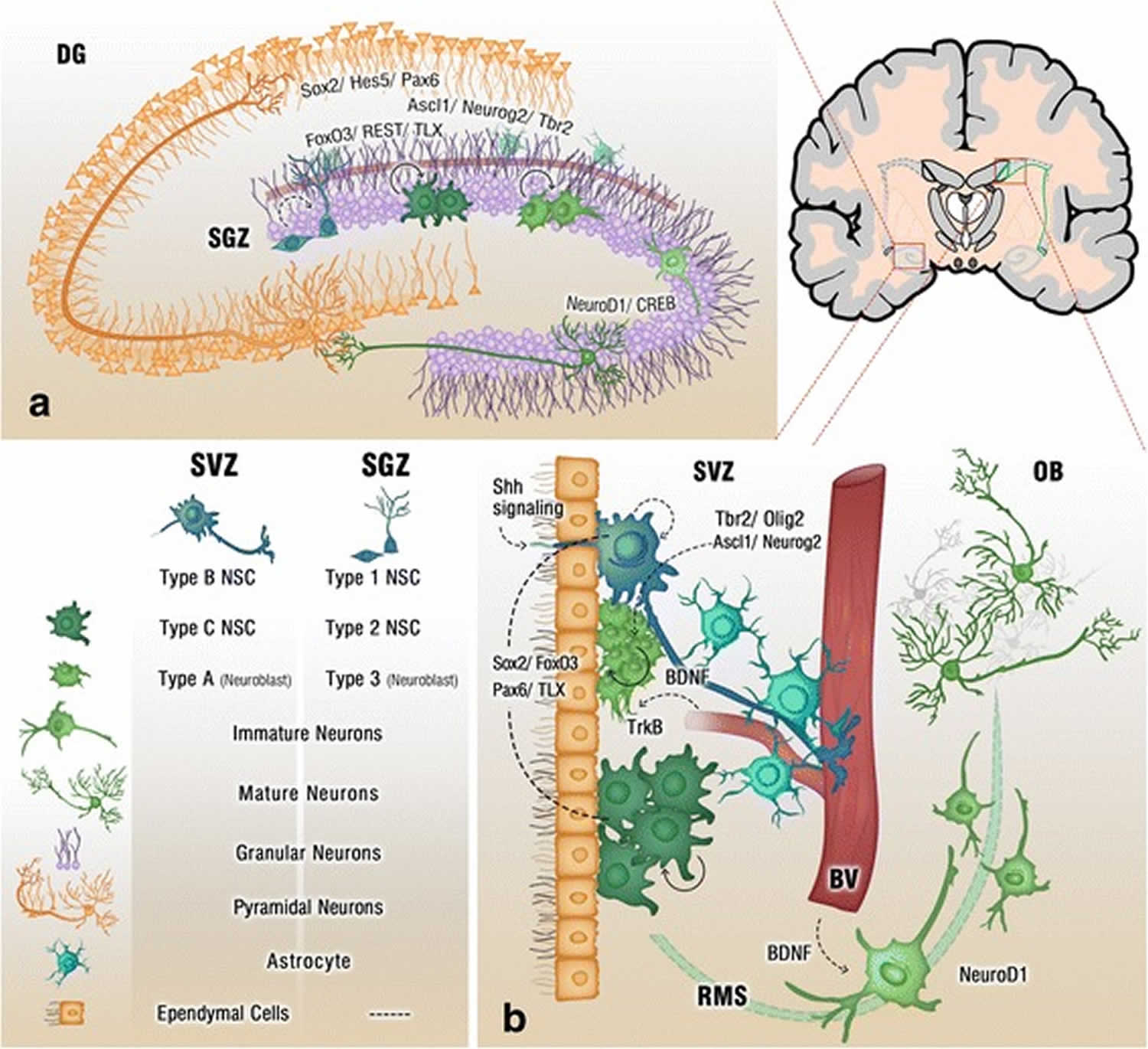
Footnote: Cross section of the adult brain showing regions of subgranular zone (SGZ) and subventricular zone (SVZ), where neurogenesis takes place. The schematic illustrates neurogenesis involving neural stem cells (NSCs) development into mature neurons and the neurogenic niches of Blood Vessels (BV), astrocytes and cilia, as well as transcription programs in the SGZ (a) and SVZ (b). Neuronal migration from the SVZ to the olfactory bulb (OB) via the rostral migratory stream (RMS) is also shown in (b)
[Source 9 ]Figure 3. Neurogenesis brain regions
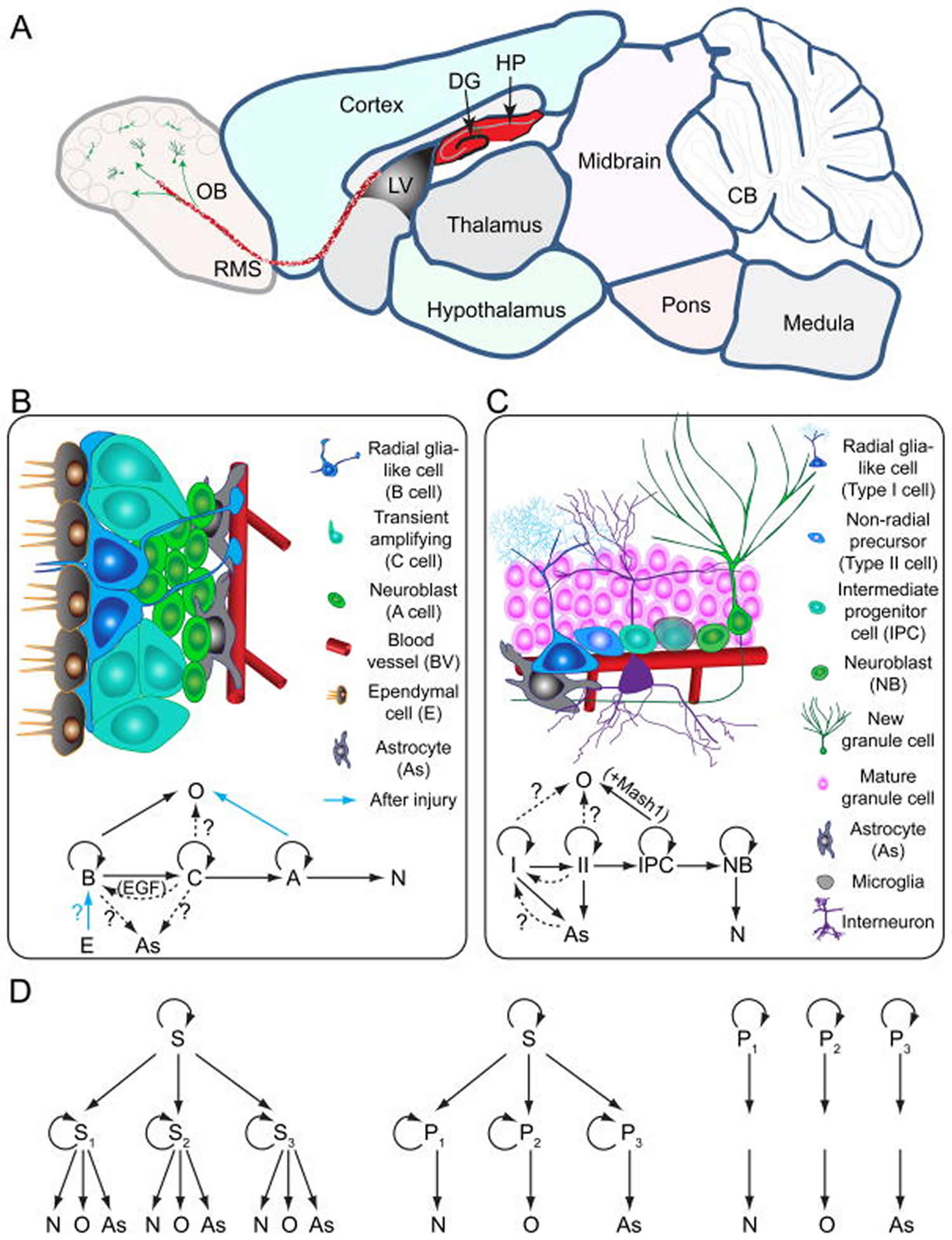
Footnotes:
Models of neural stem cells and lineage relationship in the adult dentate gyrus and subventricular zone
(A) A sagittal section view of an adult rodent brain highlighting the two restricted regions that exhibit active adult neurogenesis: dentate gyrus (DG) in the hippocampal formation (HP); the lateral ventricle (LV) to the rostral migratory stream (RMS) to the olfactory bulb (OB).
(B) A schematic illustration of the neural stem cell niche in the subventricular zone (SVZ) and a model of potential lineage relationship under basal (solid arrows) and injury conditions (blue arrows). N: immature neurons
(C) A schematic illustration of the neural stem cell niche in the subgranular zone (SGZ) in the dentate gyrus and a model of potential lineage relationship.
(D) Three lineage models of neural precursors in the adult mammalian brain. In the first model (left), adult neural stem cells (S1,2,3…) generated from primitive neural stem cells (S) are intrinsically diverse, exhibiting vastly different developmental potential depending on their regions of distribution and developmental origins. In the second model (middle), adult neural stem cells (S) are relatively homogenous and give rise to a heterogeneous population of lineage-restricted progenitors (P1,2,3…). In the third model (right), only lineage-restricted neural progenitors (P1,2,3…) are present in the adult brain; self-renewal and multi-lineage differentiation represent a collective property of a mixture of different lineage-restricted neural progenitors. N: neurons; O: oligodendrocytes; As: astrocytes.
[Source 10 ]Most neurons in the adult central nervous system are terminally differentiated and cannot be replaced when they die. However, research over the past decade has shown that small populations of new neurons are generated in the mature olfactory bulb and hippocampus. The regions identified so far in the rodent adult brain are the subventricular zone and the subgranular zone of the dentate gyrus in the hippocampus 11. Another mechanism of neuroplasticity is the modification of mature neuronal morphology, involving axonal and dendritic arborization and pruning, an increase in spine density, and synaptogenesis. At a functional level, long-term potentiation is the main mechanism mediating neuroplasticity. The transcriptional regulation of genes involved in neuroplasticity by epigenetic mechanisms also contributes to synaptic plasticity. Altogether, these processes mediated the dynamic and adaptive changes in synaptic strength 12.
Such neuroplasticity can be viewed as adaptive when associated with a gain in function 13 or as maladaptive when associated with negative consequences such as loss of function or increased injury, points illustrated by animal models and some human studies 14. Also, adaptive neuroplasticity should be distinguished from compensatory behaviors, which are behaviors that arise from mechanisms different from those operative in the distributed neural networks that typically support behavior prior to disease onset 15.
During the past few years, neuronal plasticity, and in particular adult neurogenesis, has been implicated in the beneficial effects of antidepressant drugs and electroconvulsive shock treatment. However, it has remained unclear how plasticity and neurogenesis impact mood and anxiety-related behaviors.
Although gross connectivity develops through genetically governed guidance, fine-tuning takes place through experience and activity-dependent plasticity, where neurons and connections that actively participate in network function are selected for stabilization and strengthened, whereas inactive contacts are weakened or eliminated.
Neuronal plasticity does not only involve trophic processes such as neurogenesis and synaptogenesis, but also includes atrophic processes, such as the elimination of inactive neurons and neuronal contacts. Although it is often thought that loss of neurons or synapses is harmful, elimination of connections that do not mediate useful information is, in fact, necessary for the optimal signal-to-noise ratio within the nervous system. Indeed, most of the neurons and synapses formed during development are wiped out by adulthood. Therefore, plasticity in itself does not have any particular direction; it is the experience-dependent activity within the neuronal network that determines which of the connections are strengthened and maintained and which ones are eliminated. Therefore, plasticity is adaptive when it is guided by beneficial environmental stimuli, but it can also be maladaptive, if the guiding experiences are adverse.
Neuronal plasticity is heightened during critical periods of postnatal development, which allows an efficient experience-driven fine-tuning of developing networks. After the closure of critical periods, neuronal plasticity and changes in network structure are more restricted. However, recent data indicate that several drugs used for the treatment of neuropsychiatric disorders can directly influence the plasticity and reactivate a critical period-like plasticity in the adult brain, a process known as induced plasticity.
To be translated into neuronal structure and function, neuronal activity needs molecular mediators and neurotropic factors are prime candidates for mediators between neuronal activity and plasticity. In this review, we will first introduce the role of the neurotrophin family and especially on brain-derived neurotrophic factor (BDNF) as a mediator of plasticity and drug effects. We will then discuss the role of neuronal plasticity in the mechanisms of action of drugs acting on the brain. Finally, we will review recent evidence that developmental-like plasticity, induced plasticity, can be activated in the adult brain and argue that iPlastic drugs should be combined with training, rehabilitation or psychotherapy to facilitate treatment outcome. For the role of other neurotrophic factors in neuronal plasticity, especially the family members of the glial cell line-derived neurotrophic factor (GDNF), fibroblast growth factor (FGF) and insulin-like growth factor (IGF),
Neuroplasticity in depression
Depression is a major health problem with a high prevalence and a heavy socioeconomic burden in western societies. It is associated with atrophy and impaired functioning of cortico-limbic regions involved in mood and emotion regulation 11. Despite recent advances in neuroscience research, the neurobiological mechanisms underlying the pathophysiology of depression remain poorly understood 11. It has been suggested that alterations in neurotrophins underlie impaired neuroplasticity, which may be causally related to the development and course of depression. Accordingly, mounting evidence suggests that antidepressant treatment may exert its beneficial effects by enhancing trophic signaling on neuronal and synaptic plasticity. Interestingly, currently prescribed antidepressants have been shown to increase neuroplasticity when exerting their therapeutic effects 16. However, current antidepressants still show a delayed onset of action, as well as lack of efficacy. Hence, a deeper understanding of the molecular and cellular mechanisms involved in the pathophysiology of depression, as well as in the action of antidepressants, might provide further insight to drive the development of novel fast-acting and more effective therapies.
Neuroplasticity changes in major depressive disorder
While neuroplasticity in rodents has been well documented during the last decades, the study of neuroplasticity in the human brain largely remains indirect, mostly because of methodological limitations as well as ethical constraints.
However, some alternative methods such as the assessment of the hippocampal dentate gyrus volume by MRI, magnetic resonance spectroscopy 17, and more recently the use of 14C in genomic DNA labeling 18 have provided some valuable information on human neuroplasticity. Most of the post-mortem studies have now evidenced that in adult humans, new neurons continued to be generated with a modest decline during aging 18. Although recently questioned by Sorrells et al. (2018) who suggested that human neuroplasticity could differ from other species, it has been nevertheless confirmed that in human dentate gyrus, about 700 new neurons were generated per day whatever the age 19.
Beside neurogenesis, it is also rather well-established that long-term potentiation can be induced in the human CNS with similar molecular mechanisms than those observed in rodent models 20. Human long-term potentiation has first been demonstrated in isolated cortical tissue obtained from patients undergoing surgery, and recent studies have shown that long-term potentiation-like phenomena can be obtained in the human cortex as well by using repetitive presentations of sensory stimuli while recording event-related potentials from the scalp 21.
In major depression, neuroimaging and post-mortem studies in humans indicate that structural changes are often observed in the course of this pathology. Structural MRI studies have revealed reduced hippocampal volume in individuals during a depressive episode in comparison to patients in remission 22, while increased hippocampal dendritic atrophy and cell death as well as reduced long-term potentiation and brain-derived neurotrophic factor (BDNF) expression have also been reported 23. While MRI studies were rather consistent with the observation of a reduced dentate gyrus size in patients with depression or anxiety disorders 24, post-mortem studies in depressed patients showed important disparities regarding neurogenesis, showing either no difference 25 or a decrease in the number of dentate gyrus progenitor cells 26. Although these findings make it tempting to speculate on reduced levels of neurogenesis in major depression, further investigations making use of more specific techniques are needed to better understand the dynamics of adult neurogenesis in major depression. However, other attempts to measure human brain plasticity in the course of major depression have demonstrated functional alterations such as those observed at long-term potentiation level. For example, visual-evoked potential amplitudes in the visual pathway were, compared to matched control subjects, decreased in patients with depression 27 and also in bipolar disorder patients 28.
In preclinical depression-like models as well, consistent data have reported a decrease in the proliferation and survival of hippocampal neurons when the hypothalamic pituitary adrenal axis was dysregulated. Hence, using proliferation and survival cell markers such as BrdU, Ki-67, or DCX, impaired neurogenesis was observed in rodent models of depressive-like behavior. These models included transgenic mice 29, corticosterone-induced mouse model of depression/anxiety 30, or rats subjected to chronic mild stress 31. Beside neurogenesis, numerous studies have also reported that synaptic plasticity was also greatly impaired in stress models of depression 23. Severe stress has also been shown to inhibit long-term potentiation 32 and enhance long-term depression 33 in the hippocampus and in prefrontal pyramidal cells 34.
Altogether, these data strongly suggest synaptic plasticity is strongly affected in major depression, at both structural and functional levels, and that these alterations are similar to those evidenced in rodent studies.
Effects of antidepressant therapies on neuroplasticity
Electroconvulsive therapy
Recent clinical studies showed that electroconvulsive therapy (ECT) promoted structural plasticity including increased volume and morphometric changes in the hippocampus and the amygdala along with improved clinical responses, especially in patients with a smaller hippocampal volume 35. ECT may also reduce cortical excitability and, thereby reverse increases in the excitability of cerebral cortex, during treatment-resistant depression 36. In addition, low-frequency trains of transcranial magnetic stimulation (rTMS) applied on several regions of the brain to induce long-term potentiation- and long-term depression-like changes in neuronal activity produced identical effects to those achieved with ECT in depressive patients 37.
Similar results have been demonstrated in rodent models of depression 38 and non-human primates 39.
Exercise
Exercise has also shown beneficial effects on plasticity. Although it has been proven that exercise in major depression patients reduced depressive symptoms 40, neuroplasticity per se has not yet been monitored in patients in these conditions, due to the limitations mentioned in section “Neuroplasticity changes in major depression.” However, a meta-analysis has suggested that acute aerobic, but not strength exercise, increases basal peripheral brain-derived neurotrophic factor (BDNF) concentrations, although this effect was only transient 41. A recent study showed an increase in synchronous neuronal responses during a task that requires an upregulation of cognitive control when exercise was combined with meditation 42. In preclinical studies, swimming showed antidepressant-like effects on anhedonia in stressed rodents along with a normalization of a stress-induced brain-derived neurotrophic factor (BDNF) mRNA expression decrease 43. A 21-day exercise regimen in rats transiently increased long-term potentiation in the dentate gyrus of the hippocampus 44, although recent data suggested that, if motor activity could exert positive effects on cognitive processes, it was under very controlled conditions 45 and that acute swim stress could also led to long-term depression 46.
Antidepressant treatments
Few studies have directly addressed the effects of antidepressant therapy on neuroplasticity in the human brain. Imaging studies have provided data showing that, e.g., in major depression patients taking medication for 3 years, the left hippocampus increased in volume compared to the beginning of the study 47. Interestingly, the volume of the left hippocampus has also been shown to enlarge under lithium treatment in elderly bipolar patients, probably through a neuroprotective effect 48.
Regarding neurogenesis per se, the analysis of post-mortem brains of major depression patients treated with nortriptyline and clomipramine showed an increase of neural progenitor cells and dividing cells in the dentate gyrus, as compared to healthy controls 49. In addition, the same group reported that both the fewer mature granule neurons and the smaller dentate gyrus and granule cell layer volume found in post-mortem brain tissue of depressive patients were reversed by antidepressant treatment 50. Interestingly, a meta-analysis concluded that depressed patients with a decreased hippocampal volume showed lower response/remission rates after antidepressant treatment 51.
In a rodent model of depression, both reduced hippocampal proliferation and increased cell death were reversed by chronic administration of antidepressants 52. Adult hippocampal neurogenesis was proposed to be required for the therapeutic action of antidepressants 53. Accordingly, chronic treatment with a SSRI such as fluoxetine increased hippocampal neurogenesis through the generation of newborn cells in the dentate gyrus 54. In addition, serotonin and noradrenaline reuptake inhibitor (SNRI) antidepressants, like SSRIs, also modulate neurogenesis and plasticity. Neurogenesis in the dentate gyrus of the hippocampus was increased following chronic venlafaxine administration to rats 55. Similarly, chronic venlafaxine treatment proved to be efficient in preventing the deleterious effects of restraint stress on hippocampal neurogenesis and BDNF protein expression 56. Likewise, tricyclic antidepressants (TCAs) have also been shown to modulate hippocampal neurogenesis. Clomipramine was able to counteract the stress-induced inhibition of proliferation in the hippocampus 57. Chronic imipramine and desipramine treatment increased cell proliferation in the subgranular zone 58.
In addition to their effects on neurogenesis, evidence has also been generated that antidepressants can regulate other types of plasticity. Treatments with classical antidepressants are indeed able to modulate long-term potentiation within the hippocampus 59 and chronic fluoxetine administration has also been demonstrated to increase synaptic plasticity in naive rats 60.
Overall, these data showed that antidepressants have a rather beneficial effect on neuroplasticity. However, they still show lack of efficacy in some depressed patients. Indeed, major depression is a complex disorder of which the etiology remains unclear and that cannot be simplified by neuroplasticity dysfunctions as the only targetable factor.
Summary
Growth factors and associated neurotrophic signaling play an essential role in the development and maintenance of the central nervous system 61. Accordingly, there is growing evidence that abnormal trophic support in cortico-limbic regions regulating mood and emotions may take part in the pathophysiology of depression 62. In addition, clean-cut evidence shows that antidepressants require neuroplasticity pathways to rescue the observed deficits in neuronal and synaptic plasticity often associated with mood disorders 63. However, current knowledge makes it difficult to conclude whether neuroplasticity and neurogenesis represent a cause, a consequence (or both), or an epiphenomenon of the pathological processes associated with depression. Hence, future research should focus on elucidating the exact involvement of neurotrophic signaling in the onset and course of major depression. Furthermore, mounting evidence seems to indicate that neurogenesis might not be required for the therapeutic action of antidepressants 64. In line with this hypothesis, the usage of N-methyl-d-aspartate (NMDA) receptor antagonists showed that acute induction of neuroplasticity pathways, e.g., increased brain-derived neurotrophic factor (BDNF) signaling, was sufficient to produce a robust and prolonged antidepressant effect 65. Hence, the rapid enhancement of hippocampal neuroplasticity—involving dendritic growth, spine density, and synaptic transmission—represents an original strategy to circumvent the delayed efficacy of current antidepressant drugs. Finally, the use of drugs that specifically target neurotrophic signaling should provide more insights in the role of neuroplasticity pathways in antidepressant responses. As growth factors and neurotrophins generally display a poor blood-brain barrier penetration and a short half-life in plasma 66, identification of small non-peptidic neurotrophin mimetics, albeit challenging on its own, may represent an interesting target for the development of a new class of therapeutic agents for mood-related disorders.
How does neuroplasticity work?
Neurotrophins and other growth factors
Growth factors have been proven to be strongly involved in regulating neuroplasticity by impacting upon almost all of the neuroplasticity processes, including neuronal survival, differentiation, and proliferation 67.
Brain-derived neurotrophic factor (BDNF) and neuroplasticity
Brain-derived neurotrophic factor (BDNF) is a neurotrophin involved in the growth, differentiation, and survival of neurons and has also been shown to represent an important factor in the regulation of neurogenesis and synaptic plasticity 67. It exerts its neurotrophic effects by activating the tropomyosin-related kinase receptor B (TrkB) 68. It also binds, albeit with a lower affinity, to the p75NTR receptor, which is generally known to promote proteolysis and apoptosis 69. BDNF is abundantly expressed in the mammalian brain, with the highest concentrations found in the hippocampus and cortex 70.
BDNF and neuroplasticity
Several in vitro studies have been conducted in order to unravel the effects of BDNF on neuroplasticity. Indeed, when PC12 cells transfected with TrkB were stimulated with BDNF for 48 h, neurite outgrowth was increased compared to the non-treated cells 71. Interestingly, in growth medium B27-deprived primary hippocampal cells, BDNF stimulation was able to promote dendritic outgrowth and spine formation 72 and this neuroplastic effect is probably achieved through intracellular signaling cascades 73. As such, BNDF has been shown to increase the activity of the mitogen-activated protein kinase (MAPK) cascade promoting survival in neural cell cultures 74.
In vivo evidences also support the critical role of BDNF in plasticity. In particular, mutation studies have demonstrated the role of this neurotrophin in structural and synaptic plasticity. Total BDNF deficiency is lethal and most of the mice lacking BDNF die during the second postnatal week 75. However, heterozygous BDNF knockout mice survive into adulthood and the use of these mice evidenced that BDNF was required for several forms of long-term potentiation 76. This was in agreement with data that showed that BDNF infusion in the rat hippocampus induced long-term potentiation and triggered synaptic strengthening 77. At morphological level, these mice display a specific hippocampal volume reduction 78 similarly to what was found in heterozygous TrkB mice 79 but in contrast to p75NTR-deficient mice 80, suggesting a link between hippocampal volume and BDNF-mediated TrkB signaling 81. In addition, in the hippocampus, BDNF increases the total length, but not the branching, of apical dendrites within the CA1 stratum radiatum, without affecting basal dendrites in the stratum oriens 82. However, in mutant mice in which bdnf excision mediated by Cre recombinase led to an almost total disappearance of BDNF in the brain, the volume of the hippocampus was mostly unchanged except for small changes in dendritic branches in restricted segments and signs of a modest delay in spine maturation 83. Although these data and other 84 indicate a temperate effect of BDNF in the maintenance of the cellular architecture of the adult brain, most of the studies evidenced its central role in neuronal plasticity.
Fibroblast growth factor in neuroplasticity
The fibroblast growth factor (FGF) family has been described as a major player in proliferation and maturation of neurons in the subgranular zone (SGZ) and subventricular zone (SVZ) of the hippocampal dentate gyrus 85. The FGF family is composed of 18 ligands and 4 subtypes of receptors. FGF1 is expressed mostly in neurons while FGF2 is expressed in both neurons and glial cells. FGF1 and FGF2 are the most studied ligands of this family and have been shown to be dysregulated in mood disorders 86. They can bind all four receptor subtypes in order to activate the phospholipase C-γ (PLC-γ1 MAPK and AKT pathways 86. In addition, FGF1 and FGF2 were evidenced to play a critical role in the regulation of synaptic plasticity 87.
FGF in neuroplasticity
Intracerebroventricular (ICV) injection of FGF2 induced neurogenesis in both the subgranular zone (SGZ) and subventricular zone (SVZ) 88. Mice with a complete loss-of-function FGF2 allele showed a significant decrease in newly generated neurons but no reduction in proliferating cells 89. The additional increase in cell death in the hippocampus indicated a faulty neurogenesis following FGF2 knockout 89. Conditional knockout experiments with FGF receptor 1 (FGFR1)-null mice show defective long-term potentiation and neurogenesis 90, suggesting that the FGF2-FGFR1 interaction might represent an important mediator of neurogenesis.
Vascular endothelial growth factor in neuroplasticity
Vascular endothelial growth factor (VEGF) is primarily known for its induction of angiogenesis and modulation of vascular permeability during embryogenesis and growth, as well as pathological events such as in tumorigenesis. It can bind to different receptors: receptor tyrosine kinases (VEGFR) 1 and 2 with a higher affinity for VEGFR1. In addition, mounting evidence suggests that VEGF can be considered as a potent neurotrophic factor, inducing neurogenesis, neuronal survival and proliferation, glia survival, and glia migration 91.
VEGF in neuroplasticity
Interestingly, experimental studies showed that VEGF displays robust neuroprotective effects in cell models of ischemia and hypoxia 92 as well as a positive effect on neuronal growth, maturation, and proliferation under normoxic conditions 93. A role in the development of dendrites and axons has also been described for VEGF 94. Moreover, intracerebroventricular administration of VEGF increased neuroprotection and neurogenesis in the adult rat brain after ischemia 95. More specifically, intracerebroventricular administration of VEGF increased neurogenesis in both the subgranular zone (SGZ) and subventricular zone (SVZ) of the dentate gyrus with enhanced proliferation of neurons, astroglia, and endothelial cells 96, while VEGF-B knockout mice showed impaired neurogenesis 97. Hence, the overexpression of VEGF in the hippocampus using an adeno-associated viral vector in rats resulted in increased neurogenesis and was associated with improved learning and memory 98. Interestingly, VEGF can promote neurogenesis by stimulating endothelial cells in order to release other neurotrophic factors 99. In addition, ependymal cells can synthesize VEGF leading to a stimulation of the VEGFR2 and inducing proliferation of neuronal precursors and enhanced formation of new neurons in the hippocampus 100.
Glial cell line-derived neurotrophic factor in neuroplasticity
Glial cell line-derived neurotrophic factor (GDNF) was first discovered in a glial cell line but is expressed in many brain regions. It is a member of the transforming growth factor β (TGF-β) superfamily and is important for neuronal survival especially for dopaminergic and serotonergic neurons. It binds to the GDNF-family receptors α1 (GFRα1) activating tyrosine kinase signaling 101.
Experimental studies in animal models evidenced a neuroprotective role of GDNF, and intracerebroventricular infusion of GDNF increased progenitor cell proliferation in the dentate gyrus 102 and subventricular zone (SVZ) 103. Similarly, infusion of GDNF in the striatum of rats increased progenitor cell proliferation in the hippocampus and substantia nigra 104. Moreover, GDNF induced differentiation of DG-derived neural precursors into astrocytes in vitro 105. Further, the use of an adeno-associated viral vector that induced overexpression of GDNF in the rat cortex provided neuroprotection against ischemia-induced injury 106.
Insulin-like growth factor 1 (IGF-1) in neuroplasticity
Insulin-like growth factor-1 (IGF-1) and its receptor IGF-1R are found in many tissues including the brain 107. It influences growth and differentiation processes 108. IGF-1 has been designated as a potential therapeutic target for neurodegenerative diseases such as major depression 109.
Insulin-like growth factor-1 (IGF-1) induced differentiation of neuronal precursors 110 and proved to be neuroprotective in cerebellar granule neurons in vitro 111. IGF-1 knockout mice showed a decrease in total brain size and subgranular zone (SGZ) volume, further supporting the importance of IGF-1 in neurodevelopment 112. Developmental research in mice has revealed the importance of IGF-1 in hippocampal neurogenesis and synaptogenesis 113. Furthermore, intracerebroventricular infusion of IGF-1 ameliorated the age-related decline in hippocampal neurogenesis 114, while peripheral administration of this growth factor could induce hippocampal neurogenesis in rats 115.
Nerve growth factor
Nerve growth factor (NGF) is a growth factor first described as a neurite outgrowth factor 116. Later, nerve growth factor (NGF) proved to be involved in neuronal repair and survival 117. Nerve growth factor (NGF) has been implicated in proliferation and differentiation of neuronal stem cells 118 and more recently in neurogenesis in the striatum 119 and in regulating hippocampal plasticity 120.
Examples of neuroplasticity in the clinical context
Injury: stroke, trauma and spinal cord injury
Among syndromes of human CNS (central nervous system) injury, an area in which neuroplasticity has been extensively studied, is motor recovery after stroke. Motor deficits are present in a majority of patients with stroke 121, and the degree of motor recovery can vastly influence whether or not the stroke proves disabling. This is perennially a problem of major proportions—by some estimates, 55–75% of stroke survivors still have functional limitations and reduced quality of life months after the infarct 122.
Studies of motor recovery after stroke illustrate the principle that many forms of neuroplasticity can be ongoing in parallel. Injury to a region of the motor network can result in spontaneous intra-hemispheric changes, such as in representational maps, e.g. the hand area can shift dorsally to invade the shoulder region 123 or face region 124. At the same time, the inter-hemispheric balance can shift such that the uninjured hemisphere has supranormal activity in relation to movement 125. Focal injury results in diffuse adaptive changes, including changes in the connections between network nodes 126. The molecular basis of such spontaneous adaptive changes includes a host of growth-related processes that evolve over time 127. Similar results have been described in post-stroke recovery from non-motor deficits such as neglect and language, though these rely on networks
that are differently distributed as compared with the motor system 128. Cognitive recovery after stroke has been less studied and might be more influenced by diaschisis, i.e. the remote depression of function in non-injured tissue 129. Restorative and rehabilitation post-stroke therapies produce a range of brain events that in many instances are similar to those arising during spontaneous recovery from stroke, such as a return to a normal degree of laterality. Other changes can be unique to therapy, such as new projections from neurons on the undamaged side of the brain to denervated areas of the midbrain and spinal cord 130. In some cases, the behavioral significance of such changes is understood—a shift in inter-hemispheric balance towards the uninjured hemisphere is the sign of a distressed system. In other cases, the exact behavioral significance of a pattern of post-stroke plasticity remains unclear, such as the direction of somatotopic map shift after focal cortical injury. As such, biomarkers of adaptive plasticity 131, such as those derived from transcranial magnetic stimulation and functional MRI, often have to be narrowly defined in relation to the targeted outcome. For example, a measure of brain plasticity after stroke might have very different significance at different time points or after different therapies. Other factors suggest the need for cautious interpretation of brain plasticity measures after stroke. In some cases, a stroke can affect brain function in ways outside of study hypotheses. For example, in a patient with stroke, functional MRI of the language system might be affected by reduced attention; or transcranial magnetic stimulation, by abnormal resting tone or by excess force execution. Interpretation of functional neuroimaging data always requires great care—after stroke this is all the more true, as multiple brain systems and behaviours are often affected 132.
Similar forms of adaptive neuroplasticity have been described following other forms of acute CNS injury such as traumatic brain injury 133 and spinal cord injury 134. Interestingly, after spinal cord injury, treatment-induced brain plasticity can be measured in the absence of behavioral change 135. Such data provide insight into the status of motor cortex function and might be helpful for stratifying patients for treatments, such as stem cell injections aimed at restoring voluntary motor control in patients with dense plegia after spinal cord injury. The similarity of plasticity mechanisms across divergent forms of CNS injury suggests that plasticity, as with development, uses a limited repertoire of events across numerous contexts.
Another principle is that not all plasticity has a positive impact on clinical status—in some cases, plasticity might have negative consequences. For example, new onset epilepsy is a common complication of cerebral trauma, often arising months to years after the insult. This delayed onset suggests that progressive changes in the brain, such as axonal sprouting and the formation of new connections, produce alterations in neuronal signalling and disinhibition that result in the induction of seizures 136. Other examples suggestive of maladaptive plasticity include chronic pain and allodynia following injury to a limb (such as amputation) or to CNS (dorsal spinal cord or thalamus), dystonia after various CNS injuries and autonomic dysreflexia after spinal cord injury 137. Thus, recovery from trauma or disease may reflect both adaptive and maladaptive neuroplasticity, which can occur simultaneously.
How to increase neuroplasticity
A number of promising interventions targeted towards promoting neuroplasticity are highlighted below. The examples are meant to be illustrative, rather than exhaustive. In many cases, these interventions represent application of neuroscience knowledge to rehabilitation techniques. Examples include appreciation of learning theory, Hebbian principles, task-specific training, social influences, mechanisms of verbal encoding and the interplay across brain modalities (such as influence of deafferentation on motor function). Such interventions are increasingly being applied mindful of overarching principles of neuroplasticity 138. For example, plasticity after injury is often experience dependent. Thus, interventions that aim to promote plasticity can be expected to have maximum impact when coupled with optimal training and experience. Note that measuring the impact of such experiences on behavioral outcomes might require use of domain-specific measures 139. For example, recovery of language in a patient with post-stroke aphasia is influenced by optimal language therapy 140; this language recovery is better detected with a scale that is sensitive to the language domain as compared with a global scale of overall status. Also, plasticity can be time-sensitive, occur with considerable specificity, vary with the nature of the environment and be strongly influenced by the extent of concomitant training. Salience, motivation and attention can be critical modulators of plasticity 138. Skills training illustrates a direct example of harnessing neuroplasticity to achieve clinical gains; training on selected skills has been found to improve behavioral outcomes in parallel with increased brain plasticity, in the setting of numerous forms of CNS disease. The extent to which outcomes can be improved on the backbone of such training and plasticity depends on availability of sufficient residual neural resources, regardless of the type or duration of the neurological insult 141. A major challenge, as discussed below, is to match the right patients with the right training approach.
The pursuit of neuroplasticity-based interventions benefits from strong collaborations between basic and clinical researchers 142, from preclinical investigations to clinical trials. For example, clinical scientists can better understand the limitations and assumptions in the animal models; basic researchers, in turn, can return to the lab to ask pivotal questions upon informed review of early phase human studies. Such an approach stands to provide the best understanding of plasticity mechanisms, from genes to molecules to cells to networks to behavior.
Non-invasive brain stimulation
Several forms of non-invasive brain stimulation have been examined as a means to change brain function and thereby promote neuroplasticity 143. Chief
among these is transcranial magnetic stimulation. Introduced in the mid-1980s, transcranial magnetic stimulation uses an extracranial magnetic coil to induce current in the cerebral cortex 144. Although transcranial magnetic stimulation activates a mixed population of inhibitory and excitatory cortical interneurons, in general, continuous trains of low frequency (51 Hz) repetitive transcranial magnetic stimulation or theta burst transcranial magnetic stimulation lead to suppression of cortical excitability in healthy subjects, while intermittent, bursting high frequency (41 Hz) trains of repetitive transcranial magnetic stimulation or intermittent theta burst transcranial magnetic stimulation lead to facilitation 144. Transcranial magnetic stimulation can be applied specifically and selectively to defined cortical regions, particularly when guided by neuroimaging and physiological measures 145. Transcranial direct current stimulation uses two scalp electrodes to induce low-amplitude direct currents strong enough to penetrate the brain and modify membrane potentials, thereby influencing neuronal excitability, but without triggering the depolarization of neurons 144. Both techniques can produce effects that are ‘offline’, i.e. that endure beyond the period of stimulation 146. The optimal stimulation paradigms and the best means of behavioral reinforcement require further study.
Several strategies build on the basic capacity of stimulation to modulate brain activity. Combination therapies, such as adding concomitant pharmacological or peripheral nerve stimulation, have the potential to further drive Hebbian plasticity 147 and are under study. Also, combination with imagery or behavioral intervention has the potential to increase the efficacy of neuromodulation 148. Another set of approaches builds on the model that, following stroke, cortical excitability is decreased in the ipsilesional hemisphere as a result of increased transcallosal inhibition from the contralesional hemisphere and increased in the contralesional hemisphere 149. Building on this model, enhancing excitability of the affected hemisphere using high-frequency repetitive transcranial magnetic stimulation or suppressing the contralesional hemisphere via low-frequency repetitive transcranial magnetic stimulation or cathodal transcranial direct current stimulation appears in initial studies to promote gains in motor function, at least among patients with mild to moderate impairment 150. Promising initial results have also been reported with this approach in the treatment of other neurological domains, such as aphasia 151.
The ability of repetitive transcranial magnetic stimulation to induce electrical stimulation in cortex and the hypothesis that it might produce anti-depressant effects similar to electroconvulsive therapy has resulted in several studies on its efficacy for depression. Daily prefrontal transcranial magnetic stimulation for several weeks has been shown to have significant antidepressant effects, but additional work is needed to optimize protocols and establish and improve effect sizes 152. Importantly, repetitive transcranial magnetic stimulation effects have been shown to spread to distal anatomically connected structures across specific networks, including deeper striatal and limbic structures 153. Thus, cortical stimulation might induce plastic changes in cortex directly or indirectly by modifying activity in basal ganglia networks. A key principle in neuroplasticity is the value of coupling a plasticity-promoting intervention with behavioral reinforcement; further efforts in this regard might maximize the impact of repetitive transcranial magnetic stimulation on depression.
The ability of repetitive transcranial magnetic stimulation to induce changes in cortical function has also been examined in schizophrenia. The left temporoparietal cortex is one of several regions whose overactivity is associated with the perception of external voices in hallucinating patients. Low-frequency stimulation of temporoparietal cortex has been used experimentally to inhibit cortical excitability and thereby quell severe, treatment-resistant auditory hallucinations with some success 154. However, these studies are characterized by small sample sizes and some methodological inconsistencies, making it difficult—at this point in time—to compare study findings and draw definitive conclusions about the precise mechanisms and utility of low-frequency repetitive transcranial magnetic stimulation for the treatment of hallucinations.
Deep brain stimulation
Like transcranial magnetic stimulation, deep brain stimulation uses electrical stimulation to induce neuroplasticity and produce behavioral changes, although with deep brain stimulation, electrical current is provided through implanted electrodes. Stimulation parameters such as frequency, intensity and pulse width are programmable and can be optimized 155. Two generally hypothesized mechanisms of action are that deep brain stimulation creates a functional lesion via inhibition within the stimulated region and, alternatively, that deep brain stimulation activates the neuronal network connected to the stimulated region, leading to modulation of pathological network activity 156. The former mechanism is more consistent with the immediate effects of some applications of deep brain stimulation (e.g. effects on motor function in Parkinson’s disease), while the latter may be more consistent with deep brain stimulation-induced gradual effects (e.g. circuit retraining) as seen in neuropsychiatric disorders.
Following increased use of deep brain stimulation in Parkinson’s disease and reports of its effects on emotion, interest in its application to severe, treatment-resistant mental disorders has grown. A challenge in using deep brain stimulation for disorders of mood, thought and behavior is the identification of appropriate and optimal stimulation sites. Although a primate model (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, also known as MPTP) has been useful for defining a stimulation site in Parkinson’s disorder, animal models have generally been of limited value in psychiatry. Human neuroimaging and prior lesioning studies have thus been very useful in identifying putative deep brain stimulation targets that may be appropriate for treating psychiatric disorders 157. In mood and anxiety disorders, human neuroimaging studies have generally highlighted prefrontal cortex, subcallosal cingulate cortex (Brodmann area 25), hippocampus and amygdala as dysregulated. Preliminary studies of deep brain stimulation targeting the subcallosal cortex (Brodmann area 25) and its adjacent white matter tracts have shown promise in treating depression, suggesting that disruption of the pathological limbic-cortical circuit may ameliorate treatment-resistant depression 158. Similar response rates have also been shown with stimulation of both the nucleus accumbens and anterior limb of the internal capsule 159, findings that also require further study.
Deep brain stimulation is also showing promise as a potential treatment for refractory obsessive-compulsive disorder, thought to involve overactivity of cortico-striatal-limbic circuits. Deep brain stimulation produced substantial behavioral improvements in at least half of the 60 subjects with obsessive–compulsive disorder studied over 10 years 155. The most common target in obsessive–compulsive disorder has been the internal capsule 160 extending into the ventral striatum, based on positive experiences with gamma capsulotomy and functional neuroimaging findings. Use in the related Tourette’s syndrome is more limited, but has targeted the medial thalamus and the globus pallidus internus, the latter based on studies in hyperkinetic states such as dystonia and dyskinesias of Parkinson’s disease 161.
As with many forms of plasticity, behavioral gains depend on continued therapeutic exposure. Thus, while improvement in depression and obsessive-compulsive disorder symptoms are progressive over months of chronic deep brain stimulation, they worsen progressively with termination of stimulation, although some metabolic changes linger after chronic deep brain stimulation 162. This supports the contention that deep brain stimulation can induce some long-term neuroplastic changes in neuropsychiatric disorders. Whether durable changes that survive ongoing stimulation are possible remains to be established.
Other forms of invasive brain stimulation are under study. Activity-dependent stimulation is a promising new modality for inducing plasticity. An electronic circuit that used neural activity recorded at one cortical site to trigger stimuli delivered at another site in freely behaving primates produced long-term changes in neural connections 163. The conditioning effects depended on the delay between the recorded action potentials and the stimuli, indicating a spike-timing dependent Hebbian plasticity. Negative findings have been described in a phase III trial of a different form of stimulation, epidural. Epidural motor cortex stimulation combined with physiotherapy was not found to be superior to physiotherapy alone for improvement of upper extremity motor deficits in patients with chronic stroke 164.
Neuropharmacology
The cellular and molecular events that underlie neuroplasticity occur on the backbone of specific neurochemical processes that are accessible and vulnerable to pharmacological interventions 165. Pharmacotherapies can increase neuroplasticity through molecular manipulation of numerous cellular and synaptic pathways, such as HDAC inhibitors, mTOR inhibitors and trkB agonists 166. They appear to be especially promising when used to augment circuit-specific training, i.e. to enhance experience-dependent neuroplasticity. One clear example is the ability of D-cycloserine to significantly augment the effects of exposure/extinction therapy for anxiety disorders by facilitating the activation of N-methyl-Daspartate glutamate receptors 167. A number of studies have provided mechanistic insights into these pharmacological effects. For example, Pariente et al. 168, using functional MRI, found that administration of fluoxetine to patients with stroke improved motor function in parallel with increased activation in the ipsilesional motor cortex.
Many of the above principles of plasticity extend to pharmacological approaches. For example, pharmacological targets can vary over time, and plasticity can change with experience and environment 138. In some cases, a drug has larger effects on plasticity and behavioral gains when paired with specific activities, such as with animal models of motor deficits after CNS injury 169, or with combined use of medication and cognitive behavioral psychotherapy in the treatment of human depression 170. It is important to note that the molecular mechanisms that support plasticity also have pharmacological vulnerabilities. Behavioral gains induced by plasticity-promoting pharmacological interventions can be lost, for example, with N-methyl-D-aspartate blockade or increased GABAergic tone. Indeed, many classes of drugs can retard neuroplasticity 171 and possibly reduce behavioural gains 172.
Neuroplasticity exercises
Following injury to the brain or spinal cord that induces motor deficits, many physical rehabilitation interventions have been reported to induce functional improvements. Constraint-induced movement therapy for the arm and hand 173, body weight-supported treadmill training 174, robotic devices 175, behavioral shaping, bilateral arm training 176 and task-oriented physical therapy 177 are all examples of interventions that have led to improved recovery following stroke. In some cases, functional neuroimaging studies have provided insights into mechanisms of treatment effects; for example, constraint-induced movement therapy of the upper extremity has been associated with an enlarged motor cortex map for the hand 178 as well as with bilateral increases in sensorimotor grey matter 179. However, the more complex training interventions, such as the use or robotic devices and treadmill locomotor training, generally have not improved outcomes more than conventional task related and strengthening therapies that also aim to optimize activity dependent plasticity. Likewise, among patients with incomplete spinal cord injury, training through robotic-assisted and body weight-supported techniques have improved walking only to a similar degree as standard over-ground training 180. Further research is needed to better understand how these therapies can be coordinated and optimized, especially across diverse patient groups with varied functional limitations. Practice strategies include increased repetition, sensory priming, visualization, modulation of attentional valence and reward, contextual interference, variable demand and intensity levels, blocked versus intermittent practice and various forms of feedback 181. Although these practice paradigms may enhance both skills and declarative learning in healthy subjects, their additive benefits for patients with impaired movement or cognition has been difficult to demonstrate. Note that physical rehabilitation training is not only a stand-alone therapy, but serves as an adjunct to other forms of therapy such as pharmacological and behavioural.
Aerobic exercise is a specific extension of activity-based therapies for promoting plasticity. Although the benefits of aerobic exercise in preventing or reversing cognitive and neural deterioration have yet to be fully investigated, substantial human and preclinical data support the utility of such exercise for promoting brain plasticity and improving CNS function in many conditions 182, including normal ageing and early dementia. Aerobic exercise is associated with increased neurogenesis and angiogenesis, as well as the production of neurotrophic molecules such as brain-derived neurotrophic factor and other growth factors involved in neuroprotection and the promotion of cell survival, neurite outgrowth and synaptic plasticity 183. In humans, neuroimaging studies have described a range of anatomic and functional correlates of such effects 184. Furthermore, these plasticity-promoting strategies are able to produce clinically significant changes. For example, aerobic exercise programmes lasting even just a few months significantly benefit cognitive functioning in both healthy ageing and early dementia, may be of benefit in schizophrenia 185, and have been shown to increase brain volume in a variety of regions 184 and to enhance brain network functioning 186.
Neuroplasticity training
Cognitive training can be thought of as a direct extension of physical therapy to the non-motor aspects of the human brain and so has been examined across a number of disease conditions. However, the complexities of the distributed neural systems that underlie human behavioural syndromes introduce unique challenges for the design of effective interventions. In depression and anxiety disorders, a long tradition of evidence-based cognitive-behaviour therapy is based on the principle of identifying and modifying top-down responses to maladaptive cognition, affect and behaviour 187. Evidence suggests that, as individuals learn to modify their cognitive representations and behavioural responses to distressing stimuli, widespread changes occur in frontal cognitive control systems and in limbic system activation 188.
New neuroscience-driven approaches to cognitive training are emerging and directly build on decades of animal research that have identified principles of harnessing plasticity mechanisms in the adult brain 189. For example, the cognitive deficits of schizophrenia—particularly in verbal learning and memory—are associated with illness severity and predict long-term functioning, but do not respond to currently available medications. Vinogradov et al. 190, guided by an understanding of the neurobiology of schizophrenia, performed a double-blind randomized controlled trial of intensive computerized cognitive training exercises that focus on the components of auditory and verbal processing that underlie verbal encoding. This intervention was associated with improved verbal memory in patients, as well as magnetoencephalographic evidence of increased amplitude of the M100 response to auditory stimuli, indicating plasticity in auditory cortex 191.
These findings require replication with a larger, preferably multi-site, study. Another cognitive-based, neuroscience-driven training approach aims to stimulate specific dysfunctional circuits, possibly in association with pharmacological intervention, in order to restore the integrity of frontostriatal circuitry in addiction 192.
Neuroimaging can contribute to cognitive training in a number of ways. Functional brain imaging can help to identify the neural correlates of various core mental processes such as interference control that can be targeted by cognitive training and that are relevant for a number of psychiatric disorders 193. Neuroimaging data can also be used as biomarkers, i.e. surrogate markers. A surrogate marker has been defined as ‘a laboratory measurement. . . used as a substitute for a clinically meaningful endpoint’ 194. For example, changes in functional MRI brain activation have been shown to correlate with functional gains in studies that employed cognitive training in the setting of schizophrenia 188, dyslexia 195 and depression 188, consistent with observations in studies that employed direct instructional approaches 196. Such imaging biomarkers might prove useful as predictors of clinical outcome, and a number of MRI and PET measures are under study in this regard 197. Neuroimaging can also provide molecular insights into treatment mechanism. For example, McNab et al. 198 found that cognitive training in healthy subjects was associated with changes in the density of cortical dopamine D1 receptors on PET scanning, a finding relevant to the treatment of children with attention deficit disorder 199. PET has also been used to describe changes in glucose metabolism associated with cognitive-behavioural or pharmacological treatment of depression 200. Though promising on a number of fronts, a number of issues remain to be addressed to maximize the impact of neuroimaging on cognitive training. Most imaging work has been performed on small samples, with differing approaches across labs, such as in relation to the underlying hypotheses of mechanisms of training-induced change, and further study is needed regarding the validity and reliability of neuroimaging data. Critically important is the question of whether changes in circuit strength demonstrated using neuroimaging are paralleled by meaningful behavioral changes.
The ultimate goal of cognitive training is to improve behavior by systematically harnessing neuroplasticity and driving adaptive changes in dysfunctional neural systems through carefully designed exercises. Note that cognitive training approaches have particularly broad potential, for example, as part of rehabilitation therapy of patients with focal brain injury such as stroke, where myriad cognitive syndromes exist with few treatment options, as well as in the treatment of numerous neuropsychiatric disorders such as depression and schizophrenia. Systems neuroscienceinformed cognitive training appears to be a promising treatment approach for a number of brain disorders. A key future direction for this field will be to maximize the extent to which cognitive training in one domain generalizes to others, and the extent to which such training has a meaningful impact on real-world functioning as well as the subjective experience of the individual 201.
Feedback using real-time functional magnetic resonance imaging
A central challenge to creating neuroplastic change is determining how to target plasticity to a particular brain system. Such targeting might be enabled by the ability to monitor changes in brain activation within localized brain regions in real time. Recent advances in neuroimaging and computing have enabled the development of such methods based on functional MRI-based measures of regional brain activation 202. These methods now offer the possibility of allowing individuals to view real-time measures of their own regional brain activation 203. Rapid feedback of regional activation level or of distributed patterns of brain activation might provide a novel means of instructing subjects on how to modulate their own brain function. Goals of such feedback include modulation of activity in specific brain regions in response to intrinsic or extrinsic cues, as well evaluation of the effects of various interventions.
Data suggest that subjects can indeed learn volitional control over a specific brain region. For example, healthy subjects can be taught to control brain activity within the anterior insula (Caria et al., 2007). In another study, both healthy subjects and patients with chronic pain were able to use real time functional MRI training to learn to control activation of brain regions involved in modulation of pain, which was associated with a concomitant decrease in pain perception 204. The long-term goal is to improve patient outcomes by modulating brain activity in those selected neural circuits that are most related to the target symptoms.
References- Harnessing neuroplasticity for clinical applications. Cramer SC, Sur M, Dobkin BH, O’Brien C, Sanger TD, Trojanowski JQ, Rumsey JM, Hicks R, Cameron J, Chen D, Chen WG, Cohen LG, deCharms C, Duffy CJ, Eden GF, Fetz EE, Filart R, Freund M, Grant SJ, Haber S, Kalivas PW, Kolb B, Kramer AF, Lynch M, Mayberg HS, McQuillen PS, Nitkin R, Pascual-Leone A, Reuter-Lorenz P, Schiff N, Sharma A, Shekim L, Stryker M, Sullivan EV, Vinogradov S. Brain. 2011 Jun; 134(Pt 6):1591-609. https://pdfs.semanticscholar.org/2ecc/abcfb437988c9e6d7472f58c54632772c742.pdf
- Castrén E, Antila H. Neuronal plasticity and neurotrophic factors in drug responses. Mol Psychiatry. 2017;22(8):1085–1095. doi:10.1038/mp.2017.61 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5510719
- Palatable Hyper-Caloric Foods Impact on Neuronal Plasticity. Morin Jean-Pascal, Rodríguez-Durán Luis F., Guzmán-Ramos Kioko, Perez-Cruz Claudia, Ferreira Guillaume, Diaz-Cintra Sofia, Pacheco-López Gustavo. Front. Behav. Neurosci., 14 February 2017 https://doi.org/10.3389/fnbeh.2017.00019
- Sehgal, M., Song, C., Ehlers, V. L., and Moyer, J. R. Jr. (2013). Learning to learn – intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol. Learn. Mem. 105, 186–199. doi: 10.1016/j.nlm.2013.07.008
- Castrén E, Hen R. Neuronal plasticity and antidepressant actions. Trends Neurosci. 2013;36(5):259–267. doi:10.1016/j.tins.2012.12.010 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3648595
- Synaptic activity and the construction of cortical circuits. Katz LC, Shatz CJ. Science. 1996 Nov 15; 274(5290):1133-8.
- Selective stabilisation of developing synapses as a mechanism for the specification of neuronal networks. Changeux JP, Danchin A. Nature. 1976 Dec 23-30; 264(5588):705-12.
- Neural activity and the dynamics of central nervous system development. Hua JY, Smith SJ. Nat Neurosci. 2004 Apr; 7(4):327-32.
- Shohayeb B, Diab M, Ahmed M, Ng DCH. Factors that influence adult neurogenesis as potential therapy. Transl Neurodegener. 2018;7:4. Published 2018 Feb 21. doi:10.1186/s40035-018-0109-9 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5822640/
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70(4):687-702. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3106107/
- Levy MJF, Boulle F, Steinbusch HW, van den Hove DLA, Kenis G, Lanfumey L. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology (Berl). 2018;235(8):2195–2220. doi:10.1007/s00213-018-4950-4 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6061771
- Neuronal plasticity and antidepressant actions. Castrén E, Hen R. Trends Neurosci. 2013 May; 36(5):259-67.
- Cohen LG, Celnik P, Pascual-Leone A, Corwell B, Falz L, Dambrosia J,et al. Functional relevance of cross-modal plasticity in blind humans.Nature 1997; 389: 180–3.
- Nudo RJ. Plasticity. NeuroRx 2006; 3: 420–7
- Levin MF, Kleim JA, Wolf SL. What do motor “recovery” and “compen-sation” mean in patients following stroke? Neurorehabil Neural Repair2009; 23: 313–9
- Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M. Pharmacol Rev. 2006 Mar; 58(1):115-34
- In vivo imaging of adult human hippocampal neurogenesis: progress, pitfalls and promise. Ho NF, Hooker JM, Sahay A, Holt DJ, Roffman JL. Mol Psychiatry. 2013 Apr; 18(4):404-16.
- Dynamics of hippocampal neurogenesis in adult humans. Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Boström E, Westerlund I, Vial C, Buchholz BA, Possnert G, Mash DC, Druid H, Frisén J. Cell. 2013 Jun 6; 153(6):1219-1227.
- Human Hippocampal Neurogenesis Persists throughout Aging. Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, Hen R, Mann JJ. Cell Stem Cell. 2018 Apr 5; 22(4):589-599.e5.
- Long-term potentiation and long-term depression: a clinical perspective. Bliss TV, Cooke SF. Clinics (Sao Paulo). 2011; 66 Suppl 1():3-17.
- Effects of long-term potentiation in the human visual cortex: a functional magnetic resonance imaging study. Clapp WC, Zaehle T, Lutz K, Marcar VL, Kirk IJ, Hamm JP, Teyler TJ, Corballis MC, Jancke L. Neuroreport. 2005 Dec 19; 16(18):1977-80.
- Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Kempton MJ, Salvador Z, Munafò MR, Geddes JR, Simmons A, Frangou S, Williams SC. Arch Gen Psychiatry. 2011 Jul; 68(7):675-90.
- Disorders of memory and plasticity in psychiatric disease. Pittenger C. Dialogues Clin Neurosci. 2013 Dec; 15(4):455-63.
- Structural changes in hippocampal subfields in major depressive disorder: a high-field magnetic resonance imaging study. Huang Y, Coupland NJ, Lebel RM, Carter R, Seres P, Wilman AH, Malykhin NV. Biol Psychiatry. 2013 Jul 1; 74(1):62-8.
- Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, Lesch KP. Mol Psychiatry. 2006 May; 11(5):514-22.
- Decreased numbers of progenitor cells but no response to antidepressant drugs in the hippocampus of elderly depressed patients. Lucassen PJ, Stumpel MW, Wang Q, Aronica E. Neuropharmacology. 2010 May; 58(6):940-9.
- Retinal dysfunction of contrast processing in major depression also apparent in cortical activity. Bubl E, Kern E, Ebert D, Riedel A, Tebartz van Elst L, Bach M. Eur Arch Psychiatry Clin Neurosci. 2015 Jun; 265(4):343-50.
- Evidence for impaired neocortical synaptic plasticity in bipolar II disorder. Elvsåshagen T, Moberget T, Bøen E, Boye B, Englin NO, Pedersen PØ, Andreassen OA, Dietrichs E, Malt UF, Andersson S. Biol Psychiatry. 2012 Jan 1; 71(1):68-74.
- Behavioural and neuroplastic effects of the new-generation antidepressant agomelatine compared to fluoxetine in glucocorticoid receptor-impaired mice. Païzanis E, Renoir T, Lelievre V, Saurini F, Melfort M, Gabriel C, Barden N, Mocaër E, Hamon M, Lanfumey L. Int J Neuropsychopharmacol. 2010 Jul; 13(6):759-74.
- Deep-brain magnetic stimulation promotes adult hippocampal neurogenesis and alleviates stress-related behaviors in mouse models for neuropsychiatric disorders. Zhang Y, Mao RR, Chen ZF, Tian M, Tong DL, Gao ZR, Huang M, Li X, Xu X, Zhou WH, Li CY, Wang J, Xu L, Qiu Z. Mol Brain. 2014 Feb 11; 7():11.
- The modulation of adult neuroplasticity is involved in the mood-improving actions of atypical antipsychotics in an animal model of depression. Morais M, Patrício P, Mateus-Pinheiro A, Alves ND, Machado-Santos AR, Correia JS, Pereira J, Pinto L, Sousa N, Bessa JM. Transl Psychiatry. 2017 Jun 6; 7(6):e1146.
- The stressed hippocampus, synaptic plasticity and lost memories. Kim JJ, Diamond DM. Nat Rev Neurosci. 2002 Jun; 3(6):453-62.
- Behavioural stress facilitates the induction of long-term depression in the hippocampus. Xu L, Anwyl R, Rowan MJ. Nature. 1997 May 29; 387(6632):497-500.
- Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, Morrison JH. Neuroscience. 2009 Dec 1; 164(2):798-808.
- Structural Plasticity of the Hippocampus and Amygdala Induced by Electroconvulsive Therapy in Major Depression. Joshi SH, Espinoza RT, Pirnia T, Shi J, Wang Y, Ayers B, Leaver A, Woods RP, Narr KL. Biol Psychiatry. 2016 Feb 15; 79(4):282-92.
- Anticonvulsant and antidepressant properties of electroconvulsive therapy: a proposed mechanism of action. Sackeim HA, Decina P, Prohovnik I, Malitz S, Resor SR. Biol Psychiatry. 1983 Nov; 18(11):1301-10.
- The effects of repetitive transcranial magnetic stimulation in the treatment of depression. Fitzgerald PB, Daskalakis ZJ. Expert Rev Med Devices. 2011 Jan; 8(1):85-95.
- Antidepressant-like Effects of Electroconvulsive Seizures Require Adult Neurogenesis in a Neuroendocrine Model of Depression. Schloesser RJ, Orvoen S, Jimenez DV, Hardy NF, Maynard KR, Sukumar M, Manji HK, Gardier AM, David DJ, Martinowich K. Brain Stimul. 2015 Sep-Oct; 8(5):862-7
- Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C, Spitzer G, Santarelli L, Scharf B, Hen R, Rosoklija G, Sackeim HA, Dwork AJ. J Neurosci. 2007 May 2; 27(18):4894-901.
- Neurobiological effects of exercise on major depressive disorder: A systematic review. Schuch FB, Deslandes AC, Stubbs B, Gosmann NP, Silva CT, Fleck MP. Neurosci Biobehav Rev. 2016 Feb; 61():1-11.
- Neuroplasticity – exercise-induced response of peripheral brain-derived neurotrophic factor: a systematic review of experimental studies in human subjects. Knaepen K, Goekint M, Heyman EM, Meeusen R. Sports Med. 2010 Sep 1; 40(9):765-801.
- MAP training: combining meditation and aerobic exercise reduces depression and rumination while enhancing synchronized brain activity. Alderman BL, Olson RL, Brush CJ, Shors TJ. Transl Psychiatry. 2016 Feb 2; 6():e726.
- The impacts of swimming exercise on hippocampal expression of neurotrophic factors in rats exposed to chronic unpredictable mild stress. Jiang P, Dang RL, Li HD, Zhang LH, Zhu WY, Xue Y, Tang MM. Evid Based Complement Alternat Med. 2014; 2014():729827.
- Effect of exercise, exercise withdrawal, and continued regular exercise on excitability and long-term potentiation in the dentate gyrus of hippocampus. Radahmadi M, Hosseini N, Alaei H. Brain Res. 2016 Dec 15; 1653():8-13.
- Modulation of synaptic plasticity by short-term aerobic exercise in adult mice. D’Arcangelo G, Triossi T, Buglione A, Melchiorri G, Tancredi V. Behav Brain Res. 2017 Aug 14; 332():59-63.
- The effect of acute swim stress and training in the water maze on hippocampal synaptic activity as well as plasticity in the dentate gyrus of freely moving rats: revisiting swim-induced LTP reinforcement. Tabassum H, Frey JU. Hippocampus. 2013 Dec; 23(12):1291-8.
- Effect of hippocampal and amygdala volumes on clinical outcomes in major depression: a 3-year prospective magnetic resonance imaging study. Frodl T, Jäger M, Smajstrlova I, Born C, Bottlender R, Palladino T, Reiser M, Möller HJ, Meisenzahl EM. J Psychiatry Neurosci. 2008 Sep; 33(5):423-30.
- The influence of lithium on hippocampal volume in elderly bipolar patients: a study using voxel-based morphometry. Zung S, Souza-Duran FL, Soeiro-de-Souza MG, Uchida R, Bottino CM, Busatto GF, Vallada H. Transl Psychiatry. 2016 Jun 28; 6(6):e846.
- Antidepressants increase neural progenitor cells in the human hippocampus. Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, Arango V. Neuropsychopharmacology. 2009 Oct; 34(11):2376-89.
- Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Boldrini M, Santiago AN, Hen R, Dwork AJ, Rosoklija GB, Tamir H, Arango V, John Mann J. Neuropsychopharmacology. 2013 May; 38(6):1068-77.
- Hippocampal volume predicts antidepressant efficacy in depressed patients without incomplete hippocampal inversion. Colle R, Cury C, Chupin M, Deflesselle E, Hardy P, Nasser G, Falissard B, Ducreux D, Colliot O, Corruble E. Neuroimage Clin. 2016; 12():949-955.
- Neural plasticity and proliferation in the generation of antidepressant effects: hippocampal implication. Pilar-Cuéllar F, Vidal R, Díaz A, Castro E, dos Anjos S, Pascual-Brazo J, Linge R, Vargas V, Blanco H, Martínez-Villayandre B, Pazos Á, Valdizán EM. Neural Plast. 2013; 2013():537265.
- Serotonin is required for exercise-induced adult hippocampal neurogenesis. Klempin F, Beis D, Mosienko V, Kempermann G, Bader M, Alenina N. J Neurosci. 2013 May 8; 33(19):8270-5.
- Antidepressants recruit new neurons to improve stress response regulation. Surget A, Tanti A, Leonardo ED, Laugeray A, Rainer Q, Touma C, Palme R, Griebel G, Ibarguen-Vargas Y, Hen R, Belzung C. Mol Psychiatry. 2011 Dec; 16(12):1177-88.
- A role for nuclear beta-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Mostany R, Valdizán EM, Pazos A. Neuropharmacology. 2008 Jul; 55(1):18-26.
- Synergetic effects of quetiapine and venlafaxine in preventing the chronic restraint stress-induced decrease in cell proliferation and BDNF expression in rat hippocampus. Xu H, Chen Z, He J, Haimanot S, Li X, Dyck L, Li XM. Hippocampus. 2006; 16(6):551-9.
- Repeated clomipramine treatment reversed the inhibition of cell proliferation in adult hippocampus induced by chronic unpredictable stress. Liu Q, Yu J, Mao-Ying QL, Mi WL, Li B, Wang YQ, Wang J, Wu GC. Pharmacogenomics J. 2008 Dec; 8(6):375-83.
- Antidepressants stimulate hippocampal neurogenesis by inhibiting p21 expression in the subgranular zone of the hipppocampus. Pechnick RN, Zonis S, Wawrowsky K, Cosgayon R, Farrokhi C, Lacayo L, Chesnokova V. PLoS One. 2011; 6(11):e27290.
- Chronic fluoxetine stimulates maturation and synaptic plasticity of adult-born hippocampal granule cells. Wang JW, David DJ, Monckton JE, Battaglia F, Hen R. J Neurosci. 2008 Feb 6; 28(6):1374-84.
- Repeated ECS and fluoxetine administration have equivalent effects on hippocampal synaptic plasticity. Stewart CA, Reid IC. Psychopharmacology (Berl). 2000 Feb; 148(3):217-23.
- New insights in the biology of BDNF synthesis and release: implications in CNS function. Greenberg ME, Xu B, Lu B, Hempstead BL. J Neurosci. 2009 Oct 14; 29(41):12764-7.
- The molecular neurobiology of depression. Krishnan V, Nestler EJ. Nature. 2008 Oct 16; 455(7215):894-902.
- Stress, depression, and neuroplasticity: a convergence of mechanisms. Pittenger C, Duman RS. Neuropsychopharmacology. 2008 Jan; 33(1):88-109.
- The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, Almeida OF, Sousa N. Mol Psychiatry. 2009 Aug; 14(8):764-73, 739.
- NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. Nature. 2011 Jun 15; 475(7354):91-5.
- A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Ochs G, Penn RD, York M, Giess R, Beck M, Tonn J, Haigh J, Malta E, Traub M, Sendtner M, Toyka KV. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000 Jun; 1(3):201-6.
- BDNF and synaptic plasticity, cognitive function, and dysfunction. Lu B, Nagappan G, Lu Y. Handb Exp Pharmacol. 2014; 220():223-50.
- p75 and Trk: a two-receptor system. Chao MV, Hempstead BL. Trends Neurosci. 1995 Jul; 18(7):321-6.
- TrkB inhibition as a therapeutic target for CNS-related disorders. Boulle F, Kenis G, Cazorla M, Hamon M, Steinbusch HW, Lanfumey L, van den Hove DL. Prog Neurobiol. 2012 Aug; 98(2):197-206.
- Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Ernfors P, Wetmore C, Olson L, Persson H. Neuron. 1990 Oct; 5(4):511-26.
- Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. Cazorla M, Prémont J, Mann A, Girard N, Kellendonk C, Rognan D. J Clin Invest. 2011 May; 121(5):1846-57.
- p11 mediates the BDNF-protective effects in dendritic outgrowth and spine formation in B27-deprived primary hippocampal cells. Park SW, Nhu le H, Cho HY, Seo MK, Lee CH, Ly NN, Choi CM, Lee BJ, Kim GM, Seol W, Lee JG, Kim YH. J Affect Disord. 2016 May 15; 196():1-10.
- Excitatory actions of GABA increase BDNF expression via a MAPK-CREB-dependent mechanism–a positive feedback circuit in developing neurons. Obrietan K, Gao XB, Van Den Pol AN. J Neurophysiol. 2002 Aug; 88(2):1005-15.
- ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. Hetman M, Hsuan SL, Habas A, Higgins MJ, Xia Z. J Biol Chem. 2002 Dec 20; 277(51):49577-84.
- Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Ernfors P, Lee KF, Jaenisch R. Nature. 1994 Mar 10; 368(6467):147-50.
- Aarse J, Herlitze S, Manahan-Vaughan D. The requirement of BDNF for hippocampal synaptic plasticity is experience-dependent. Hippocampus. 2016;26:739–751
- Control of synaptic consolidation in the dentate gyrus: mechanisms, functions, and therapeutic implications. Bramham CR. Prog Brain Res. 2007; 163():453-71.
- Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Magariños AM, Li CJ, Gal Toth J, Bath KG, Jing D, Lee FS, McEwen BS. Hippocampus. 2011 Mar; 21(3):253-64.
- Haploinsufficiency in trkB and/or trkC neurotrophin receptors causes structural alterations in the aged hippocampus and amygdala. von Bohlen und Halbach O, Minichiello L, Unsicker K. Eur J Neurosci. 2003 Oct; 18(8):2319-25.
- Implications of p75NTR for dentate gyrus morphology and hippocampus-related behavior revisited. Dokter M, Busch R, Poser R, Vogt MA, von Bohlen Und Halbach V, Gass P, Unsicker K, von Bohlen Und Halbach O. Brain Struct Funct. 2015; 220(3):1449-62.
- BDNF effects on dendritic spine morphology and hippocampal function. von Bohlen Und Halbach O, von Bohlen Und Halbach V. Cell Tissue Res. 2018 Sep; 373(3):729-741.
- ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Alonso M, Medina JH, Pozzo-Miller L. Learn Mem. 2004 Mar-Apr; 11(2):172-8.
- Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. J Neurosci. 2010 Feb 3; 30(5):1739-49.
- Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. Baquet ZC, Gorski JA, Jones KR. J Neurosci. 2004 Apr 28; 24(17):4250-8.
- Fibroblast growth factor-2 signaling in neurogenesis and neurodegeneration. Woodbury ME, Ikezu T. J Neuroimmune Pharmacol. 2014 Mar; 9(2):92-101.
- The fibroblast growth factor family: neuromodulation of affective behavior. Turner CA, Watson SJ, Akil H. Neuron. 2012 Oct 4; 76(1):160-74.
- Interactions between cholinergic and fibroblast growth factor receptors in brain trophism and plasticity. Di Liberto V, Mudo G, Fuxe K, Belluardo N. Curr Protein Pept Sci. 2014; 15(7):691-702.
- The FGF-2/FGFRs neurotrophic system promotes neurogenesis in the adult brain. Mudò G, Bonomo A, Di Liberto V, Frinchi M, Fuxe K, Belluardo N. J Neural Transm (Vienna). 2009 Aug; 116(8):995-1005.
- Fibroblast growth factor-2 deficiency causes defects in adult hippocampal neurogenesis, which are not rescued by exogenous fibroblast growth factor-2. Werner S, Unsicker K, von Bohlen und Halbach O. J Neurosci Res. 2011 Oct; 89(10):1605-17.
- Fibroblast growth factor receptor-1 is required for long-term potentiation, memory consolidation, and neurogenesis. Zhao M, Li D, Shimazu K, Zhou YX, Lu B, Deng CX. Biol Psychiatry. 2007 Sep 1; 62(5):381-90.
- VEGF ligands and receptors: implications in neurodevelopment and neurodegeneration. Carmeliet P, Ruiz de Almodovar C. Cell Mol Life Sci. 2013 May; 70(10):1763-78.
- Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Jin KL, Mao XO, Greenberg DA. Proc Natl Acad Sci U S A. 2000 Aug 29; 97(18):10242-7.
- Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Khaibullina AA, Rosenstein JM, Krum JM. Brain Res Dev Brain Res. 2004 Jan 31; 148(1):59-68.
- VEGF is required for dendritogenesis of newly born olfactory bulb interneurons. Licht T, Eavri R, Goshen I, Shlomai Y, Mizrahi A, Keshet E. Development. 2010 Jan; 137(2):261-71.
- VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. J Clin Invest. 2003 Jun; 111(12):1843-51.
- Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Proc Natl Acad Sci U S A. 2002 Sep 3; 99(18):11946-50.
- Vascular endothelial growth factor-B (VEGFB) stimulates neurogenesis: evidence from knockout mice and growth factor administration. Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA. Dev Biol. 2006 Jan 15; 289(2):329-35.
- VEGF, a mediator of the effect of experience on hippocampal neurogenesis. During MJ, Cao L. Curr Alzheimer Res. 2006 Feb; 3(1):29-33.
- Angiogenesis in refractory depression: A possible phenotypic target to avoid the blood brain barrier. Yamada MK. Drug Discov Ther. 2016; 10(2):74-8.
- VEGF regulates antidepressant effects of lamotrigine. Sun R, Li N, Li T. Eur Neuropsychopharmacol. 2012 Jun; 22(6):424-30.
- Role of trophic factors GDNF, IGF-1 and VEGF in major depressive disorder: A comprehensive review of human studies. Sharma AN, da Costa e Silva BF, Soares JC, Carvalho AF, Quevedo J. J Affect Disord. 2016 Jun; 197():9-20.
- Stroke-induced progenitor cell proliferation in adult spontaneously hypertensive rat brain: effect of exogenous IGF-1 and GDNF. Dempsey RJ, Sailor KA, Bowen KK, Türeyen K, Vemuganti R. J Neurochem. 2003 Nov; 87(3):586-97.
- Intracerebral infusion of glial cell line-derived neurotrophic factor promotes striatal neurogenesis after stroke in adult rats. Kobayashi T, Ahlenius H, Thored P, Kobayashi R, Kokaia Z, Lindvall O. Stroke. 2006 Sep; 37(9):2361-7.
- Progenitor proliferation in the adult hippocampus and substantia nigra induced by glial cell line-derived neurotrophic factor. Chen Y, Ai Y, Slevin JR, Maley BE, Gash DM. Exp Neurol. 2005 Nov; 196(1):87-95.
- GDNF facilitates differentiation of the adult dentate gyrus-derived neural precursor cells into astrocytes via STAT3. Boku S, Nakagawa S, Takamura N, Kato A, Takebayashi M, Hisaoka-Nakashima K, Omiya Y, Inoue T, Kusumi I. Biochem Biophys Res Commun. 2013 May 17; 434(4):779-84.
- Recombinant adeno-associated virus vector expressing glial cell line-derived neurotrophic factor reduces ischemia-induced damage. Tsai TH, Chen SL, Chiang YH, Lin SZ, Ma HI, Kuo SW, Tsao YP. Exp Neurol. 2000 Dec; 166(2):266-75.
- Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. Aberg ND, Brywe KG, Isgaard J. ScientificWorldJournal. 2006 Jan 18; 6():53-80.
- Insulin-like Growth Factors in a clinical setting: Review of IGF-I. Frysak Z, Schovanek J, Iacobone M, Karasek D. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2015 Sep; 159(3):347-51.
- Szczesny E, Slusarczyk J, Glombik K, Budziszewska B, Kubera M, Lason W, Basta-Kaim A. Possible contribution of IGF-1 to depressive disorder. Pharmacol Rep. 2013;65:1622–1631
- Insulin-like growth factor-I and neurogenesis in the adult mammalian brain. Anderson MF, Aberg MA, Nilsson M, Eriksson PS. Brain Res Dev Brain Res. 2002 Mar 31; 134(1-2):115-22.
- Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. D’Mello SR, Galli C, Ciotti T, Calissano P. Proc Natl Acad Sci U S A. 1993 Dec 1; 90(23):10989-93.
- Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. Neuron. 1995 Apr; 14(4):717-30.
- Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. O’Kusky JR, Ye P, D’Ercole AJ. J Neurosci. 2000 Nov 15; 20(22):8435-42.
- Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Lichtenwalner RJ, Forbes ME, Bennett SA, Lynch CD, Sonntag WE, Riddle DR. Neuroscience. 2001; 107(4):603-13.
- Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. Aberg MA, Aberg ND, Hedbäcker H, Oscarsson J, Eriksson PS. J Neurosci. 2000 Apr 15; 20(8):2896-903.
- Outgrowth of sympathetic adrenergic neurons in mice treated with a nerve-growth factor (NGF). Olson L. Z Zellforsch Mikrosk Anat. 1967; 81(2):155-73.
- Intranasal delivery of nerve growth factor to protect the central nervous system against acute cerebral infarction. Zhao HM, Liu XF, Mao XW, Chen CF. Chin Med Sci J. 2004 Dec; 19(4):257-61.
- Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Cattaneo E, McKay R. Nature. 1990 Oct 25; 347(6295):762-5.
- Intranasal nerve growth factor enhances striatal neurogenesis in adult rats with focal cerebral ischemia. Zhu W, Cheng S, Xu G, Ma M, Zhou Z, Liu D, Liu X. Drug Deliv. 2011 Jul; 18(5):338-43.
- NGF is essential for hippocampal plasticity and learning. Conner JM, Franks KM, Titterness AK, Russell K, Merrill DA, Christie BR, Sejnowski TJ, Tuszynski MH. J Neurosci. 2009 Sep 2; 29(35):10883-9.
- Rathore S, Hinn A, Cooper L, Tyroler H, Rosamond W. Characterizationof incident stroke signs and symptoms: findings from the atheroscler-osis risk in communities study. Stroke 2002; 33: 2718–21
- Levin MF, Kleim JA, Wolf SL. What do motor “recovery” and “compen-sation” mean in patients following stroke? Neurorehabil Neural Repair2009; 23: 313–9.
- Muellbacher W, Richards C, Ziemann U, Wittenberg G, Weltz D,Boroojerdi B, et al. Improving hand function in chronic stroke. ArchNeurol 2002; 59: 1278–82.
- Cramer SC, Crafton KR. Somatotopy and movement representation sitesfollowing cortical stroke. Exp Brain Res 2006; 168: 25–32
- Murase N, Duque J, Mazzocchio R, Cohen LG. Influence of interhemi-spheric interactions on motor function in chronic stroke. Ann Neurol2004; 55: 400–9
- Sharma N, Baron JC, Rowe JB. Motor imagery after stroke: relating out-come to motor network connectivity. Ann Neurol 2009b; 66: 604–16
- Wieloch T, Nikolich K. Mechanisms of neural plasticity following braininjury. Curr Opin Neurobiol 2006; 16: 258–64
- Raymer AM, Beeson P, Holland A, Kendall D, Maher LM, Martin N,et al. Translational research in aphasia: from neuroscience to neuror-ehabilitation. J Speech Lang Hear Res 2008; 51: S259–75
- Baillieux H, De Smet HJ, Dobbeleir A, Paquier PF, De Deyn PP, Marien P.Cognitive and affective disturbances following focal cerebellar damagein adults: a neuropsychological and SPECT study. Cortex 2010; 46:869–79.
- Chen P, Goldberg D, Kolb B, Lanser M, Benowitz L. Inosine inducesaxonal rewiring and improves behavioral outcome after stroke. ProcNatl Acad Sci USA 2002a; 99: 9031–6
- Milot MH, Cramer SC. Biomarkers of recovery after stroke. Curr OpinNeurol 2008; 21: 654–9
- Krakauer JW. Motor learning: its relevance to stroke recovery and neu-rorehabilitation. Curr Opin Neurol 2006; 19: 84–90
- Belanger HG, Vanderploeg RD, Curtiss G, Warden DL. Recent neuroima-ging techniques in mild traumatic brain injury. J Neuropsychiatry ClinNeurosci 2007; 19: 5–20.
- Rosenzweig ES, Courtine G, Jindrich DL, Brock JH, Ferguson AR,Strand SC, et al. Extensive spontaneous plasticity of corticospinal pro-jections after primate spinal cord injury. Nat Neurosci 2010; 13:1505–10
- Cramer SC, Orr EL, Cohen MJ, Lacourse MG. Effects of motor imagerytraining after chronic, complete spinal cord injury. Exp Brain Res2007b; 177: 233–42
- Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy followingcortical injury: cellular and molecular mechanisms as targets for poten-tial prophylaxis. Epilepsia 2009; 50 (Suppl 2): 30–40
- KarlA, Birbaumer N, Lutzenberger W, Cohen LG, Flor H. Reorganizationof motor and somatosensory cortex in upper extremity amputees withphantom limb pain. J Neurosci 2001; 21: 3609–18
- Kleim JA, Jones TA. Principles of experience-dependent neural plasticity:implications for rehabilitation after brain damage. J Speech Lang HearRes 2008; 51: S225–39
- Cramer SC, Koroshetz WJ, Finklestein SP. The case for modality-specificoutcome measures in clinical trials of stroke recovery-promotingagents. Stroke 2007a; 38: 1393–5
- Bhogal S, Teasell R, Speechley M. Intensity of aphasia therapy, impact onrecovery. Stroke 2003; 34: 987–93
- Riley J, Le V, Der-Yeghiaian L, See J, Newton J, Ward N, et al. Anatomyof Stroke Injury Predicts Gains from Therapy. Stroke 2011; 42: 421–6
- Hachinski V, Donnan GA, Gorelick PB, Hacke W, Cramer SC, Kaste M,et al. Stroke: working toward a prioritized world agenda. Stroke 2010;41: 1084–99
- Plow EB, Carey JR, Nudo RJ, Pascual-Leone A. Invasive cortical stimula-tion to promote recovery of function after stroke: a critical appraisal.Stroke 2009; 40: 1926–31
- Wagner T, Valero-Cabre A, Pascual-Leone A. Noninvasive human brainstimulation. Annu Rev Biomed Eng 2007; 9: 527–65
- Kleim J, Kleim E, Cramer S. Systematic assessment of training-inducedchanges in corticospinal output to hand using frameless stereotaxictranscranial magnetic stimulation. Nature Protocols 2007; 2: 1675–84
- Reis J, Schambra HM, Cohen LG, Buch ER, Fritsch B, Zarahn E, et al.Noninvasive cortical stimulation enhances motor skill acquisition overmultiple days through an effect on consolidation. Proc Natl Acad SciUSA 2009; 106: 1590–5
- Conforto AB, Ferreiro KN, Tomasi C, dos Santos RL, Moreira VL,Marie SK, et al. Effects of somatosensory stimulation on motor func-tion after subacute stroke. Neurorehabil Neural Repair 2010; 24:263–72.
- Edwards DJ, Krebs HI, Rykman A, Zipse J, Thickbroom GW,Mastaglia FL, et al. Raised corticomotor excitability of M1 forearmarea following anodal tDCS is sustained during robotic wrist therapyin chronic stroke. Restor Neurol Neurosci 2009; 27: 199–207
- Nowak DA, Grefkes C, Ameli M, Fink GR. Interhemispheric competitionafter stroke: brain stimulation to enhance recovery of function of theaffected hand. Neurorehabil Neural Repair 2009; 23: 641–56
- Fregni F, Pascual-Leone A. Technology insight: noninvasive brain stimu-lation in neurology-perspectives on the therapeutic potential of rTMSand tDCS. Nat Clin Pract Neurol 2007; 3: 383–93
- Martin P, Naeser M, Theoret H, Tormos J, Nicholas M, Kurland J, et al.Transcranial magnetic stimulation as a complementary treatment foraphasia. Semin Speech Lang 2004; 25: 181–91
- Padberg F, George MS. Repetitive transcranial magnetic stimulation ofthe prefrontal cortex in depression. Exp Neurol 2009; 219: 2–13
- Speer AM, Benson BE, Kimbrell TK, Wassermann EM, Willis MW,Herscovitch P, et al. Opposite effects of high and low frequencyrTMS on mood in depressed patients: relationship to baseline cerebralactivity on PET. J Affect Disord 2009; 115: 386–94
- Vercammen A, Knegtering H, Bruggeman R, Westenbroek HM,Jenner JA, Slooff CJ, et al. Effects of bilateral repetitive transcranialmagnetic stimulation on treatment resistant auditory-verbal hallucin-ations in schizophrenia: a randomized controlled trial. Schizophr Res2009; 114: 172–9
- Denys D, Mantione M. Deep brain stimulation in obsessive-compulsivedisorder. Prog Brain Res 2009; 175: 419–27
- Johnson MD, Miocinovic S, McIntyre CC, Vitek JL. Mechanisms andtargets of deep brain stimulation in movement disorders.Neurotherapeutics 2008; 5: 294–308
- Greenberg BD, Rauch SL, Haber SN. Invasive circuitry-based neurother-apeutics: stereotactic ablation and deep brain stimulation for OCD.Neuropsychopharmacology 2010b; 35: 317–36
- Lozano AM, Mayberg HS, Giacobbe P, Hamani C, Craddock RC,Kennedy SH. Subcallosal cingulate gyrus deep brain stimulation fortreatment-resistant depression. Biol Psychiatry 2008; 64: 461–7
- Bewernick BH, Hurlemann R, Matusch A, Kayser S, Grubert C,Hadrysiewicz B, et al. Nucleus accumbens deep brain stimulation de-creases ratings of depression and anxiety in treatment-resistant depres-sion. Biol Psychiatry 2010; 67: 110–6.
- Greenberg BD, Gabriels LA, Malone DA Jr, Rezai AR, Friehs GM,Okun MS, et al. Deep brain stimulation of the ventral internal cap-sule/ventral striatum for obsessive-compulsive disorder: worldwide ex-perience. Mol Psychiatry 2010a; 15: 64–79
- Larson PS. Deep brain stimulation for psychiatric disorders.Neurotherapeutics 2008; 5: 50–8
- Lujan JL, Chaturvedi A, McIntyre CC. Tracking the mechanism of deepbrain stimulation for neuropsychiatric disorders. Front Biosci 2008; 13:5892–904.
- Jackson A, Mavoori J, Fetz EE. Long-term motor cortex plasticity inducedby an electronic neural implant. Nature 2006; 444: 56–60
- Levy R, Benson R, Winstein C. for the Everest Study InvestigatorsCorticalStimulation for Upper-Extremity Hemiparesis from Ischemic Stroke:Everest Study Primary Endpoint Results. International StrokeConference. New Orleans, LA, 2008
- Floel A, Cohen LG. Recovery of function in humans: cortical stimulationand pharmacological treatments after stroke. Neurobiol Dis 2010; 37:243–51
- Potter WB, O’Riordan KJ, Barnett D, Osting SM, Wagoner M, Burger C,et al. Metabolic regulation of neuronal plasticity by the energy sensorAMPK. PLoS ONE 2010; 5: e8996
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K,Zimand E, et al. Cognitive enhancers as adjuncts to psychotherapy:use of D-cycloserine in phobic individuals to facilitate extinction offear. Arch Gen Psychiatry 2004; 61: 1136–44
- Pariente J, Loubinoux I, Carel C, Albucher J, Leger A, Manelfe C, et al.Fluoxetine modulates motor performance and cerebral activation ofpatients recovering from stroke. Ann Neurol 2001; 50: 718–29
- Garcia-Alias G, Barkhuysen S, Buckle M, Fawcett JW. ChondroitinaseABC treatment opens a window of opportunity for task-specific re-habilitation. Nat Neurosci 2009; 12: 1145–51
- DeRubeis RJ, Siegle GJ, Hollon SD. Cognitive therapy versus medicationfor depression: treatment outcomes and neural mechanisms. Nat RevNeurosci 2008; 9: 788–96
- Butefisch C, Davis B, Wise S, Sawaki L, Kopylev L, Classen J, et al.Mechanisms of use-dependent plasticity in the human motor cortex.Proc Natl Acad Sci USA 2000; 97: 3661–5
- Lazar RM, Fitzsimmons BF, Marshall RS, Mohr JP, Berman MF.Midazolam challenge reinduces neurological deficits after transient is-chemic attack. Stroke 2003; 34: 794–6.
- Wolf SL, Newton H, Maddy D, Blanton S, Zhang Q, Winstein CJ, et al.The Excite Trial: relationship of intensity of constraint induced move-ment therapy to improvement in the wolf motor function test. RestorNeurol Neurosci 2007; 25: 549–62
- Duncan PW, Sullivan KJ, Behrman AL, Azen SP, Wu SS, Nadeau SE,et al. Protocol for the Locomotor Experience Applied Post-stroke (LEAPS) trial: a randomized controlled trial. BMC Neurol2007; 7: 39
- Lo AC, Guarino PD, Richards LG, Haselkorn JK, Wittenberg GF,Federman DG, et al. Robot-assisted therapy for long-term upper-limbimpairment after stroke. N Engl J Med 2010; 362: 1772–83
- Lin KC, Chen YA, Chen CL, Wu CY, Chang YF. The effects of bilateralarm training on motor control and functional performance in chronicstroke: a randomized controlled study. Neurorehabil Neural Repair2010; 24: 42–51
- Jonsdottir J, Cattaneo D, Recalcati M, Regola A, Rabuffetti M,Ferrarin M, et al. Task-oriented biofeedback to improve gait in indi-viduals with chronic stroke: motor learning approach. NeurorehabilNeural Repair 2010; 24: 478–85
- Sawaki L, Butler AJ, Xiaoyan L, Wassenaar PA, Mohammad YM,Blanton S, et al. Constraint-induced movement therapy results inincreased motor map area in subjects 3 to 9 months after stroke.Neurorehabil Neural Repair 2008; 22: 505–13
- Gauthier LV, Taub E, Perkins C, Ortmann M, Mark VW, Uswatte G.Remodeling the brain: plastic structural brain changes produced bydifferent motor therapies after stroke. Stroke 2008; 39: 1520–5
- Dobkin BH. Confounders in rehabilitation trials of task-oriented training:lessons from the designs of the EXCITE and SCILT multicenter trials.Neurorehabil Neural Repair 2007; 21: 3–13
- Subramanian SK, Massie CL, Malcolm MP, Levin MF. Does provision ofextrinsic feedback result in improved motor learning in the upper limbpoststroke? A systematic review of the evidence. Neurorehabilitationand Neural Repair 2010; 24: 113–24
- Hillman CH, Erickson KI, Kramer AF. Be smart, exercise your heart: ex-ercise effects on brain and cognition. Nature Reviews Neuroscience2008; 9: 58–65.
- Rhyu IJ, Bytheway JA, Kohler SJ, Lange H, Lee KJ, Boklewski J, et al.Effects of aerobic exercise training on cognitive function and corticalvascularity in monkeys. Neuroscience 2010; 167: 1239–48
- Pajonk FG, Wobrock T, Gruber O, Scherk H, Berner D, Kaizl I, et al.Hippocampal plasticity in response to exercise in schizophrenia. ArchGen Psychiatry 2010; 67: 133–43
- Kramer AF, Erickson KI. Capitalizing on cortical plasticity: influence ofphysical activity on cognition and brain function. Trends Cogn Sci2007; 11: 342–8.
- Colcombe SJ, Kramer AF, Erickson KI, Scalf P, McAuley E, Cohen NJ,et al. Cardiovascular fitness, cortical plasticity, and aging. Proc NatlAcad Sci USA 2004; 101: 3316–21
- Beck AT. The current state of cognitive therapy: a 40-year retrospective.Arch Gen Psychiatry 2005; 62: 953–9
- Farb NA, Anderson AK, Mayberg H, Bean J, McKeon D, Segal ZV.Minding one’s emotions: mindfulness training alters the neural expres-sion of sadness. Emotion 2010; 10: 25–33
- Buonomano DV, Merzenich MM. Cortical plasticity: from synapses tomaps. Annu Rev Neurosci 1998; 21: 149–86
- Vinogradov S, Luks TL, Schulman BJ, Simpson GV. Deficit in a neuralcorrelate of reality monitoring in schizophrenia patients. Cereb Cortex2008; 18: 2532–9
- Dale CL, Findlay AM, Adcock RA, Vertinski M, Fisher M, Genevsky A,et al. Timing is everything: neural response dynamics during syllableprocessing and its relation to higher-order cognition in schizophreniaand healthy comparison subjects. Int J Psychophysiol 2010; 75:183–93
- Packard MG. Anxiety, cognition, and habit: a multiple memory systemsperspective. Brain Res 2009; 1293: 121–8.
- Persson J, Reuter-Lorenz PA. Gaining control: training executive functionand far transfer of the ability to resolve interference. Psychol Sci 2008;19: 881–8
- Temple R. A regulatory authority’s opinion about surrogate endpoints.In: Nimmo W, Tucker G, editors. Clinical Measurement in DrugEvaluation. New York: J. Wiley; 1995.
- Temple E, Deutsch GK, Poldrack RA, Miller SL, Tallal P, Merzenich MM,et al. Neural deficits in children with dyslexia ameliorated by behavioralremediation: evidence from functional MRI. Proc Natl Acad Sci USA2003; 100: 2860–5.
- Keller TA, Just MA. Altering cortical connectivity: remediation-inducedchanges in the white matter of poor readers. Neuron 2009; 64:624–31
- Rosenberg PB, Hillis AE. Biomarkers for Alzheimer’s disease: ready for thenext step. Brain 2009; 132 (Pt 8): 2002–4
- McNab F, Varrone A, Farde L, Jucaite A, Bystritsky P, Forssberg H, et al.Changes in cortical dopamine D1 receptor binding associated withcognitive training. Science 2009; 323: 800–2
- Klingberg T, Fernell E, Olesen PJ, Johnson M, Gustafsson P, Dahlstrom K,et al. Computerized training of working memory in children withADHD–a randomized, controlled trial. J Am Acad Child AdolescPsychiatry 2005; 44: 177–86
- Kennedy SH, Konarski JZ, Segal ZV, Lau MA, Bieling PJ, McIntyre RS,et al. Differences in brain glucose metabolism between responders toCBT and venlafaxine in a 16-week randomized controlled trial. Am JPsychiatry 2007; 164: 778–88.
- Green CS, Bavelier D. Exercising your brain: a review of human brainplasticity and training-induced learning. Psychol Aging 2008; 23:692–701
- Cox RW, Jesmanowicz A, Hyde JS. Real-time functional magnetic reson-ance imaging. Magn Reson Med 1995; 33: 230–6.
- deCharms RC. Applications of real-time fMRI. Nat Rev Neurosci 2008; 9:720–9.
- deCharms RC, Maeda F, Glover GH, Ludlow D, Pauly JM, Soneji D,et al. Control over brain activation and pain learned by usingreal-time functional MRI. Proc Natl Acad Sci USA 2005; 102:18626–31





{kind=link}