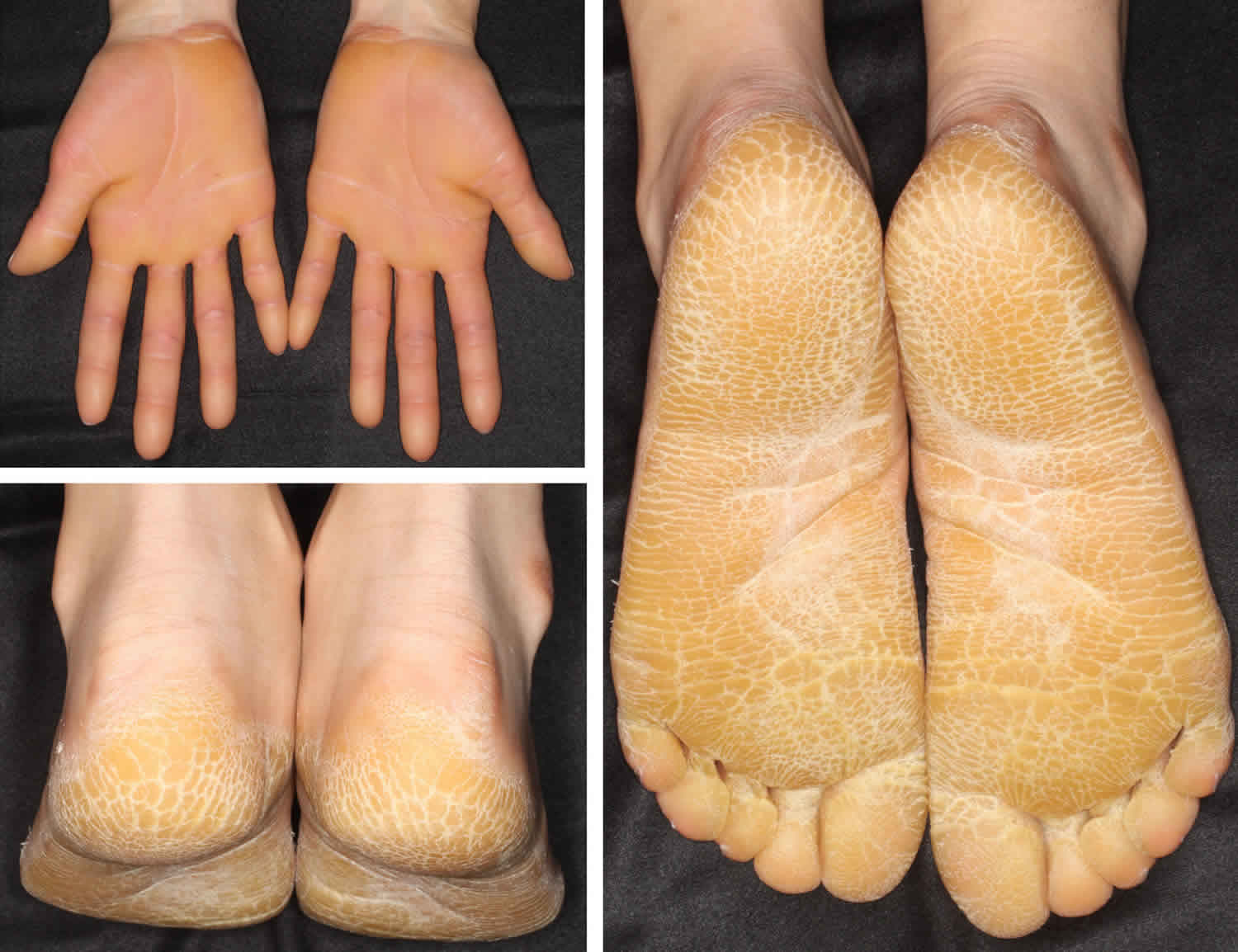
Palmoplantar keratodermas
Palmoplantar keratoderma also sometimes known as ‘keratosis palmaris et plantaris’, is a medical term that means marked thickening of the skin on the soles of the feet and palms of the hands.
Classification of palmoplantar keratodermas depends on whether or not it is inherited, and its clinical features:
- Diffuse keratodermas affect most of the palms and soles.
- Focal keratodermas mainly affect pressure areas.
- Punctate-type keratoderma results in tiny bumps on the palms and soles.
- Most often the abnormal skin involves only the palms and soles (non-transgradient palmoplantar keratoderma) but sometimes it extends on to the top of the hands and feet as well (transgradient).
In some rare forms of keratoderma other organs in the body may be affected in addition to the skin, and the keratoderma can be a marker of this internal abnormality.
Palmoplantar keratoderma types
Hereditary palmoplantar keratodermas
Hereditary palmoplantar keratoderma is a heterogeneous group of disorders characterized by hyperkeratosis of the palm and the sole skin 1. Hereditary palmoplantar keratoderma are divided into four groups – diffuse, focal, striate and punctate palmoplantar keratoderma – according to the clinical patterns of the hyperkeratotic lesions. Each group includes simple palmoplantar keratoderma, without associated features, and palmoplantar keratoderma with associated features, such as involvement of nails, teeth and other organs. palmoplantar keratoderma have been classified by a clinically based descriptive system. In recent years, many causative genes of palmoplantar keratoderma have been identified, which has confirmed and/or rearranged the traditional classifications.
Figure 1. Hereditary palmoplantar keratoderma
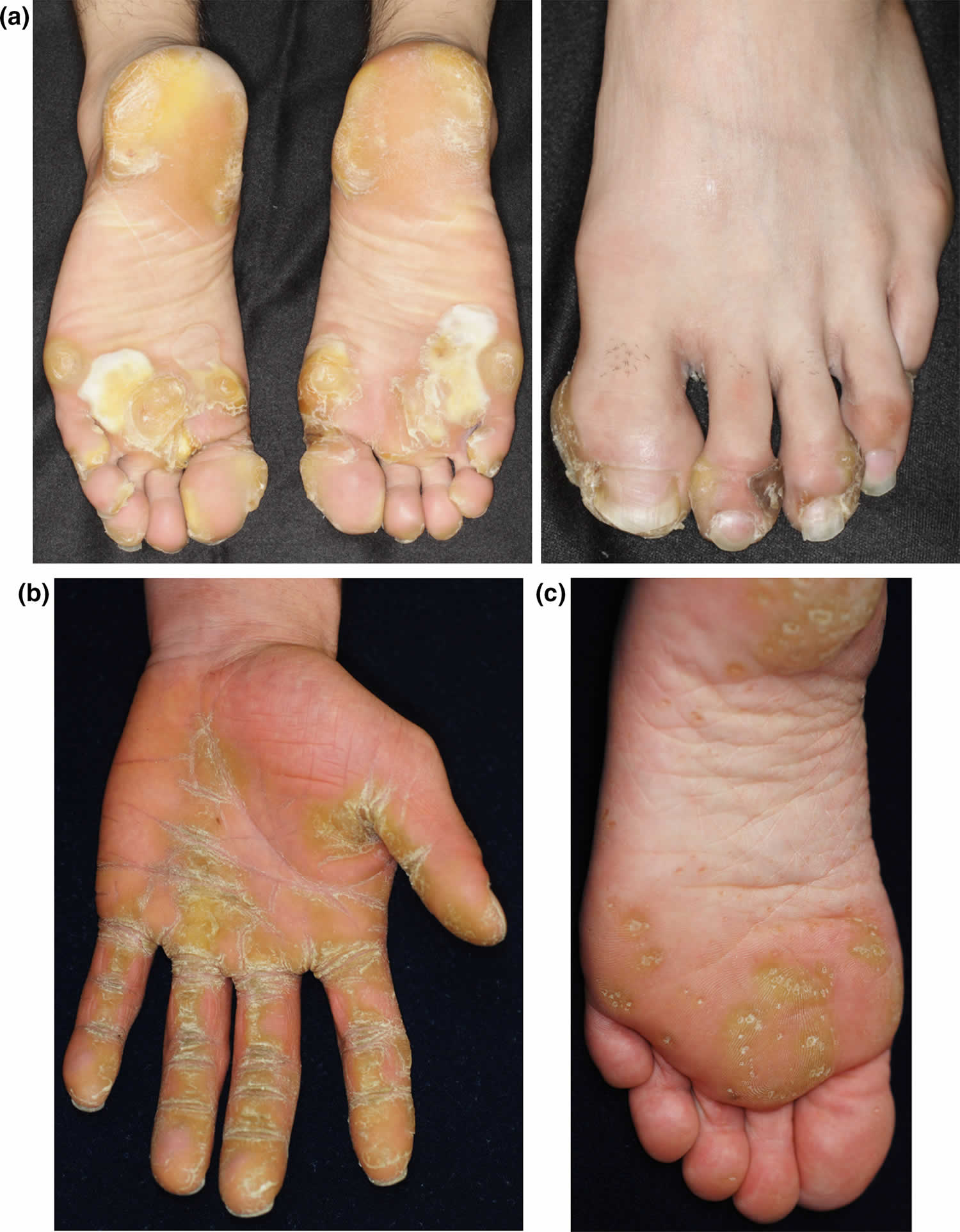
Footnote: (a) Focal palmoplantar keratoderma, caused by a KRT6C mutation, showing painful circumscribed hyperkeratosis, such as calluses on the bodyweight‐loading area of the soles 2. (b) Striate palmoplantar keratoderma, caused by a DSG1 mutation, showing skin thickening, with a prominent linear pattern along the flexor aspects of the fingers 3. (c) Buschke–Fischer–Brauer palmoplantar keratoderma, caused by an AAGAB mutation, showing multiple tiny punctate keratoses on the palms and soles 4.
[Source 1 ]Diffuse hereditary palmoplantar keratodermas
- Unna-Thost type (autosomal dominant)
- Vorner’s type (autosomal dominant)
- Mal de Meleda type (autosomal dominant or recessive)
- Huriez syndrome (autosomal dominant)
- Olmsted syndrome (unknown inheritance pattern)
- Vohwinkel syndrome (autosomal dominant)
- Palmoplantar keratoderma with sensorineural deafness (mitochondrial inheritance)
- Bart-Pumphrey syndrome (autosomal dominant)
- Hidrotic ectodermal dysplasia (autosomal dominant)
- Papillon-Lefevre syndrome (autosomal recessive)
- Diffuse palmoplantar keratoderma with woolly hair and arrythmogenic cardiomyopathy (autosomal recessive)
Focal hereditary palmoplantar keratodermas
- Palmoplantar keratoderma striata/areata type (autosomal dominant)
- Hereditary painful callosities (autosomal dominant)
- Howel-Evans syndrome or tylosis (autosomal dominant)
- Richner-Hanhart syndrome (autosomal recessive)
- Pachyonychia congenita (autosomal dominant)
- Striate palmoplantar keratoderma with woolly hair and dilated cardiomyopathy (autosomal recessive)
Punctate palmoplantar keratodermas
- Punctate keratoderma (autosomal dominant)
- Filiform keratoderma (autosomal dominant)
- Marginal keratoderma (acrokeratoelastoidosis, autosomal dominant)
Acquired palmoplantar keratodermas
Acquired palmoplantar keratodermas may be focal or diffuse. They may arise in association with a variety of different skin and internal conditions.
- An inflammatory skin condition (eg, eczema or psoriasis)
- Infections
- Medications and toxins
- An internal cancer
- A systemic inflammatory disease
- Circulation problems
- Sun damage.
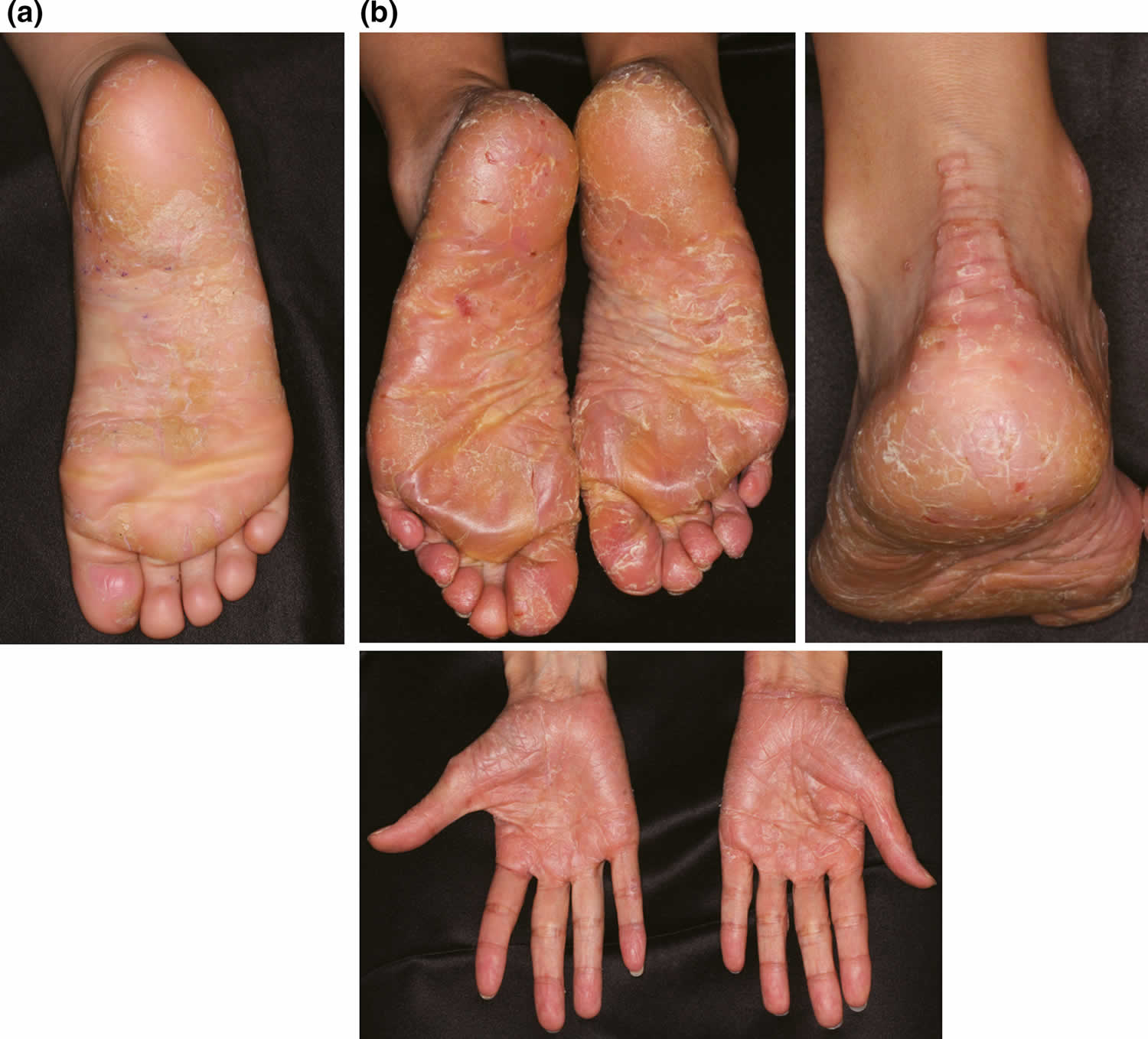
In the clinic, acquired palmoplantar keratoderma are important in the differential diagnoses of hereditary palmoplantar keratoderma, especially for hereditary palmoplantar keratoderma without associated features. Acquired palmoplantar keratoderma usually occur later in life and may be due to many causes, such as keratoderma climactericum, drugs, malnutrition, chemicals, systemic disease, malignancy, dermatoses, infections and idiopathies 5. For example, acquired palmoplantar keratoderma due to contact dermatitis (Figure 2a) and psoriasis vulgaris confined to the palmoplantar area (Figure 2b) are comparatively common and are sometimes difficult to distinguish from hereditary palmoplantar keratoderma. A skin biopsy is essential in diagnosing these cases. Lack of a family history is not necessarily evidence of an acquired palmoplantar keratoderma, because autosomal recessive palmoplantar keratoderma can appear sporadically from parent carriers and because autosomal dominant palmoplantar keratoderma can also occur sporadically by de novo mutations.
Figure 2. Acquired palmoplantar keratoderma
Palmoplantar keratoderma causes
Palmoplantar keratodermas may be inherited (hereditary) or more commonly, acquired.
- Hereditary palmoplantar keratoderma: the condition runs in families and is passed down or from one or both parent(s) to their children
- Acquired palmoplantar keratoderma: the condition is not inherited and occurs as a result of a change in the health or the environment of the affected person
The hereditary palmoplantar keratodermas are caused by a gene abnormality that results in abnormal skin protein (keratin). They may be inherited either by an autosomal dominant or autosomal recessive pattern.
- Autosomal dominant palmoplantar keratodermas are likely to occur in every generation of a family. If one parent is affected there is a 50% chance that each child will be affected.
- Autosomal recessive palmoplantar keratodermas occur less commonly within an affected family. This is because both parents need to pass on an abnormal gene to the child for it to be affected. People with one affected gene only do not have the condition themselves but carry the abnormal gene and are referred to as ‘carriers’ of the disease. They may pass on the abnormal gene to their children but the children will only be affected if their other parent also carries an abnormal gene and passes it on to the same child.
Figure 3. Palmoplantar keratodermas autosomal dominant inheritance pattern
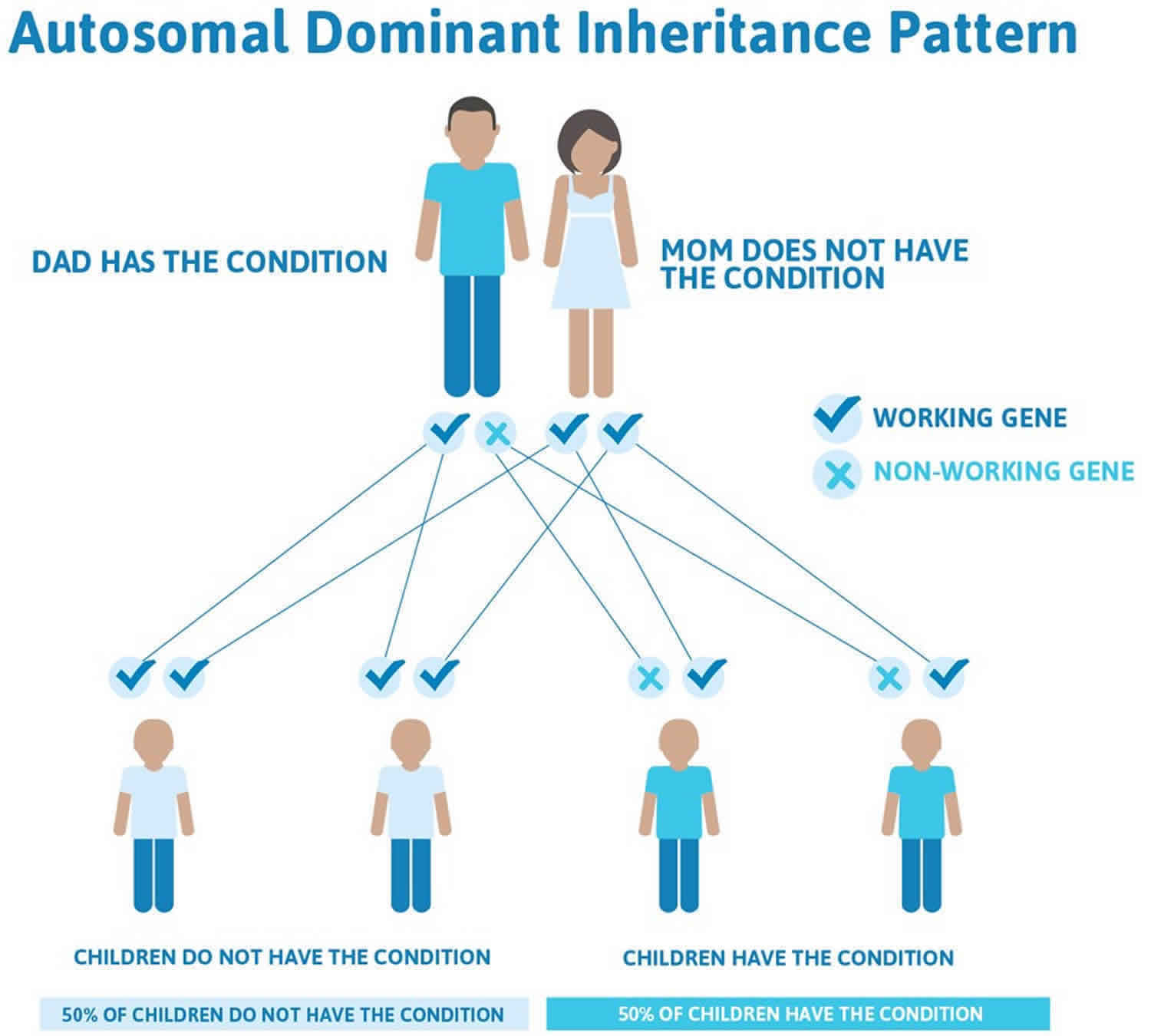
Figure 4. Palmoplantar keratodermas autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Palmoplantar keratoderma symptoms
Focal palmoplantar keratoderma
Painful circumscribed hyperkeratosis, like calluses, on the bodyweight‐loading area of the soles is characteristic of focal palmoplantar keratoderma (Figure 1a). Focal palmoplantar keratoderma is known for its major symptom of pachyonychia congenita, caused by heterozygous mutations of KRT6A, KRT6B, KRT16 or KRT17 6. While hypertrophic nail dystrophy and several ectodermal features are complicated in pachyonychia congenita patients, specific mutations in KRT16 cause focal palmoplantar keratoderma with or without minimal changes in the nails and other ectodermal tissues 7. Mutations in KRT6C and a specific mutation in DSG1 also cause focal palmoplantar keratoderma with or without minimal changes in the nails and other ectodermal tissues 8.
In 2015, a gain‐of‐function mutation in TRPV3 was identified as the cause of focal palmoplantar keratoderma in a Chinese family by whole‐genome sequencing 9. TRPV3 belongs to a large family of calcium‐permeable transient receptor potential ion channel membrane proteins, and the mutation in TRPV3 was suggested to disrupt the balance between keratinocyte proliferation and differentiation 9. Several mutations in TRPV3 have also been reported as the cause of Olmsted syndrome, which shows mutilating diffuse palmoplantar keratoderma and perioral hyperkeratotic plaques, supporting the importance of TRPV3 activity in keratinocyte differentiation 9.
Striate palmoplantar keratoderma
Striate palmoplantar keratoderma shows skin thickening, which is prominent in a linear pattern along the flexor aspects of the fingers and over pressure points on the soles (Figure 1b). It shows autosomal dominant inheritance and is divided into three types according to the responsible gene: DSG1, DSP and KRT1 10.
Heterozygous mutations in DSG1 cause striate, diffuse and focal palmoplantar keratoderma 11. DSG1 is a major component of desmosomes in the upper layer of the epidermis, and loss of DSG1 expression on the cell membrane leads to weakened intercellular adhesion. Thus, histopathological features of palmoplantar keratoderma with DSG1 mutations show characteristic clues of varying degrees of intercellular space enlargement and partial separation of keratinocytes in the spinous and granular cell layers 12.
In 2013, Samuelov et al. 13 reported that patients who had homozygous DSG1 mutations causing striate palmoplantar keratoderma via consanguineous marriage revealed a new syndrome, comprising severe dermatitis, multiple allergies and metabolic wasting (SAM) syndrome. The clinical manifestations were congenital ichthyosiform erythroderma, focal and striate palmoplantar keratoderma, and hypotrichosis. They had multiple food allergies, elevated immunoglobulin E levels and recurrent infections with malabsorption. Regarding the pathoetiology, the lack of membrane expression of DSG1 may result in a compromised epidermal barrier, resulting in sensitization to multiple environmental allergens. SAM syndrome belongs to a group of diseases, including Netherton syndrome and peeling skin syndrome caused by corneodesmosin deficiency, in which congenital insufficiency of the stratum corneum barrier causes erythroderma and multiple allergies. Recently, milder cases of SAM syndrome were also reported with palmoplantar keratoderma, dermatitis and multiple allergies but without hypotrichosis, metabolic wasting and severe recurrent infection 14.
Punctate palmoplantar keratoderma
Punctate palmoplantar keratoderma is clinically classified into three groups 15:
- Buschke–Fischer–Brauer type
- Porokeratotic type
- Acrokeratoelastoidosis.
All show autosomal dominant inheritance, whereas the clinical manifestations of each are different and characteristic. Buschke–Fischer–Brauer type palmoplantar keratoderma shows multiple tiny punctate keratoses on the palms and soles (Figure 1c) 16. It appears during late childhood to adolescence, and the lesions increase in number with advancing age and form larger lesions. Heterozygous mutations in AAGAB, encoding α‐ and γ‐adaptin‐binding protein p34, and COL14A1, were identified as causes of Buschke–Fischer–Brauer palmoplantar keratoderma 17. Porokeratotic‐type palmoplantar keratoderma shows tiny keratotic spines on the palms and soles, beginning during an individual’s early 20s, and histological examination reveals columnar parakeratosis 15. Acrokeratoelastoidosis is characterized by small keratotic papules that primarily involve the margins of the hands and feet, appearing during adolescence or adult life, and histological findings reveal degeneration of elastic fibers 18. The gene(s) responsible for porokeratotic‐type palmoplantar keratoderma and acrokeratoelastoidosis remain unknown.
Diffuse palmoplantar keratoderma without transgrediens
Diffuse palmoplantar keratoderma shows diffuse hyperkeratosis over the palms and soles. One of the key points for a clinical diagnosis is the presence or absence of transgrediens, an expansion of the disease phenotype to the dorsal surfaces of the hands and feet, inner wrists and the Achilles tendon area. Diffuse palmoplantar keratoderma without transgrediens include Vörner palmoplantar keratoderma and diffuse palmoplantar keratoderma caused by heterozygous distinct mutation of DSG1 19. Both are autosomal dominant traits but can be distinguished by histological findings and the responsible genes. Vörner palmoplantar keratoderma is diffuse palmoplantar keratoderma (Figure 5) caused by mutations in KRT1 or KRT9 and histologically shows epidermolytic hyperkeratosis 20. Unna–Thost palmoplantar keratoderma was recognized as a non‐epidermolytic form of diffuse palmoplantar keratoderma that resembles Vörner palmoplantar keratoderma clinically. However, Küster et al. 21 investigated the offspring of the original family diagnosed by Unna and Thost and found epidermolytic hyperkeratosis by histology. Genetic testing revealed the p.R162W mutation in KRT9 in the original family evaluated by Unna and Thost and the p.N160I mutation in KRT9 in the original family evaluated by Vörner, and both mutations are located on the coil‐1A segment at the beginning of the central rod domain of KRT9 20. Thus, it seems likely that Unna–Thost palmoplantar keratoderma and Vörner palmoplantar keratoderma are the same entity.
Diffuse palmoplantar keratoderma caused by DSG1 mutations show enlarged intercellular spaces and partial separation of keratinocytes in spinous and granular cell layers on histology.3, 8 Mutations in DSG1 show clinical heterogeneity; striate palmoplantar keratoderma, diffuse palmoplantar keratoderma, focal palmoplantar keratoderma and SAM syndrome, described below, are all caused by different mutations of DSG1 13.
Figure 5. Vorner palmoplantar keratoderma
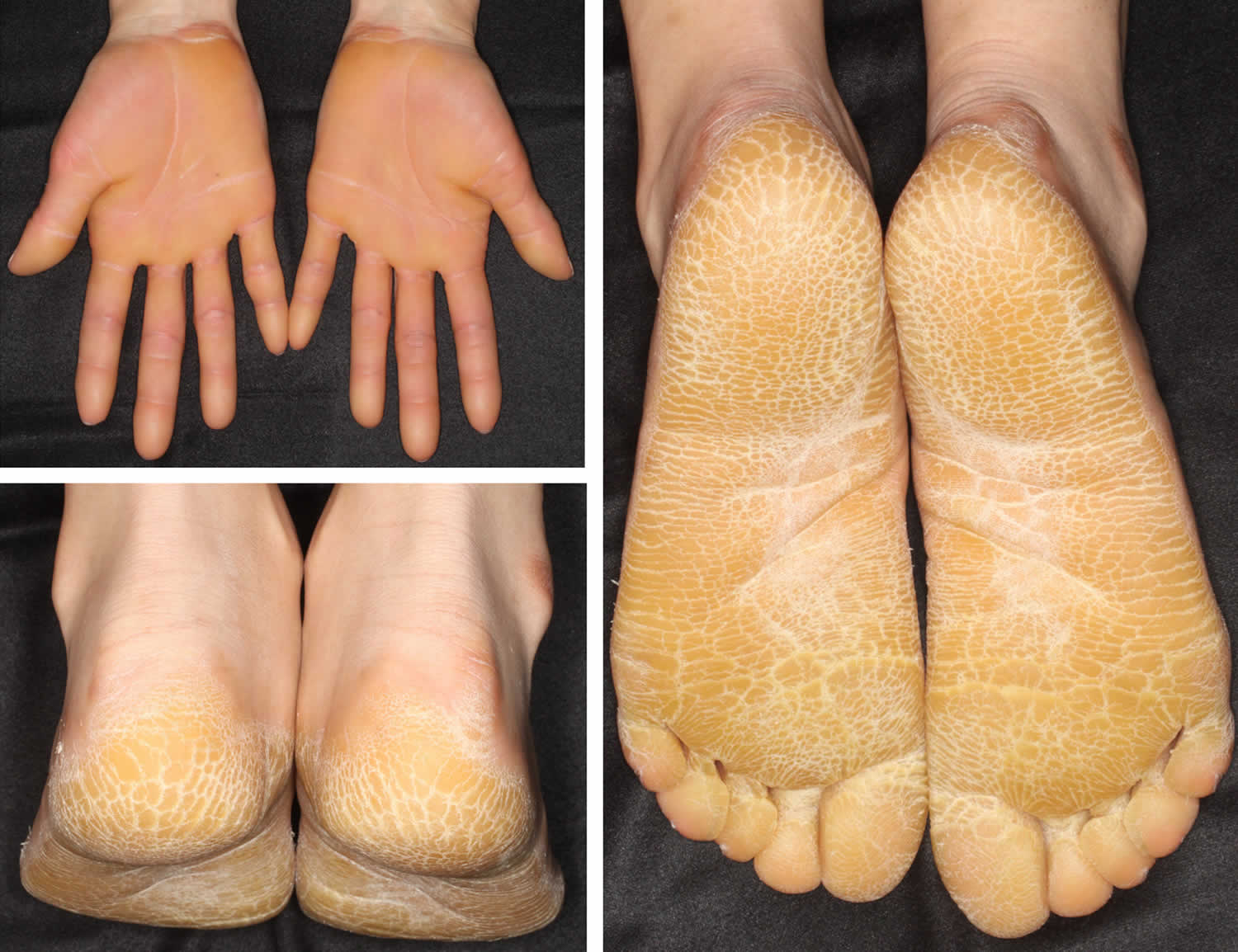
Footnote: Vörner palmoplantar keratoderma showing diffuse hyperkeratosis without transgrediens.
Diffuse palmoplantar keratoderma with transgrediens
Diffuse palmoplantar keratoderma with transgrediens include several diseases: mal de Meleda, acral keratoderma, and palmoplantar keratoderma of Nagashima, Bothnian, Gamborg‐Nielsen, Greither and Sybert 22. Among these, Nagashima palmoplantar keratoderma and Bothnian palmoplantar keratoderma are specifically characterized by a whitish spongy change in palmoplantar hyperkeratotic skin upon water exposure 23.
Nagashima palmoplantar keratoderma shows autosomal recessive behavior and is the most common palmoplantar keratoderma in Japan and China, estimated at 1.2/10 000 and 3.1/10 000, respectively, according to a cohort study 23. Nagashima palmoplantar keratoderma was first described as a palmoplantar keratoderma showing a similar distribution to, but a considerably milder disease phenotype than, mal de Meleda by Masaji Nagashima in the Japanese published work in 1977 24. Mal de Meleda is the most severe and progressive hyperkeratosis among the diffuse palmoplantar keratoderma and leads to constricting bands, spontaneous amputation and/or flexion contractures (Figure 6) 25. Nagashima palmoplantar keratoderma shows a non‐progressive, mild hyperkeratosis with skin redness and does not show constricting bands, spontaneous amputation or flexion contractures (Figure 7a) 22. In 2013, Kubo et al. 23 identified, by whole‐exome sequencing, biallelic loss‐of‐function mutations in SERPINB7, which encodes a cytoplasmic member of the serine protease inhibitor superfamily, as a cause of Nagashima palmoplantar keratoderma, confirming that Nagashima palmoplantar keratoderma is genetically distinct from mal de Meleda, which is caused by biallelic mutations in SLURP1 26. The whitish spongy change upon water exposure in Nagashima palmoplantar keratoderma has been suggested to result from enhanced water permeation into the stratum corneum, damaged by overactivated proteases due to the lack of SERPINB7 (Figure 7b) 23.
Bothnian palmoplantar keratoderma was first described in 1994 as an autosomal dominant form of diffuse non‐epidermolytic palmoplantar keratoderma, which has a high prevalence of 0.3–0.55% in the two northernmost provinces of Sweden, situated to the west and northwest of the Gulf of Bothnia 27. Bothnian palmoplantar keratoderma resembles Nagashima palmoplantar keratoderma in terms of its mild hyperkeratosis with transgrediens and the whitish spongy changes upon water exposure (Figure 8) 27. In 2013, Blaydon et al. 28 identified heterozygous missense mutations in AQP5, encoding water‐channel protein AQP5, in affected members of seven Swedish families, three British families and one Scottish family. It was suggested from protein modeling that the amino acid substitution found in Bothnian palmoplantar keratoderma may increase the diameter of the constriction point of the water channel of AQP5.24 Thus, the Bothnian palmoplantar keratoderma sequence variants could have a direct influence on AQP5 gating and/or water flow through the channel 28.
Whitish spongy changes in the palms are also observed in other disease conditions, such as aquagenic keratoderma or aquagenic wrinkling of the palms 29, a rare condition characterized by excessive wrinkling, palmar edema and whitish papules after brief exposure to water. This condition has been reported mostly in patients and carriers for cystic fibrosis, caused by mutations of CFTR 29. There are several hypotheses as to the disease mechanism of aquagenic wrinkling of the palms, including dysfunction of the TRPV4 channel in the keratinocytes of cystic fibrosis patients that results in dysregulated water influx through eccrine ducts 29.
Gamborg‐Nielsen palmoplantar keratoderma shows a similar morphological phenotype to mal de Meleda but has been reported to have less severe hyperkeratosis and no nail deformities or distant keratosis, except for the knuckle pads. Thus, the two disorders have been considered to be independent diseases 30. In 2014, Zhao et al. 31 performed genetic analysis of 15 individuals with Gamborg‐Nielsen palmoplantar keratoderma from nine families. Of these, 14 were described previously by Gamborg‐Nielsen et al. and were born in the northernmost counties of Sweden 30. They found a homozygous mutation, c.43T>C, of SLURP1 in 14 individuals from the northernmost counties of Sweden and a compound heterozygous mutation, c.280T>A and c.43T>C, of SLURP1 in one individual from southern Sweden. Because the c.43T>C mutation had been reported in mal de Meleda, Gamborg‐Nielsen palmoplantar keratoderma is considered an allelic variant of mal de Meleda.26 The individual with the compound heterozygous mutation of c.280T>A and c.43T>C in SLURP1 showed whitish spongy changes upon water exposure, typically not observed in Gamborg‐Nielsen palmoplantar keratoderma or mal de Meleda 31.
Greither palmoplantar keratoderma and Sybert palmoplantar keratoderma are both autosomal dominant forms of diffuse palmoplantar keratoderma with transgrediens and progressive features with age 32. Sybert palmoplantar keratoderma shows more severe and a wider range of hyperkeratosis than does Greither palmoplantar keratoderma 32. Greither palmoplantar keratoderma is caused by a KRT1 mutation – Greither palmoplantar keratoderma, Vörner palmoplantar keratoderma, striate palmoplantar keratoderma, epidermolytic hyperkeratosis, ichthyosis histrix and cyclic ichthyosis with epidermolytic hyperkeratosis show clinical heterogeneity of KRT1 mutations – but the gene locus for Sybert palmoplantar keratoderma is unknown 33. Acral keratoderma is an autosomal recessive palmoplantar keratoderma and shows a characteristic morphology and distribution of hyperkeratosis: diffuse and striate hyperkeratosis of the palms and soles and linear hyperkeratotic lesions over the elbows, knees and Achilles tendon area 34.
Figure 6. Mal de Meleda diffuse palmoplantar keratoderma
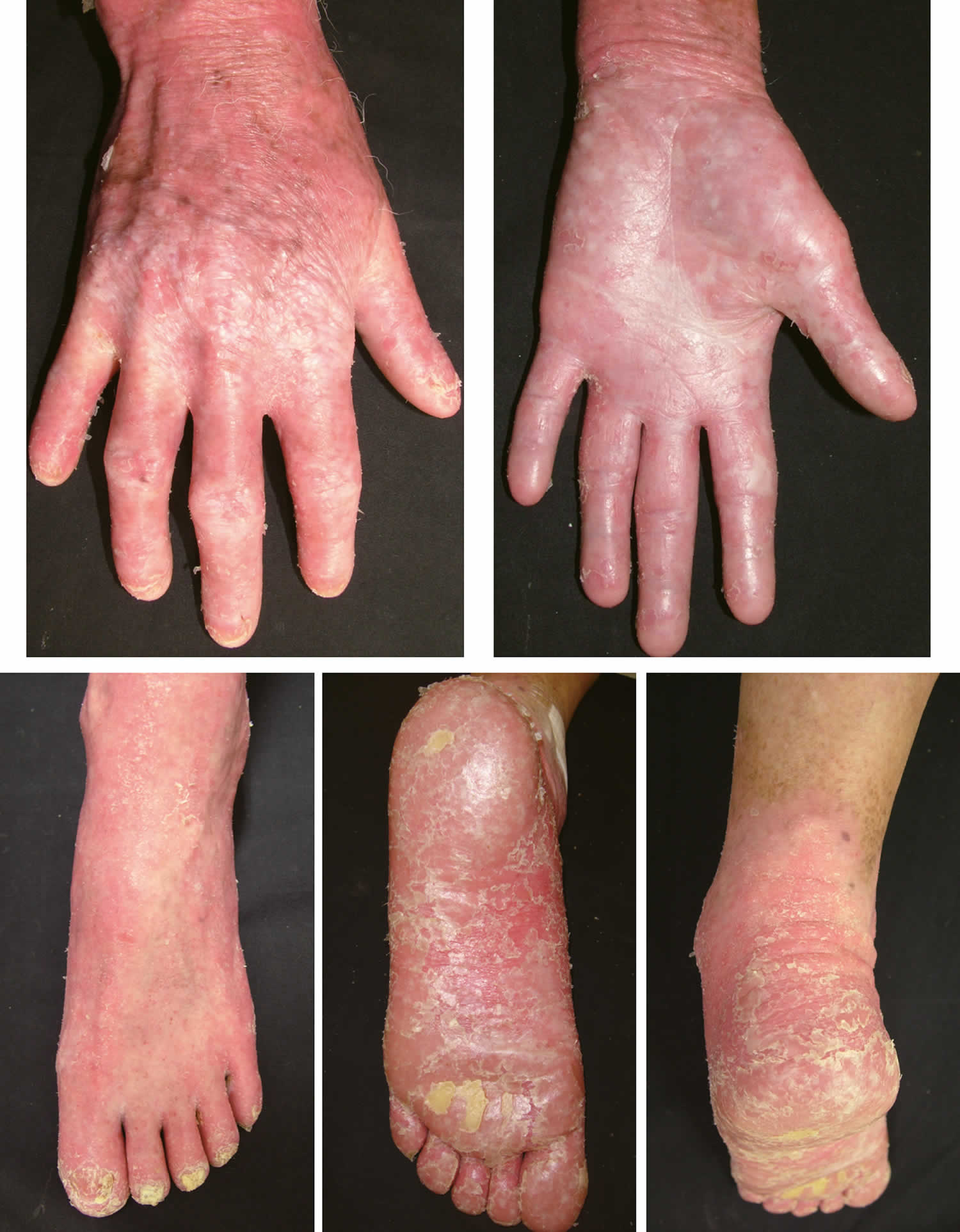
Footnote: Mal de Meleda showing the most severe hyperkeratosis with transgrediens and leading to flexion contractures.
[Source 35 ]Figure 7. Nagashima palmoplantar keratoderma
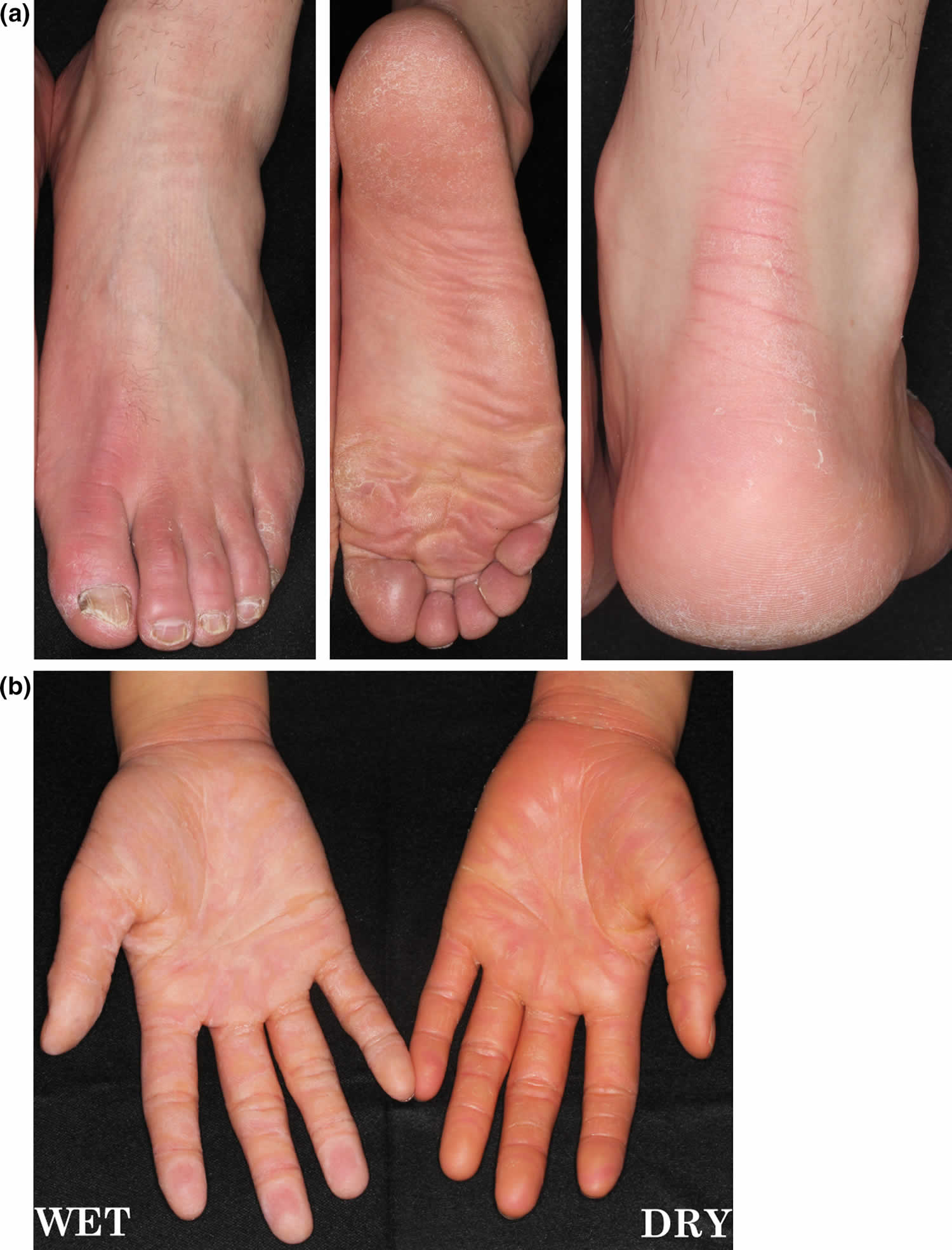
Footnote: (a) Nagashima palmoplantar keratoderma showing mild hyperkeratosis with skin redness and transgrediens. (b) Right hand showing whitish spongy changes after 5 min water exposure.
Figure 8. Bothnian palmoplantar keratoderma
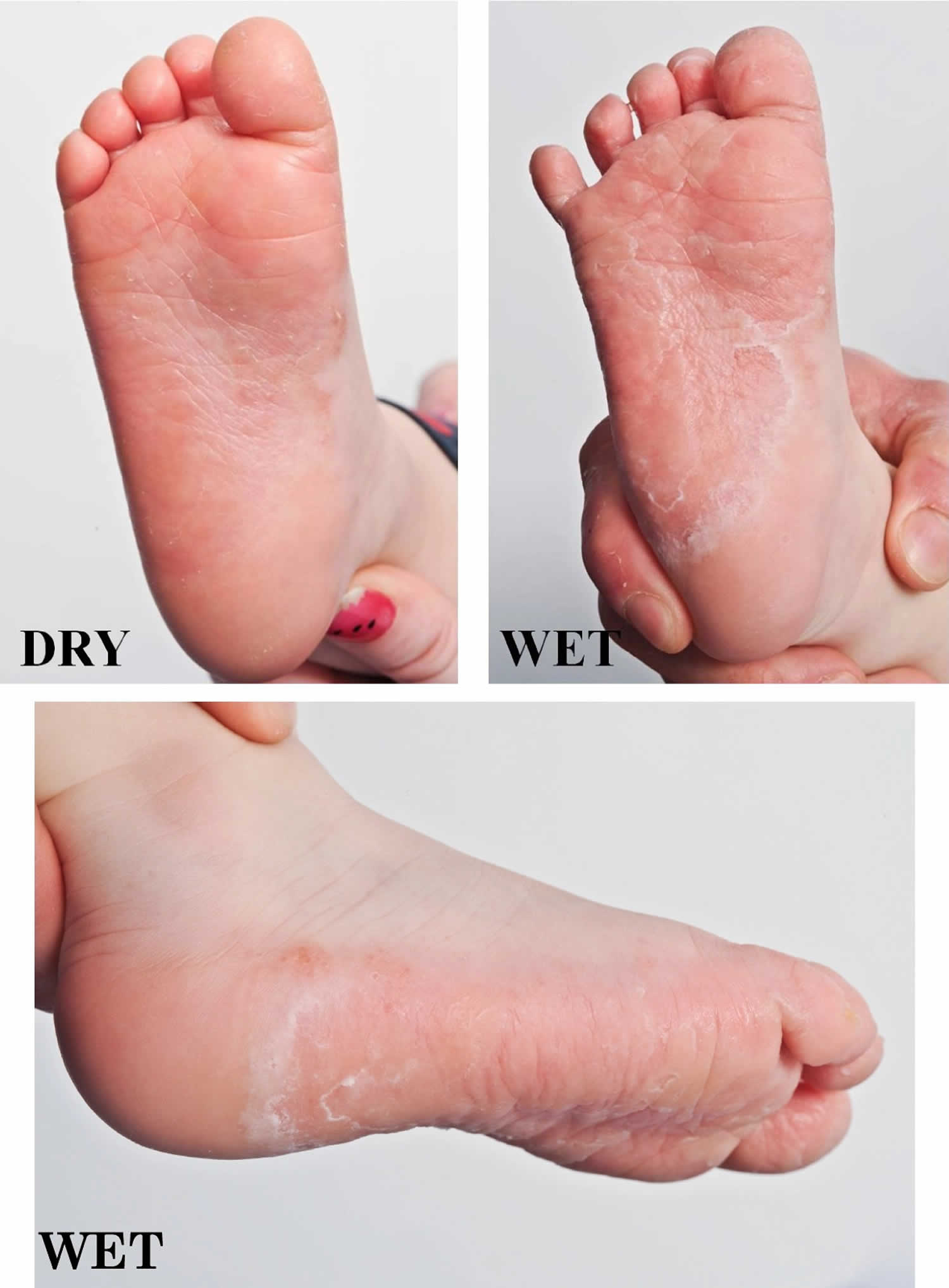
Footnote: Bothnian palmoplantar keratoderma resembles Nagashima palmoplantar keratoderma in showing mild hyperkeratosis with transgrediens and whitish spongy changes upon water exposure.
[Source 36 ]Palmoplantar keratoderma diagnosis
Careful observations, history taking, skin biopsy and genetic testing are important in diagnosing hereditary palmoplantar keratoderma.
Palmoplantar keratoderma treatment
The following treatments soften the thickened skin and makes it less noticeable.
- Emollients
- Keratolytic agents (eg, 6% salicylic acid, 70% propylene glycol, 30% water)
- Topical retinoids
- Topical vitamin D ointment (calcipotriol)
- Oral retinoids (acitretin).
- Sakiyama, T. and Kubo, A. (2016), Hereditary palmoplantar keratoderma “clinical and genetic differential diagnosis”. J Dermatol, 43: 264-274. doi:10.1111/1346-8138.13219 https://doi.org/10.1111/1346-8138.13219
- Kubo A, Oura Y, Hirano T et al. Collapse of the keratin filament network through the expression of mutant keratin 6c observed in a case of focal plantar keratoderma. J Dermatol 2013; 40: 553–557.
- Nomura T, Mizuno O, Miyauchi T et al. Striate palmoplantar keratoderma: report of a novel DSG1 mutation and atypical clinical manifestations. J Dermatol Sci 2015; pii: S0923‐1811(15)30056‐6. doi: 10.1016/j.jdermsci.2015.10.004
- Nomura T, Moriuchi R, Takeda M et al. Low‐dose etretinate shows promise in management of punctate palmoplantar keratoderma type 1: case report and review of the published work. J Dermatol 2015; 42: 889–892.
- Patel S, Zirwas M, English JC 3rd. Acquired palmoplantar keratoderma. Am J Clin Dermatol 2007; 8: 1–11.
- Eliason MJ, Leachman SA, Feng BJ et al. A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita. J Am Acad Dermatol 2012; 67: 680–686.
- Smith FJ, Fisher MP, Healy E et al. Novel keratin 16 mutations and protein expression studies in pachyonychia congenita type 1 and focal palmoplantar keratoderma. Exp Dermatol 2000; 9: 170–177.
- Wilson NJ, Messenger AG, Leachman SA et al. Keratin K6c mutations cause focal palmoplantar keratoderma. J Invest Dermatol 2010; 130: 425–429.
- He Y, Zeng K, Zhang X et al. A gain‐of‐function mutation in TRPV3 causes focal palmoplantar keratoderma in a Chinese family. J Invest Dermatol 2015; 135: 907–909.
- Whittock NV, Smith FJ, Wan H et al. Frameshift mutation in the V2 domain of human keratin 1 results in striate palmoplantar keratoderma. J Invest Dermatol 2002; 118: 838–844.
- Milingou M, Wood P, Masouye I et al. Focal palmoplantar keratoderma caused by an autosomal dominant inherited mutation in the desmoglein 1 gene. Dermatology 2006; 212: 117–122.
- Bergman R, Hershkovitz D, Fuchs D et al. Disadhesion of epidermal keratinocytes: a histologic clue to palmoplantar keratodermas caused by DSG1 mutations. J Am Acad Dermatol 2010; 62: 107–113.
- Samuelov L, Sarig O, Harmon RM et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet 2013; 45: 1244–1248.
- Schlipf NA, Vahlquist A, Teigen N et al. Whole exome sequencing identifies novel autosomal recessive DSG1 mutations associated with mild SAM syndrome. Br J Dermatol 2015; doi: 10.1111/bjd.14079.
- Brown FC. Punctate keratoderma. Arch Dermatol 1971; 104: 682–683.
- Brauer A. Über eine besondere Form des hereditären Keratoms (keratoderma disseminatum hereditarium palmare et plantare). Arch Dermatol Syph 1913; 114: 211–236.
- Giehl KA, Eckstein GN, Pasternack SM et al. Nonsense mutations in AAGAB cause punctate palmoplantar keratoderma type Buschke‐Fischer‐Brauer. Am J Hum Genet 2012; 91: 754–759.
- Costa O. Akrokerato‐elastoidosis (a hitherto underscribed skin disease). Dermatologica 1953; 107: 164–168.
- Keren H, Bergman R, Mizrachi M et al. Diffuse nonepidermolytic palmoplantar keratoderma caused by a recurrent nonsense mutation in DSG1. Arch Dermatol 2005; 141: 625–628.
- Küster W, Reis A, Hennies HC. Epidermolytic palmoplantar keratoderma of Vorner: re‐evaluation of Vorner’s original family and identification of a novel keratin 9 mutation. Arch Dermatol Res 2002; 294: 268–272.
- Küster W, Becker A. Indication for the identity of palmoplantar keratoderma type Unna‐Thost with type Vorner. Thost’s family revisited 110 years later. Acta Derm Venereol 1992; 72: 120–122.
- Kabashima K, Sakabe J, Yamada Y et al. “Nagashima‐type” keratosis as a novel entity in the palmoplantar keratoderma category. Arch Dermatol 2008; 144: 375–379.
- Kubo A, Shiohama A, Sasaki T et al. Mutations in SERPINB7, encoding a member of the serine protease inhibitor superfamily, cause Nagashima‐type palmoplantar keratosis. Am J Hum Genet 2013; 93: 945–956.
- Nagashima M. Palmoplantar Keratoses. In Handbook of Human Genetics, Volume 9. Tokyo: Igaku Shoin, 1977.
- Hovorka O, Ehlers E. Mal de Meleda. Arch Dermatol Res 1897; 40: 251–256.
- Fischer J, Bouadjar B, Heilig R et al. Mutations in the gene encoding SLURP‐1 in Mal de Meleda. Hum Mol Genet 2001; 10: 875–880.
- Lind L, Lundstrom A, Hofer PA et al. The gene for diffuse palmoplantar keratoderma of the type found in northern Sweden is localized to chromosome 12q11‐q13. Hum Mol Genet 1994; 3: 1789–1793.
- Blaydon DC, Lind LK, Plagnol V et al. Mutations in AQP5, encoding a water‐channel protein, cause autosomal‐dominant diffuse nonepidermolytic palmoplantar keratoderma. Am J Hum Genet 2013; 93: 330–335.
- Park L, Khani C, Tamburro J. Aquagenic wrinkling of the palms and the potential role for genetic testing. Pediatr Dermatol 2012; 29: 237–242.
- Kastl I, Anton‐Lamprecht I, Gamborg Nielsen P. Hereditary palmoplantar keratosis of the Gamborg Nielsen type. Clinical and ultrastructural characteristics of a new type of autosomal recessive palmoplantar keratosis. Arch Dermatol Res 1990; 282: 363–370.
- Zhao L, Vahlquist A, Virtanen M et al. Palmoplantar keratoderma of the Gamborg‐Nielsen type is caused by mutations in the SLURP1 gene and represents a variant of Mal de Meleda. Acta Derm Venereol 2014; 94: 707–710.
- Sybert VP, Dale BA, Holbrook KA. Palmar‐plantar keratoderma. A clinical, ultrastructural, and biochemical study. J Am Acad Dermatol 1988; 18: 75–86.
- Gach JE, Munro CS, Lane EB et al. Two families with Greither’s syndrome caused by a keratin 1 mutation. J Am Acad Dermatol 2005; 53: S225–S230.
- Nesbitt LT Jr, Rothschild H, Ichinose H et al. Acral keratoderma. Arch Dermatol 1975; 111: 763–768.
- Sakabe J, Kabashima‐Kubo R, Kubo A et al. A Japanese case of Mal de Meleda with SLURP1 mutation. J Dermatol 2014; 41: 764–765.
- Abdul‐Wahab A, Takeichi T, Liu L et al. Autosomal dominant diffuse non‐epidermolytic palmoplantar keratoderma due to a recurrent mutation in aquaporin‐5. Br J Dermatol 2015; doi: 10.1111/bjd.13931





{kind=link}