What is paraganglioma
Paraganglioma (also known as extra-adrenal pheochromocytoma) is a type of noncancerous (benign) neuroendocrine tumor that occurs in autonomic ganglia called paraganglia. Paraganglia are small organs that mainly consist of neuroendocrine cells derived from the embryonic neural crest that have the ability to synthesize and secrete catecholamines [adrenaline, noradrenaline and dopamine] 1. Paragangliomas can arise from any anatomical location with chromaffin-containing autonomic tissue from the base of the skull to the pelvis. Paragangliomas have been previously described in the head and neck regions, jugulotympanic area, bladder, and other locations leading to varying clinical presentations with the majority of cases commonly remaining asymptomatic 2. Paragangliomas are usually found in the head, neck, thorax and abdomen. However, a type of paraganglioma known as pheochromocytoma develops in the adrenal glands. Adrenal glands are located on top of each kidney and produce hormones in response to stress. Most people with paraganglioma develop only one tumor in their lifetime.
Pheochromocytoma is a rare neuroendocrine tumor that arises from chromaffin cells that are found in the adrenal medulla. Note the difference between the location of the chromaffin cells tumor, as defined by the World Health Organization, inside the adrenal glands chromaffin cells tumor is called pheochromocytoma (also known as intra-adrenal paraganglioma) and outside of adrenal gland chromaffin cells tumor is called paraganglioma 3. The adrenal glands make important hormones called catecholamines. Adrenaline (epinephrine) and noradrenaline (norepinephrine) are two types of catecholamines that help control heart rate, blood pressure, blood sugar, and the way the body reacts to stress. Sometimes a pheochromocytoma will release extra adrenaline and noradrenaline into the blood and cause signs or symptoms of disease.
Extra-adrenal paragangliomas, nowadays often referred to as only paragangliomas, are classified as sympathetic or parasympathetic depending on the type of paraganglia in which they have their origin. Sympathetic paragangliomas arise from chromaffin cells of paraganglia along the sympathetic chains and are usually located in the chest, abdomen, or pelvis (Figure 1). Parasympathetic paragangliomas arise from the glomera that are distributed along parasympathetic nerves in the head, neck, and upper mediastinum and are therefore also referred to as head and neck paragangliomas (Figure 2).
Pheochromocytomas and paragangliomas are rare tumors. Their prevalence is unknown but has been estimated to lie between 1:6500 and 1:2500 in the United States 4. Autopsy series have revealed a higher prevalence of about 1:2000, suggesting that many tumors remain undiagnosed 5. The annual incidence has been reported to be 2 to 10 cases per million 6. The tumors may occur in all ages but have the highest incidence between 40 and 50 years, with an approximately equal sex distribution 7. In 693 unselected pheochromocytoma/paraganglioma patients, about 69% of the patients had pheochromocytoma, 15% had sympathetic paraganglioma, and 22% had parasympathetic paraganglioma (some had a combination of tumors), providing an approximate measure of the relative incidence of the different tumor types 8.
Figure 1. Paraganglioma tumor locations
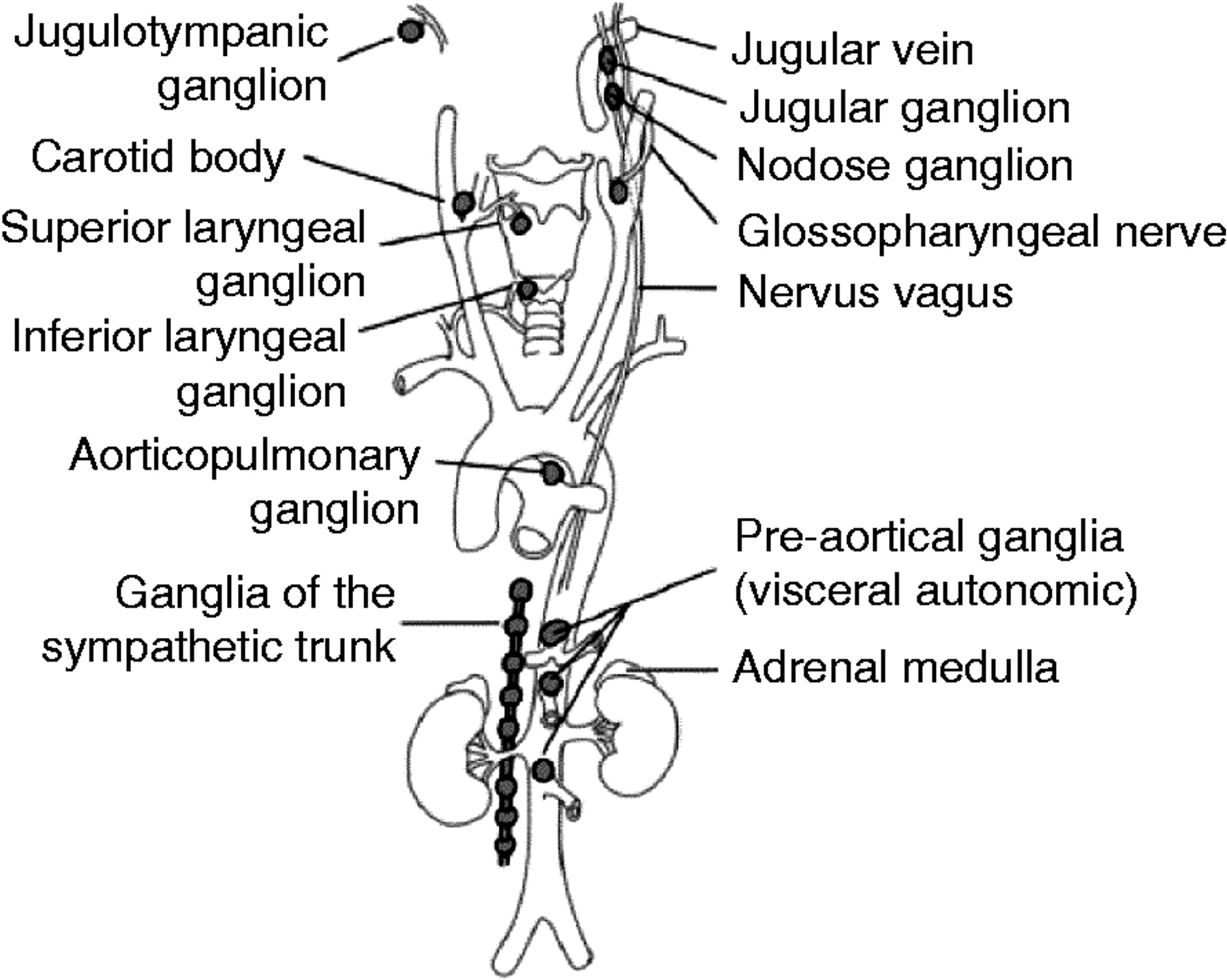 Note: Pheochromocytomas arise in the medulla of the adrenal gland, whereas sympathetic paragangliomas arise along the sympathetic chains in the pelvis, abdomen, and chest. Parasympathetic paraganglioma arise along the parasympathetic nerves in the head, neck, and mediastinum, the most common location being the carotid body.
Note: Pheochromocytomas arise in the medulla of the adrenal gland, whereas sympathetic paragangliomas arise along the sympathetic chains in the pelvis, abdomen, and chest. Parasympathetic paraganglioma arise along the parasympathetic nerves in the head, neck, and mediastinum, the most common location being the carotid body.Pheochromocytomas and sympathetic paragangliomas are very similar histologically as well as functionally 3. They generally produce large amounts of catecholamines, mainly adrenaline and noradrenaline, at rates many times higher than normal, resulting in a high concentration of these fight-or-flight response causing hormones in the bloodstream 9. The tumors usually cause hypertension, which may be either paroxysmal or sustained. Typical symptoms are recurring episodes of headache, sweating, and palpitations. Other symptoms may include anxiety, tremors, nausea, pallor, and abdominal or chest pain. Up to 10% of the patients have only minor or no signs of clinical symptoms and an increasing number of tumors are incidentally found during imaging studies 10. In other cases, the tumors can cause severe cardiovascular or neurological manifestations such as shock, heart failure, seizures, and stroke, which can become life threatening and also obstruct a correct diagnosis 11.
Parasympathetic paragangliomas are histologically similar to pheochromocytomas and sympathetic paragangliomas 1, but whereas the latter two tumor forms are almost always clinically functional, parasympathetic paragangliomas are usually not 3. They typically have no or only a low production of catecholamines 12 and commonly present as a slow-growing, painless cellular mass 3. Consequently, many patients are non-symptomatic. However, depending on site, the space occupation by the tumors may cause symptoms such as pain, hearing disturbances, hoarseness, and dysphagia.
The majority of pheochromocytomas and paragangliomas are benign. Malignancy is defined as the presence of distant metastases 3 and occurs in ∼5–13% of pheochromocytomas 7, 15–23% of sympathetic paragangliomas 7, and 2–20% (depending on site) of parasympathetic paragangliomas 7. The most common sites for metastasis are bone, liver, and lung tissue 13. Currently, malignancy cannot be predicted with certainty, although some histological or gene expression features might be suggestive of malignancy 14. The prognosis of malignant PCC and paraganglioma is poor, with a 5-year mortality rate >50% 13. There is currently no effective or curative treatment, but surgery, chemotherapy, and radiotherapy are beneficial in some patients.
The majority of paragangliomas are sporadic which means paragangliomas and pheochromocytomas occur in people with no history of the tumors in their families and appear not to be inherited. In about 25 percent of people develop a paraganglioma or pheochromocytoma as part of a hereditary syndrome that may affect other organs and tissues in the body 15. Researchers have identified several types of hereditary paraganglioma-pheochromocytoma. Each type is distinguished by its genetic cause. People with types 1, 2, and 3 typically develop paragangliomas in the head or neck region. People with type 4 usually develop extra-adrenal paragangliomas in the abdomen and are at higher risk for malignant tumors that metastasize. The other types are very rare. Hereditary paraganglioma-pheochromocytoma is typically diagnosed in a person’s 30s. However, the tumors often are not associated with any syndromes, in which case the condition is called nonsyndromic paraganglioma or pheochromocytoma.
Paragangliomas and pheochromocytomas can occur in individuals with certain inherited disorders, such as von-Hippel Lindau syndrome, Carney-Stratakis syndrome, and certain types of multiple endocrine neoplasia (MEN). These other disorders feature additional tumor types and have different genetic causes. Some . These cases are designated as sporadic.
Paragangliomas and pheochromocytomas can also occur in the following two other very rare syndromes:
- The Carney triad of extra-adrenal paraganglioma, gastrointestinal stromal tumor 16 and pulmonary chondroma.
- The Carney-Stratakis dyad of paraganglioma and gastrointestinal stromal tumor 17.
Other genetic causes of pheochromocytoma and paraganglioma are being studied. For example, truncating germline mutations in the transmembrane-encoding gene TMEM127 on chromosome 2q11 have been shown to be present in approximately 30% of affected patients with familial disease and in about 3% of patients with apparently sporadic pheochromocytomas without a known genetic cause 18. TMEM127 is a negative regulator of mammalian target of rapamycin (mTOR) effector proteins.
Pheochromocytomas and paragangliomas arise from neural crest tissue. Neural crest tissue develops into sympathetic and parasympathetic paraganglia.
Sympathetic paraganglia include the following:
- The adrenal medulla.
- The organ of Zuckerkandl near the aortic bifurcation.
- Other paraganglia along the distribution of the sympathetic nervous system.
Parasympathetic paraganglia include the following:
- The carotid body.
- Other paraganglia along the cervical and thoracic branches of the vagus and glossopharyngeal nerves.
The sympathetic nervous system controls the “fight-or-flight” response, a series of changes in the body due to hormones released in response to stress. Although most sympathetic paragangliomas are pheochromocytomas, some are found outside the adrenal glands, usually in the abdomen, and are called extra-adrenal paragangliomas. Most sympathetic paragangliomas, including pheochromocytomas, produce hormones called catecholamines, such as epinephrine (adrenaline), norepinephrine (noradrenaline) and dopamine. These excess catecholamines can cause signs and symptoms such as high blood pressure (hypertension), episodes of rapid heartbeat (palpitations), headaches, or sweating.
Most paragangliomas are associated with ganglia of the parasympathetic nervous system, which controls involuntary body functions such as digestion and saliva formation. Parasympathetic paragangliomas, typically found in the head and neck, usually do not produce hormones. However, large tumors may cause signs and symptoms such as coughing, hearing loss in one ear, or difficulty swallowing.
Although most paragangliomas and pheochromocytomas are noncancerous, some can become cancerous (malignant) and spread to other parts of the body (metastasize). Extra-adrenal paragangliomas become malignant more often than other types of paraganglioma or pheochromocytoma.
Paraganglioma tumor can affect people of any age but most often shows up between the ages of 30 and 50. Paraganglioma tumor is often slow growing and noncancerous (benign). But paraganglioma can invade nearby parts of the body, become cancerous (malignant) and spread distantly (metastasize).
With about half of paraganglioma tumors, the abnormal cells produce hormones known as catecholamines or adrenaline, which is the fight-or-flight hormone. This may induce high blood pressure, a rapid heartbeat, flushed skin, sweating, headache and tremors.
Surgery to remove paraganglioma tumor is usually the first treatment choice for a paraganglioma, if feasible. If left untreated, a paraganglioma can result in severe or life-threatening damage and progress to the point where surgical treatment isn’t an option. In people with paraganglioma cancer and distantly spread metastatic paraganglioma, medicine and other treatments can help control the disease and symptoms and even extend survival.
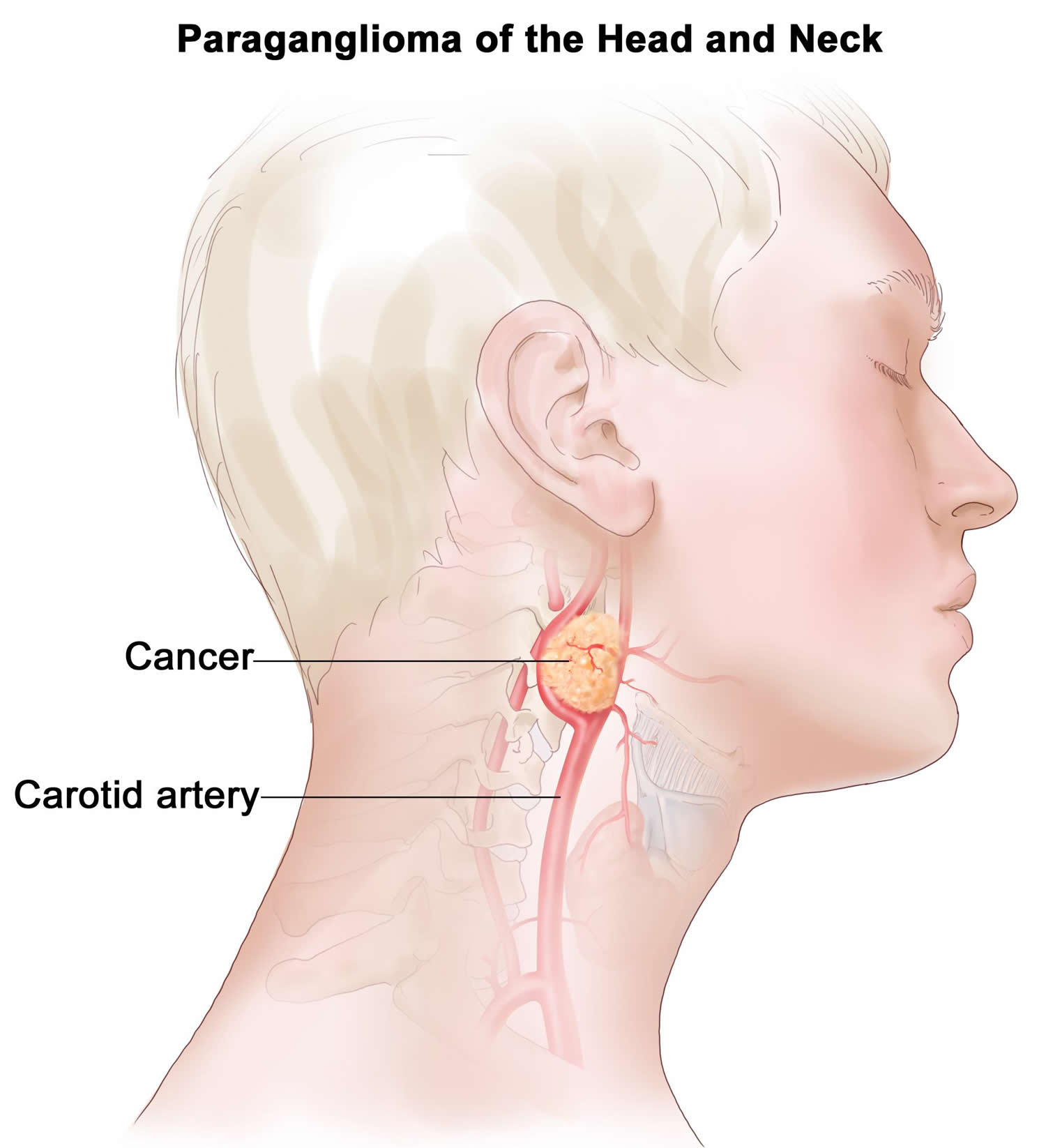
Paraganglioma neck and head
Head and neck paragangliomas often referred to as glomus tumors, are rare neuroendocrine tumors, originating from paraganglia associated with automic branches of the lower cranial nerves 19. Head and neck paragangliomas account for about 0.6% of all head and neck tumors and usually present between the 4th and 6th decades of life. Head and neck paragangliomas mostly arise from paraganglia at the carotid bifurcation, in or around the jugular bulb, in the cervical tract of the vagus, or within the temporal bone. Head and neck paragangliomas are generally slow-growing, but they infiltrate vascular structures and anatomically complex regions of the skull base. A high percentage of head and neck paragangliomas (over 30%) arises on the basis of genetic predisposition 19. Germline mutations in the succinate-ubiquinone oxidoreductase (succinate dehydrogenase, SDH) subunits are the most frequently involved in head and neck paraganglioma predisposition. The corresponding genes encode subunits (SDHA, SDHB, SDHC, SDHD) or cofactors (SDHAF2) of Complex II succinate dehydrogenase. This complex of the mitochondrial respiratory chain is responsible for the enzymatic oxidation of succinate to fumarate in the tricarboxylic acid cycle and for electron transport to coenzyme Q 20.
Head and neck paragangliomas range in spectrum from small lesions (usually detected in paraganglioma mutation carriers) to large unresectable masses, often invading cranial nerves and sometimes the brainstem. Head and neck paragangliomas are almost always nonsecreting tumors (up to 95%), and thus, they are often discovered on imaging studies or revealed by symptoms of cervical mass and/or compression or infiltration of adjacent structures (eg, hearing loss, tinnitus, cervical mass, dysphagia, and cranial nerve palsies) 21.
At the present time, there are no reliable cytological, histological, immunohistochemical, or molecular criteria for malignancy 22. The diagnosis of malignancy remains strictly based on the finding of tumor loci where paraganglial cells are not usually present, such as the lymph nodes, lung, bone, or liver. In the absence of metastases at initial presentation, the diagnosis can only be made when metastases are discovered, which may often be too late if a patient did not undergo appropriate follow-up or if treatment options failed.
Head and neck paragangliomas are often nonsecreting tumors (up to 70%–95% based on whether methoxytyramine or metanephrines are measured) and thus often discovered on imaging studies or revealed by the presence of cervical/skull base masses with or without compression or infiltration of adjacent structures (eg, hearing loss, tinnitus, cervical mass, dysphagia, and cranial nerve palsies). In nonsecreting cases, accurate diagnosis of an head and neck paraganglioma is made by thorough clinical examination together with MRI and paraganglioma-specific functional imaging studies 23.
At present, surgery is the only therapeutic option, but in some case radical removal may be difficult or impossible, depending on location and stage of progression 24. When surgical eradication is not possible, radio- and chemotherapy are used, but only partial responses are observed even with targeted radiotherapy 25.
Figure 2. Paraganglioma neck
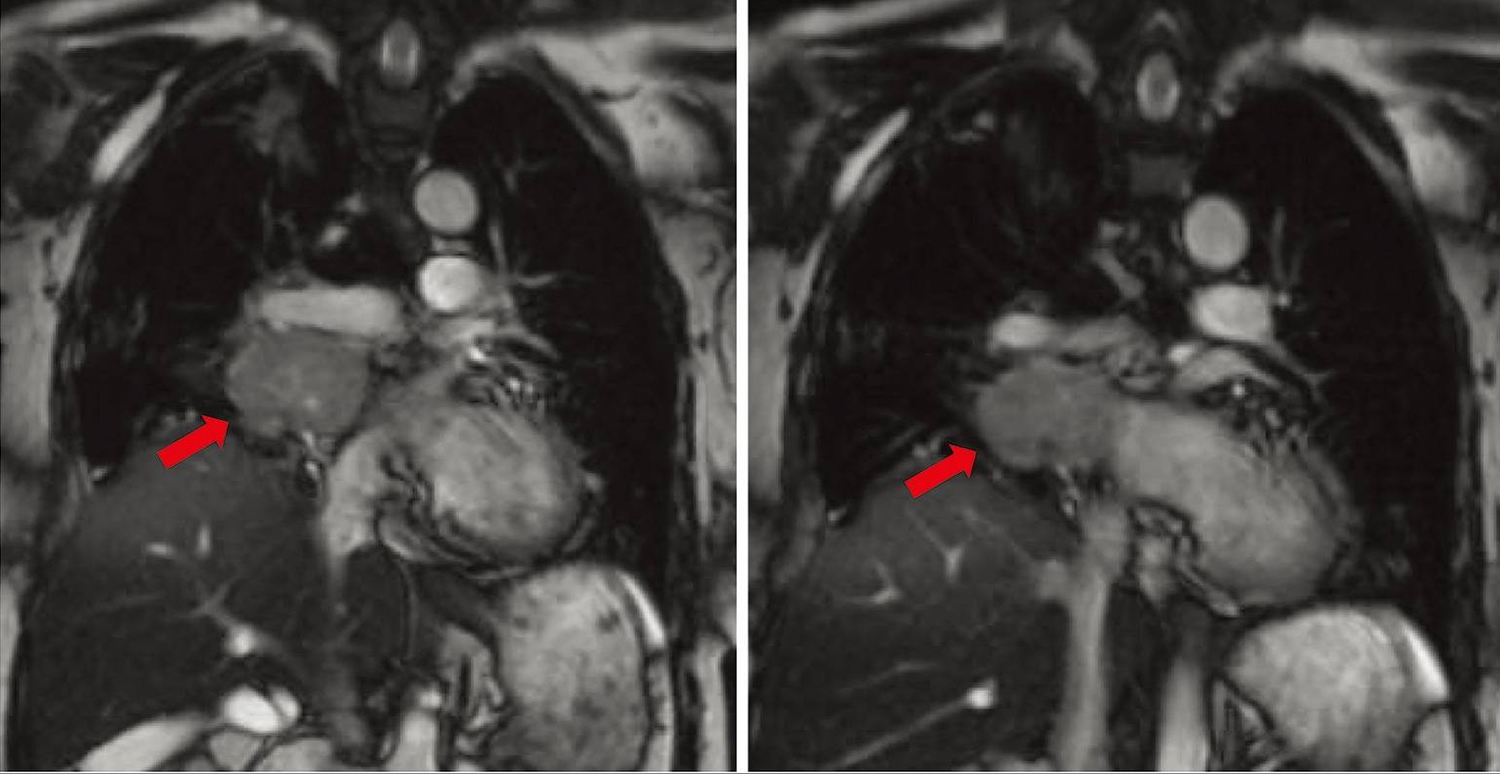
Cardiac paraganglioma
Only 2% of all chromaffin cell tumors occur as thoracic paraganglioma 26. Thoracic paraganglioma can be in the posterior mediastinum arising from para-aortic sympathetic ganglia or less commonly in the middle mediastinum, arising from cardiac structures themselves, which are appropriately designated as cardiac paraganglioma. Cardiac paraganglioma is an example of rare paraganglioma in that it specifically originated from a cardiac structure, with such tumors being reported to occur in the thorax in only 1–2% of all paraganglioma 27.
Cardiac paraganglioma usually originate from the left atrium and less commonly arise from the aortic root. Rarer still, very few cardiac paragangliomas have been reported to arise from other cardiac chambers such as the right atrium or left ventricle 28.
The clinical presentation of cardiac paraganglioma differs based on the functional status of the tumor. Presenting symptoms of functional cardiac paraganglioma are well reported with key symptoms such as hypertension, palpitations, headache, and sweating being most prevalent 29. Non-functional paraganglioma are much more subtle in clinical presentation and are typically asymptomatic until the tumor compromises cardiac function due to local mass effect. Several reported cases have illustrated this effect, describing tumors obstructing the left-ventricular outflow tract or even directly occluding coronary artery blood flow 30. In these instances, presenting symptoms included chest pain, dyspnea on exertion, fatigue, and cardiac murmur on physical exam. The tumors were then detected on subsequent cardiac angiography or other imaging during clinical work-up of these symptoms 31. Local mass effect from tumors on nearby respiratory system structures may also cause pulmonary symptoms such as cough, hemoptysis, and stridor. Similarly, mass effect on the esophagus may present as dysphagia.
Cardiac paraganglioma are typically benign and are best treated by surgical resection. This is most often achieved through median sternotomy with cardiopulmonary bypass. However, surgical excision of these tumors is challenging due to their intimate connection with surrounding vital structures, hypervascularity with increased risk of severe hemorrhage, and difficulty in accessing the tumor. The most common surgical approach to resect cardiac paraganglioma is median sternotomy with the use of cardiopulmonary bypass. An extensive literature review of cardiac paraganglioma from 1974–2014 by Wang et al. 32 indicated that the majority of the cases were resected through this approach. They also reported 15 cases being resected through a thoracotomy incision and only four cases not requiring the use of cardiopulmonary bypass 32. One case report indicated an attempted resection of a cardiac paraganglioma arising from the left atrium by thoracotomy, but converted to median sternotomy intraoperatively 33. Another report utilizing a thoracotomy approach resulted in severe bleeding leading to intraoperative cardiac arrest. The authors of this case recommended use of cardiopulmonary bypass as the main takeaway from their case 34. The case presented by the authors is rare in the fact that the tumor was successfully resected through a thoracotomy incision and although cardiopulmonary bypass was available on standby throughout the procedure, it was not required.
Due to the hypervascular nature of cardiac paraganglioma, some attention has been given to preoperative arterial embolization to minimize intraoperative bleeding. In our case, the patient received right internal mammary artery embolization after the first unsuccessful attempt at tumor resection. In the review of seven cardiac paragangliomas by Ramlawi et al 35, one patient received preoperative arterial embolization and the authors acknowledge the utility of preoperative arterial embolization in avoiding massive intraoperative hemorrhage from previous experience at their institution. Additionally, one other case report described successful embolization of a large vessel of the left coronary artery found to be supplying a large intrapericardial paraganglioma 36.
Surgical resection of cardiac paraganglioma is considered a high-risk procedure due to increased risk of intraoperative hemorrhage from the highly vascularized nature of these tumors and frequent extensive adhesions to surrounding vital structures. Intraoperative mortality has been reported to be 3.1% by Wang et al.’s 32 review of 158 cases of cardiac paragangliomas. An additional 2.5% mortality was reported for the immediate postoperative course. A review of individual case reports from 2014–2016, two years since Wang et al.’s review, shows no intraoperative mortality reported from 11 cases 37. Among these cases, one instance of intraoperative cardiac arrest occurred due to severe hemorrhage and one unexpected intraoperative circulatory collapse, however both ended with successful outcomes 38.
After recovery from surgery, benign cardiac paragangliomas that are completely resected with negative margins have been associated with favorable outcomes in survival with low rates of tumor recurrence 33. For those surviving the immediate post-operative course, a 1-year survival of 98.2% and 5-year survival of 78.2% has been reported 32.
Figure 3. Cardiac paraganglioma (magnetic resonance angiography [MRA] of the chest showing the location, size, and extent of right hilar tumor. It is noted to abut the right pulmonary vein and pulmonary artery. Red arrows indicate the cardiac paraganglioma tumor mass)
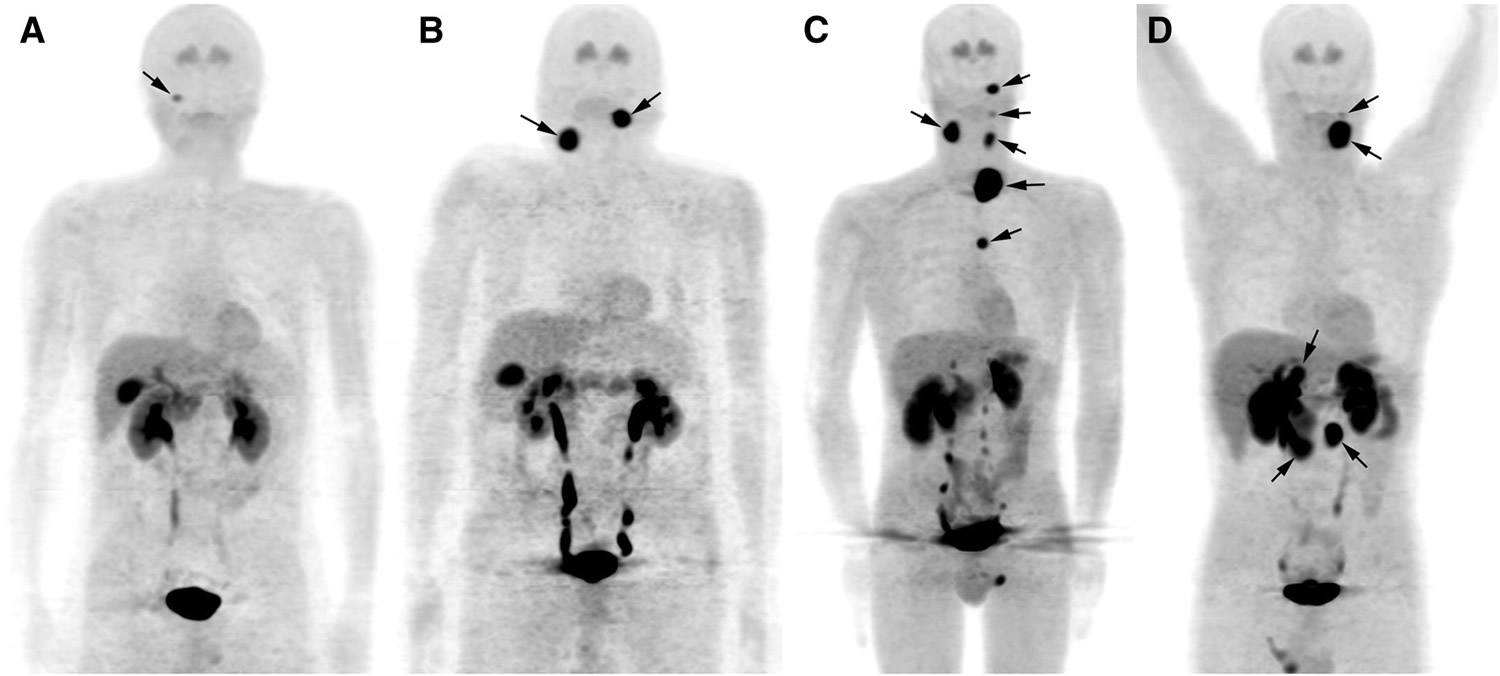
Paraganglioma cancer
The incidence of malignant paraganglioma is rare and is estimated to be 93 per 400 million people 39. To date, the only reliable criterion for malignancy is the presence of metastatic spread of chromaffin cells in tissues that normally do not contain such cells 40.
Laboratory testing includes 24-hour urine collection and measurement of urinary fractionated metanephrines and catecholamines. Testing should be done for patients with no symptoms related to excessive catecholamine secretion. The sensitivity of this urine collection study has been reported to be 84% for norepinephrine (noradrenaline), 74% total metanephrines, 18% dopamine, and 14% epinephrine (adrenaline), respectively 41. However, Chen et al 42 reported greater sensitivities of 97% for urinary metanephrines and 86% for urinary catecholamines. Histological studies are required for definitive diagnosis. Previous studies described a Zellballen appearance in which chief cell nests are surrounded by sustentacular supporting cells 43. Tumor markers include neuron-specific enolase (92.1% sensitivity), chromogranin (84.2%), and met-enkephalin (73%) 44. Histological studies have also denoted a negative keratin marker and the appearance of S-100 and glial fibrillary acid protein (GFAP) in the surrounding sustentacular cells 43.
The standard of care for cases of early stage mediastinal paraganglioma is complete surgical resection. Currently, there are no standardized treatment guidelines for malignant mediastinal paraganglioma 45. For cases of metastatic disease, systemic therapy may be employed to decrease tumor burden. Averbuch et al 46 demonstrated the efficacy of combination chemotherapy with 750 mg/m2 cyclophosphamide, 1.4 mg/m2 vincristine, and 600 mg/m2 dacarbazine in the treatment of malignant paraganglioma. A long-term follow-up study of 18 patients recommended the use of the cyclophosphamide+vincristine+dacarbazine regimen in patients with malignant paraganglioma or pheochromocytoma resulting in 11% (2/18) complete response rate and 44% (8/18) partial response rate 46. Although many studies have demonstrated partial remissions and complete remissions in some cases, there are no data of chemotherapy regimens prolonging survival in cases of metastatic disease 47.
Liver-directed radioembolization of Y90 microsphere resins is used for local radiation exposure using the altered arterial network supplying the tumor. This local irradiation allows for a concentrated emission of target blood vessels while maintaining tolerable toxicity levels in the surrounding parenchyma 48. Y90 radioembolization has been described as an emerging form of in situ cytoreductive treatment of hepatic metastasis of neuroendocrine tumors with variable response rates 49.
Paraganglioma survival rate
The prognosis (chance of recovery) and treatment options depend on the following:
- Whether the paraganglioma tumor is benign or malignant.
- Whether the paraganglioma tumor is in one area only or has spread to other places in the body.
- Whether there are signs or symptoms caused by a higher-than-normal amount of catecholamines.
- Whether the paraganglioma tumor has just been diagnosed or has recurred (come back).
There are three ways that cancer spreads in the body.
Cancer can spread through tissue, the lymph system, and the blood:
- Tissue. The cancer spreads from where it began by growing into nearby areas.
- Lymph system. The cancer spreads from where it began by getting into the lymph system. The cancer travels through the lymph vessels to other parts of the body.
- Blood. The cancer spreads from where it began by getting into the blood. The cancer travels through the blood vessels to other parts of the body.
Cancer may spread from where it began to other parts of the body.
When cancer spreads to another part of the body, it is called metastasis. Cancer cells break away from where they began (the primary tumor) and travel through the lymph system or blood.
Lymph system. The cancer gets into the lymph system, travels through the lymph vessels, and forms a tumor (metastatic tumor) in another part of the body.
Blood. The cancer gets into the blood, travels through the blood vessels, and forms a tumor (metastatic tumor) in another part of the body.
The metastatic tumor is the same type of cancer as the primary tumor. For example, if pheochromocytoma spreads to the bone, the cancer cells in the bone are actually pheochromocytoma cells. The disease is metastatic pheochromocytoma, not bone cancer.
A retrospective chart review of 30 pediatric patients, from 1975 to 2005 showed that those who were classified as having benign disease had a 100% survival rate as compared to those with malignant disease, who had 5-, 10-, and 15-year survival rates of 78, 62, and 31%, respectively 50.
Ludwig et al. 51 reported on a 100% overall survival rate and 95% 5-year disease free survival, based on the lower malignancy rate of 7% in their patient population and their ability to achieve negative microscopic margins in all resections. Recent survival data from the EAPPR of the patients who were diagnosed in the pediatric age group show that 6% (8 patients) of those with hereditary disease (144 patients) died, with a follow-up mean range of 10–19 years (range 0–53 years) 52. Three of the patients had Von Hippel–Lindau, three had SDHB mutations, two had neurofibromatosis type 1, and one SDHA with the cause of death being metastases in seven and cardiac failure in one patient. All 33 patients with sporadic disease, followed for a mean of 10 years (range 1–45 years) were alive at subsequent follow-ups. The overall mean life expectancy of hereditary disease was 62 years. Life expectancy was greatly reduced with SDHB-associated disease, at 47 years, while patients with Von Hippel–Lindau had the lowest life expectancy at 27 years.
Long-term follow-up of these tumors is essential due to recurrence, which has been noted to occur anywhere between 1 and 14 years following initial presentation 52 in small pediatric case series. In contrast, data from the EAPPR showed that 38% of the pediatric registrants (n = 177) had recurrences after a mean time period of 25 years 52, with a reported recurrence rate of 12–38% 52. The incidence of recurrent tumors increased with time, from 25% at 9 years to 50% at 31 years 52. The types of recurrent tumors were extra-adrenal (18%) and contralateral (13%) more so than same side (16%) ones 52. Recurrences were significantly more common in patients with germline mutations than those with sporadic disease and tended to recur 10 years earlier, with a latency period of 23 versus 33 years, respectively 52. The mutations seen with these recurrent tumors were associated with Von Hippel–Lindau and SDHD mutations. Within these gene-specific mutations, SDHD mutations had a recurrent tumor after 18 years of latency versus 21 years for Von Hippel–Lindau mutations. Head and neck paragangliomas recurred in 4% of pediatric patients and were caused by SDHD mutations at initial diagnosis and during recurrences. Seven percent of patients had a third recurrence, with a time interval of 1–20 years (mean 5 years) from second to third tumor; they all had germline mutations. The prevalence of malignancy was highest in SDHB mutation-positive individuals, with extra-adrenal and thoracic paragangliomas posing added risk for malignancy 52.
In a retrospective study of 263 patients with pheochromocytomas or paragangliomas, 125 were found to have metastatic disease, of which 32 patients presented before 20 years of age (42). Of those, 72% (23 patients) had a germline mutation in SDHB, 9.4% (3 patients) had an SDHD mutation, and 6.3% (2 patients) had a Von Hippel–Lindau mutation, with the absence of a known mutation in the remainder (4 patients). The study that established plasma methoxytyramine as a biomarker for metastatic pheochromocytomas and paragangliomas also recognized the association between extra-adrenal disease, tumor size >5 cm and SDHB mutation carriers associated with a high risk of malignancy 53.
Long-term follow-up on patients with hereditary pheochromocytomas and paragangliomas cannot be stressed enough given the lifelong risk of recurrence and metastatic disease. Laboratory testing with serum/urine metanephrines should be performed yearly and patients should undergo imaging studies intermittently and as clinically indicated based upon symptoms and/or positive laboratory testing 51 at follow-up visits. Smaller pediatric and adult case series recommend follow-up at 6 weeks and between 6 months and 1 year following initial surgery, then annually 51.
The different characteristics of known mutations may change the follow-up frequency and surveillance emphasis. For example, SDHB mutations have high risk of metastasis 54, Von Hippel–Lindau and SDHD mutation carriers have high recurrence rates 52, and SDHA and TMEM127 have now been identified to confer added risks of malignancy. Recommendations of the EAPPR are to perform annual surveillance for the first 3 years after initial diagnosis of mutation carriers, this being the time frame where malignancy is apparent unless diagnosed at presentation, followed by lifelong follow-up. However, since malignancy can occur much later in life, constant and frequent follow-up is advisable in such patients.
Paraganglioma symptoms
Patients with pheochromocytomas and sympathetic paragangliomas may present with symptoms of excess catecholamine production, including the following:
- Hypertension.
- Headache.
- Perspiration.
- Forceful palpitations.
- Tremor.
- Facial pallor.
These symptoms are often paroxysmal, although sustained hypertension between paroxysmal episodes occurs in 50% to 60% of patients with pheochromocytoma 55. Episodes of hypertension can be variable in frequency, severity, and duration and are often extremely difficult to manage medically. Hypertensive crisis can lead to cardiac arrhythmias, myocardial infarction, and even death.
Patients are often very symptomatic from excess catecholamine secretion. Symptoms of catecholamine excess can be spontaneous or induced by a variety of events, including the following:
- Strenuous physical exertion.
- Trauma.
- Labor and delivery.
- Anesthesia induction.
- Surgery or other invasive procedures, including direct instrumentation of the tumor (e.g., fine-needle aspiration).
- Foods high in tyramine (e.g., red wine, chocolate, and cheese).
- Urination (e.g., bladder wall tumor, which is rare).
Phenoxybenzamine (blocks alpha receptors) is an effective treatment for catecholamine excess and metyrosine (blocks catecholamine synthesis) can be added if needed.
Parasympathetic paragangliomas do not secrete catecholamines and usually present as a neck mass with symptoms related to compression or are incidentally discovered on an imaging study performed for an unrelated reason.
Paraganglioma diagnosis
If your primary doctor thinks you may have a paraganglioma, he or she may refer you to an expert in cancer (medical oncologist), an expert in conditions of the endocrine system (endocrinologist) or a surgeon.
You may need a number of tests:
- 24-hour urine test. You may be asked to collect a urine sample every time you urinate during a 24-hour period. This will be used to assess hormone excretion into urine. You will be given written instructions on how to store, label and return the samples.
- Blood test. You may have blood drawn for laboratory testing, including an assessment of hormone levels. Talk to your doctor about special preparations, such as fasting or skipping a medication. Do not skip a dose without specific instructions from your doctor.
- Imaging tests. Your doctor may order one or more imaging tests to define as accurately as possible the location of the tumor. These studies may include an MRI, CT scan and specialized nuclear medicine imaging such as a PET scan.
- Genetic testing. Paragangliomas sometimes run in families, associated with altered genes that can be transmitted from parent to child. Your doctor may have your blood tested for this. And if an altered gene is detected, your doctor may recommend that you see a genetic counselor.
Note that examining tumor tissue under a microscope cannot with certainty determine whether a tumor is cancerous.
People with paragangliomas need care by an experienced, multidisciplinary team. Ask your doctor if he or she regularly treats people with this condition, as most doctors rarely (if ever) encounter paragangliomas and are unfamiliar with the best approaches to diagnosing and treating this rare tumor. Under such conditions, it’s important to seek a second opinion from a team that specializes in the care of people with rare neuroendocrine tumors such as paragangliomas.
Genetic testing may be recommended by a genetic counselor for patients who:
- Have a personal or family history of traits linked with inherited pheochromocytoma or paraganglioma syndrome.
- Have tumors in both adrenal glands.
- Have more than one tumor in one adrenal gland.
- Have signs or symptoms of extra catecholamines being released into the blood or malignant (cancerous) paraganglioma.
- Are diagnosed before age 40.
Genetic testing is sometimes recommended for patients with pheochromocytoma who:
- Are aged 40 to 50 years.
- Have a tumor in one adrenal gland.
- Do not have a personal or family history of an inherited syndrome.
When certain gene changes are found during genetic testing, the testing is usually offered to family members who are at risk but do not have signs or symptoms.
Genetic testing is not recommended for patients older than 50 years.
Further tests to find out if pheochromocytoma and paraganglioma have spread
After pheochromocytoma and paraganglioma have been diagnosed, tests are done to find out if the tumor has spread to other parts of the body.
The extent or spread of cancer is usually described as stage. It is important to know whether the cancer has spread in order to plan treatment. The following tests and procedures may be used to determine if the tumor has spread to other parts of the body:
- CT scan (CAT scan): A procedure that makes a series of detailed pictures of areas inside the body, such as the neck, chest, abdomen, and pelvis, taken from different angles. The pictures are made by a computer linked to an x-ray machine. A dye may be injected into a vein or swallowed to help the organs or tissues show up more clearly. The abdomen and pelvis are imaged to detect tumors that release catecholamine. This procedure is also called computed tomography, computerized tomography, or computerized axial tomography.
- MRI (magnetic resonance imaging): A procedure that uses a magnet, radio waves, and a computer to make a series of detailed pictures of areas inside the body such as the neck, chest, abdomen, and pelvis. This procedure is also called nuclear magnetic resonance imaging (NMRI).
- MIBG scan : A procedure used to find neuroendocrine tumors, such as pheochromocytoma and paraganglioma. A very small amount of a substance called radioactive MIBG is injected into a vein and travels through the bloodstream. Neuroendocrine tumor cells take up the radioactive MIBG and are detected by a scanner. Scans may be taken over 1-3 days. An iodine solution may be given before or during the test to keep the thyroid gland from absorbing too much of the MIBG.
- Octreotide scan : A type of radionuclide scan used to find certain tumors, including tumors that release catecholamine. A very small amount of radioactive octreotide (a hormone that attaches to certain tumors) is injected into a vein and travels through the bloodstream. The radioactive octreotide attaches to the tumor and a special camera that detects radioactivity is used to show where the tumors are in the body.
- FDG-PET scan (fluorodeoxyglucose-positron emission tomography scan): A procedure to find malignant tumor cells in the body. A small amount of FDG, a type of radioactive glucose (sugar), is injected into a vein. The PET scanner rotates around the body and makes a picture of where glucose is being used in the body. Malignant tumor cells show up brighter in the picture because they are more active and take up more glucose than normal cells do.
Paraganglioma and Pheochromocytoma Staging
There is no standard staging system for pheochromocytoma and paraganglioma. Pheochromocytoma and paraganglioma are described as localized, regional, or metastatic.
Localized pheochromocytoma and paraganglioma
The tumor is found in one or both adrenal glands (pheochromocytoma) or in one area only (paraganglioma).
Regional pheochromocytoma and paraganglioma
Cancer has spread to lymph nodes or other tissues near where the tumor began.
Metastatic pheochromocytoma and paraganglioma
The cancer has spread to other parts of the body, such as the liver, lungs, bone, or distant lymph nodes.
Recurrent Pheochromocytoma and Paraganglioma
Recurrent pheochromocytoma or paraganglioma is cancer that has recurred (come back) after it has been treated. The cancer may come back in the same place or in another part of the body.
Paraganglioma treatment
Treatment of a paraganglioma depends on where the abnormal growth is located and whether it is cancerous. A paraganglioma is considered cancerous (malignant) if it spreads to other parts of the body (metastasizes). This is a rare occurrence in paragangliomas.
Cells from a cancerous (malignant) paraganglioma can migrate to lymph nodes, bone, liver, lungs or elsewhere. In people with this type of paraganglioma, it’s critical to treat any elevated hormone levels and other signs and symptoms.
Treatment options include:
- Medications. Your doctor will first attempt to control symptoms caused by your paraganglioma, such as high blood pressure, if you have the type of paraganglioma that produces hormones. Drugs that usually help people with this condition include alpha blockers and beta blockers. In people with hormone-producing paragangliomas, it’s critical to control blood pressure before surgery or other therapy.
- Surgery. Surgery to remove the tumor is generally the first choice for treatment, if feasible. Even if the tumor has spread, surgery is often used to reduce its size. If paraganglioma tissue that produces hormones is disturbed, as happens during surgery, hormones in the tumor can be released and cause serious problems, such as elevated blood pressure and pulse. To prevent that, your doctor will have you prepare for surgery by taking medication to block the hormone’s effects for 7 to 14 days before surgery. You may also be asked to record your blood pressure multiple times a day, to follow a high-sodium diet and to report your blood pressure results to your medical team. Paraganglioma surgery is done under general anesthesia. It’s important that specialized surgeons and anesthesiologists be involved. You’ll likely need to stay in the hospital for several days after surgery. Even after successful removal of the tumor during surgery, tumors may recur. So you’ll need to visit your doctor regularly for follow-up appointments.
- Radiation therapy. Radiation therapy is sometimes used to help control tumor growth and improve your symptoms if other options, such as surgery, aren’t advisable. If the tumor is in the head or near vital nerves or blood vessels, a technique called stereotactic body radiation therapy may be used to limit damage to nearby healthy tissue.Radiation therapy is a cancer treatment that uses high-energy x-rays or other types of radiation to kill cancer cells or keep them from growing. There are two types of radiation therapy:
- External radiation therapy uses a machine outside the body to send radiation toward the cancer.
- Internal radiation therapy uses a radioactive substance sealed in needles, seeds, wires, or catheters that are placed directly into or near the cancer.The way the radiation therapy is given depends on the type of cancer being treated and whether it is localized, regional, metastatic, or recurrent. External radiation therapy and 131I-MIBG therapy are used to treat pheochromocytoma.Pheochromocytoma is sometimes treated with 131I-MIBG, which carries radiation directly to tumor cells. 131I-MIBG is a radioactive substance that collects in certain kinds of tumor cells, killing them with the radiation that is given off. The 131I-MIBG is given by infusion. Not all pheochromocytomas take up 131I-MIBG, so a test is done first to check for this before treatment begins.
- Thermal ablation therapy. Under some circumstances, specialized interventional radiologists may destroy tumor deposits with an approach called thermal ablation therapy.
- Clinical trials. You may be eligible for a clinical trial testing an experimental treatment.
- Watchful waiting. If paraganglioma tumor is slow growing, your doctor may monitor your condition during regular follow-up appointments even if you aren’t undergoing therapy.
Patients with pheochromocytoma and paraganglioma that cause signs or symptoms are treated with drug therapy.
Drug therapy begins when pheochromocytoma or paraganglioma is diagnosed. This may include:
- Drugs that keep the blood pressure normal. For example, one type of drug called alpha-blockers stops noradrenaline from making small blood vessels more narrow. Keeping the blood vessels open and relaxed improves blood flow and lowers blood pressure.
- Drugs that keep the heart rate normal. For example, one type of drug called beta-blockers stops the effect of too much noradrenaline and slows the heart rate.
- Drugs that block the effect of extra hormones made by the adrenal gland.
Drug therapy is often given for one to three weeks before surgery.
Localized Paraganglioma and Pheochromocytoma
Treatment of localized benign paraganglioma or pheochromocytoma is usually surgery to completely remove the tumor. If the tumor is in the adrenal gland, the entire adrenal gland is removed.
Regional Paraganglioma and Pheochromocytoma
Treatment of paraganglioma or pheochromocytoma that has spread to nearby organs or lymph nodes is surgery to completely remove the tumor. Nearby organs that the cancer has spread to, such as the kidney, liver, part of a major blood vessel, and lymph nodes, may also be removed.
Metastatic Paraganglioma and Pheochromocytoma
Treatment of metastatic paraganglioma or pheochromocytoma may include the following:
- Surgery to completely remove the cancer, including tumors that have spread to distant parts of the body.
- Palliative therapy, to relieve symptoms and improve the quality of life, including:
- Surgery to remove as much cancer as possible.
- Combination chemotherapy.
- Radiation therapy with 131I-MIBG.
- External radiation therapy to areas (such as bone) where cancer has spread and cannot be removed by surgery.
- Embolization (treatment to block an artery that supplies blood to a tumor).
- Ablation therapy using radiofrequency ablation or cryoablation for tumors in the liver or bone.
- A clinical trial of targeted therapy with a tyrosine kinase inhibitor.
- A clinical trial of internal radiation therapy using a new radioactive substance.
Recurrent Paraganglioma and Pheochromocytoma
Treatment of recurrent paraganglioma or pheochromocytoma may include the following:
- Surgery to completely remove the cancer.
- When surgery to remove the cancer is not possible, palliative therapy to relieve symptoms and improve the quality of life, including:
- Combination chemotherapy.
- Targeted therapy.
- Radiation therapy using 131I-MIBG.
- External radiation therapy to areas (such as bone) where cancer has spread and cannot be removed by surgery.
- Ablation therapy using radiofrequency ablation or cryoablation.
- McNichol AM. 2001 Differential diagnosis of pheochromocytomas and paragangliomas. Endocrine Pathology 12 407–415. doi:10.1385/EP:12:4:407
- Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, Young WF Jr. J Clin Endocrinol Metab. 2001 Nov; 86(11):5210-6. https://www.ncbi.nlm.nih.gov/pubmed/11701678/
- DeLellis RA, Lloyd RV, Heitz PU & Eng C Eds 2004 World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Endocrine Organs. pp 147–166. Lyon, France: IARC Press
- Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV & Pacak K. 2010 The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 39 775–783. doi:10.1097/MPA.0b013e3181ebb4f0
- McNeil AR, Blok BH, Koelmeyer TD, Burke MP & Hilton JM. 2000 Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Australian and New Zealand Journal of Medicine 30 648–652. doi:10.1111/j.1445-5994.2000.tb04358.x
- Ariton M, Juan CS & AvRuskin TW. 2000 Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocrine Practice 6 249–252.
- Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, Mori L, Pignataro V, Bernini G, Giache V, Bacca A et al. 2009 Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. Journal of Clinical Endocrinology and Metabolism 94 1541–1547. doi:10.1210/jc.2008-2419
- Cascon A, Pita G, Burnichon N, Landa I, Lopez-Jimenez E, Montero-Conde C, Leskela S, Leandro-Garcia LJ, Leton R, Rodriguez-Antona C et al. 2009b Genetics of pheochromocytoma and paraganglioma in Spanish patients. Journal of Clinical Endocrinology and Metabolism 94 1701–1705. doi:10.1210/jc.2008-2756
- Karagiannis A, Mikhailidis DP, Athyros VG & Harsoulis F. 2007 Pheochromocytoma: an update on genetics and management. Endocrine-Related Cancer 14 935–956. doi:10.1677/ERC-07-0142
- Kopetschke R, Slisko M, Kilisli A, Tuschy U, Wallaschofski H, Fassnacht M, Ventz M, Beuschlein F, Reincke M, Reisch N et al. 2009 Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. European Journal of Endocrinology 161 355–361. doi:10.1530/EJE-09-0384
- Sibal L, Jovanovic A, Agarwal SC, Peaston RT, James RA, Lennard TW, Bliss R, Batchelor A & Perros P. 2006 Phaeochromocytomas presenting as acute crises after beta blockade therapy. Clinical Endocrinology 65 186–190. doi:10.1111/j.1365-2265.2006.02571.x
- van Duinen N, Steenvoorden D, Kema IP, Jansen JC, Vriends AH, Bayley JP, Smit JW, Romijn JA & Corssmit EP. 2010 Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. Journal of Clinical Endocrinology and Metabolism 95 209–214. doi:10.1210/jc.2009-1632
- Chrisoulidou A, Kaltsas G, Ilias I & Grossman AB. 2007 The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocrine-Related Cancer 14 569–585. doi:10.1677/ERC-07-0074
- Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, Brennan M & Ghossein RA. 2008 Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery 143 759–768. doi:10.1016/j.surg.2008.02.007
- Amar L, Bertherat J, Baudin E, et al.: Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 23 (34): 8812-8, 2005 https://www.ncbi.nlm.nih.gov/pubmed/16314641
- Carney JA: Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 74 (6): 543-52, 1999 https://www.ncbi.nlm.nih.gov/pubmed/10377927
- Carney JA, Stratakis CA: Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 108 (2): 132-9, 2002 https://www.ncbi.nlm.nih.gov/pubmed/11857563
- Qin Y, Yao L, King EE, et al.: Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 42 (3): 229-33, 2010 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2998199/
- Taïeb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR Jr, et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev. 2014. October;35: 795–819. doi: 10.1210/er.2014-1026 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4167435/
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000. February 4;287: 848–851 https://www.ncbi.nlm.nih.gov/pubmed/10657297
- Taïeb D, Kaliski A, Boedeker CC, et al. Current Approaches and Recent Developments in the Management of Head and Neck Paragangliomas. Endocrine Reviews. 2014;35(5):795-819. doi:10.1210/er.2014-1026. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4167435/
- Malignant pheochromocytomas and paragangliomas: a diagnostic challenge. Gimm O, DeMicco C, Perren A, Giammarile F, Walz MK, Brunaud L. Langenbecks Arch Surg. 2012 Feb; 397(2):155-77. https://www.ncbi.nlm.nih.gov/pubmed/22124609/
- Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr, Endocrine Society. J Clin Endocrinol Metab. 2014 Jun; 99(6):1915-42. https://www.ncbi.nlm.nih.gov/pubmed/24893135/
- Lack EE, Lloyd RV, Carney JA, Woodruff JM; Association of Directors of Anatomic and Surgical Pathology. Recommendations for reporting of extra-adrenal paragangliomas. Mod Pathol. 2003. August;16: 833–855. doi: 10.1097/01.MP.0000081050.89276.CA
- Fliedner SM, Lehnert H, Pacak K. Metastatic paraganglioma. Semin Oncol. 2010. December;37: 627–637. doi: 10.1053/j.seminoncol.2010.10.017 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3018803/
- Surgical treatment of cardiac pheochromocytomas. Orringer MB, Sisson JC, Glazer G, Shapiro B, Francis I, Behrendt DM, Thompson NW, Lloyd RV. J Thorac Cardiovasc Surg. 1985 May; 89(5):753-7. https://www.ncbi.nlm.nih.gov/pubmed/3990325/
- Orringer MB, Sisson JC, Glazer G, et al. Surgical treatment of cardiac pheochromocytomas. J Thorac Cardiovasc Surg 1985;89:753-7 https://www.ncbi.nlm.nih.gov/pubmed/3990325
- Echocardiography in the evaluation of cardiac pheochromocytoma. Mandak JS, Benoit CH, Starkey RH, Nassef LA Jr. Am Heart J. 1996 Nov; 132(5):1063-6. https://www.ncbi.nlm.nih.gov/pubmed/8892789/
- Brown ML, Zayas GE, Abel MD, et al. Mediastinal paragangliomas: the Mayo Clinic experience. Ann Thorac Surg 2008;86:946-51. 10.1016/j.athoracsur.2008.04.105 https://www.ncbi.nlm.nih.gov/pubmed/18721588
- Hayek ER, Hughes MM, Speakman ED, et al. Cardiac paraganglioma presenting with acute myocardial infarction and stroke. Ann Thorac Surg 2007;83:1882-4. 10.1016/j.athoracsur.2006.12.023 https://www.ncbi.nlm.nih.gov/pubmed/17462425
- Marinho B, Malangatana G, Almeida J, et al. Acute coronary syndrome as clinical presentation of cardiac paraganglioma. Rev Port Cir Cardiotorac Vasc 2011;18:153-5 https://www.ncbi.nlm.nih.gov/pubmed/23596618
- Wang JG, Han J, Jiang T, et al. Cardiac paragangliomas. J Card Surg 2015;30:55-60. 10.1111/jocs.12455 https://www.ncbi.nlm.nih.gov/pubmed/25331372
- El-Ashry AA, Cerfolio RJ, Singh SP, et al. Cardiac paraganglioma. J Card Surg 2015;30:135-9. 10.1111/jocs.12479 https://www.ncbi.nlm.nih.gov/pubmed/25533017
- Yamamoto Y, Kodama K, Yamato H, et al. Successful Removal of Giant Intrapericardial Paraganglioma via Posterolateral Thoracotomy. Case Rep Surg 2014;2014:308462. 10.1155/2014/308462 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4241561/
- Ramlawi B, David EA, Kim MP, et al. Contemporary surgical management of cardiac paragangliomas. Ann Thorac Surg 2012;93:1972-6. 10.1016/j.athoracsur.2012.02.040 https://www.ncbi.nlm.nih.gov/pubmed/22537533
- Brichon PY, Chavanon O, Thony F, et al. An intrapericardial paraganglioma with embolization of a large vessel from the left coronary artery. Eur J Cardiothorac Surg 2014;45:e234. 10.1093/ejcts/ezu110 https://www.ncbi.nlm.nih.gov/pubmed/24639450
- Adrogue HE, Sinaiko AR. Prevalence of hypertension in junior high school-aged children: effect of new recommendations in the 1996 Updated Task Force Report. Am J Hypertens (2001) 14(5 Pt 1):412–4.10.1016/S0895-7061(00)01277-2 https://www.ncbi.nlm.nih.gov/pubmed/11368459
- Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, Jin R, et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol (2010) 34(5):636–44.10.1097/PAS.0b013e3181d6150d https://www.ncbi.nlm.nih.gov/pubmed/20305538
- Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Welander J, Söderkvist P, Gimm O. Endocr Relat Cancer. 2011 Dec; 18(6):R253-76. http://erc.endocrinology-journals.org/content/18/6/R253.long
- Posterior mediastinal paragangliomas: a report of three patients with peculiar tumours. Ayadi-Kaddour A, Braham E, Ismail O, Smati B, Djilani H, El Mezni F. Respirology. 2009 Apr; 14(3):459-61. https://www.ncbi.nlm.nih.gov/pubmed/19192222/
- Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18(6):R253–276. doi: 10.1530/ERC-11-0170. https://www.ncbi.nlm.nih.gov/pubmed/22041710
- Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39(6):775–783. doi: 10.1097/MPA.0b013e3181ebb4f0 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3419007/
- Ayadi-Kaddour A, Braham E, Ismail O, Smati B, Djilani H, El Mezni F. Posterior mediastinal paragangliomas: a report of three patients with peculiar tumours. Respirology. 2009;14(3):459–461. doi: 10.1111/j.1440-1843.2008.01475.x https://www.ncbi.nlm.nih.gov/pubmed/19192222
- Moran CA, Suster S, Fishback N, Koss MN. Mediastinal paragangliomas. A clinicopathologic and immunohistochemical study of 16 cases. Cancer. 1993;72(8):2358–2364. doi: 10.1002/1097-0142(19931015)72:8<2358::AID-CNCR2820720811>3.0.CO;2-B https://www.ncbi.nlm.nih.gov/pubmed/8402449
- Prades CA, Atassi B, Nazeer H. Metastatic Malignant Paraganglioma: A Case Report and Review of Literature. World Journal of Oncology. 2017;8(3):92-95. doi:10.14740/wjon1033w. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5650004/
- Averbuch SD, Steakley CS, Young RC, Gelmann EP, Goldstein DS, Stull R, Keiser HR. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 1988;109(4):267–273. doi: 10.7326/0003-4819-109-4-267 https://www.ncbi.nlm.nih.gov/pubmed/3395037
- Nomura K, Kimura H, Shimizu S, Kodama H, Okamoto T, Obara T, Takano K. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab. 2009;94(8):2850–2856. doi: 10.1210/jc.2008-2697 https://www.ncbi.nlm.nih.gov/pubmed/19470630
- Fishbein L, Bonner L, Torigian DA, Nathanson KL, Cohen DL, Pryma D, Cengel KA. External beam radiation therapy (EBRT) for patients with malignant pheochromocytoma and non-head and -neck paraganglioma: combination with 131I-MIBG. Horm Metab Res. 2012;44(5):405–410. doi: 10.1055/s-0032-1308992 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4357844/
- Memon K, Lewandowski RJ, Mulcahy MF, Riaz A, Ryu RK, Sato KT, Gupta R. et al. Radioembolization for neuroendocrine liver metastases: safety, imaging, and long-term outcomes. Int J Radiat Oncol Biol Phys. 2012;83(3):887–894. doi: 10.1016/j.ijrobp.2011.07.041 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3297703/
- Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pham TH, Moir C, Thompson GB, Zarroug AE, Hamner CE, Farley D, van Heerden J, Lteif AN, Young WF Jr. Pediatrics. 2006 Sep; 118(3):1109-17. https://www.ncbi.nlm.nih.gov/pubmed/16951005/
- Ludwig AD, Feig DI, Brandt ML, Hicks MJ, Fitch ME, Cass DL. Recent advances in the diagnosis and treatment of pheochromocytoma in children. Am J Surg (2007) 194(6):792–6; discussion 796–7.10.1016/j.amjsurg.2007.08.028 https://www.ncbi.nlm.nih.gov/pubmed/18005773
- Bausch B, Wellner U, Bausch D, Schiavi F, Barontini M, Sanso G, et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr Relat Cancer (2014) 21(1):17–25.10.1530/ERC-13-0415 http://erc.endocrinology-journals.org/content/21/1/17.long
- Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer (2012) 48(11):1739–49.10.1016/j.ejca.2011.07.016 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3372624/
- King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol (2011) 29(31):4137–42.10.1200/JCO.2011.34.6353 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3208535/
- Lenders JW, Eisenhofer G, Mannelli M, et al.: Phaeochromocytoma. Lancet 366 (9486): 665-75, 2005 Aug 20-26 https://www.ncbi.nlm.nih.gov/pubmed/16112304




{kind=link}