What is retinoblastoma
Retinoblastoma is a type of cancer that forms in the retina (the light-sensitive tissue at the back of the eye). Retinoblastoma usually occurs in young children, and can affect one or both eyes.
Retinoblastoma is a rare disease. Only about 200 to 300 children are diagnosed with retinoblastoma each year in the United States. It is more common in infants and very young children than in older children. The average age of children when they are diagnosed is 2. Although it can occur at any age, it rarely occurs in children older than 6. Retinoblastoma may be in one eye or in both eyes. In some cases the disease is inherited from a parent.
- About 3 out of 4 children with retinoblastoma have a tumor in only one eye. In about 1 case in 4, both eyes are affected.
Retinoblastoma occurs about equally in boys and girls and in different races and ethnicities. It also occurs equally in the right or left eye.
Overall, more than 9 out of 10 children with retinoblastoma are cured, but the outlook is not nearly as good if the cancer has spread outside of the eye.
Retinoblastoma is a serious, life-threatening disease. However, with early diagnosis and timely treatment, in most cases, a child’s eyesight and life can be saved.
Certain factors affect prognosis (chance of recovery) and treatment options.
The prognosis (chance of recovery) and treatment options depend on the following:
- Whether the cancer is in one or both eyes.
- The size and number of tumors.
- Whether the tumor has spread to the area around the eye, to the brain, or to other parts of the body.
- Whether there are symptoms at the time of diagnosis, for trilateral retinoblastoma.
- The age of the child.
- How likely it is that vision can be saved in one or both eyes.
- Whether a second type of cancer has formed.
Retinoblastoma occurs in heritable and nonheritable forms.
A child is thought to have the heritable form of retinoblastoma when one of the following is true:
- There is a family history of retinoblastoma.
- There is a certain mutation (change) in the RB1 gene. The mutation in the RB1 gene may be passed from the parent to the child or it may occur in the egg or sperm before conception or soon after conception.
- There is more than one tumor in the eye or there is a tumor in both eyes.
- There is a tumor in one eye and the child is younger than 1 year.
After heritable retinoblastoma has been diagnosed and treated, new tumors may continue to form for a few years. Regular eye exams to check for new tumors are usually done every 2 to 4 months for at least 28 months.
Nonheritable retinoblastoma is retinoblastoma that is not the heritable form. Most cases of retinoblastoma are the nonheritable form.
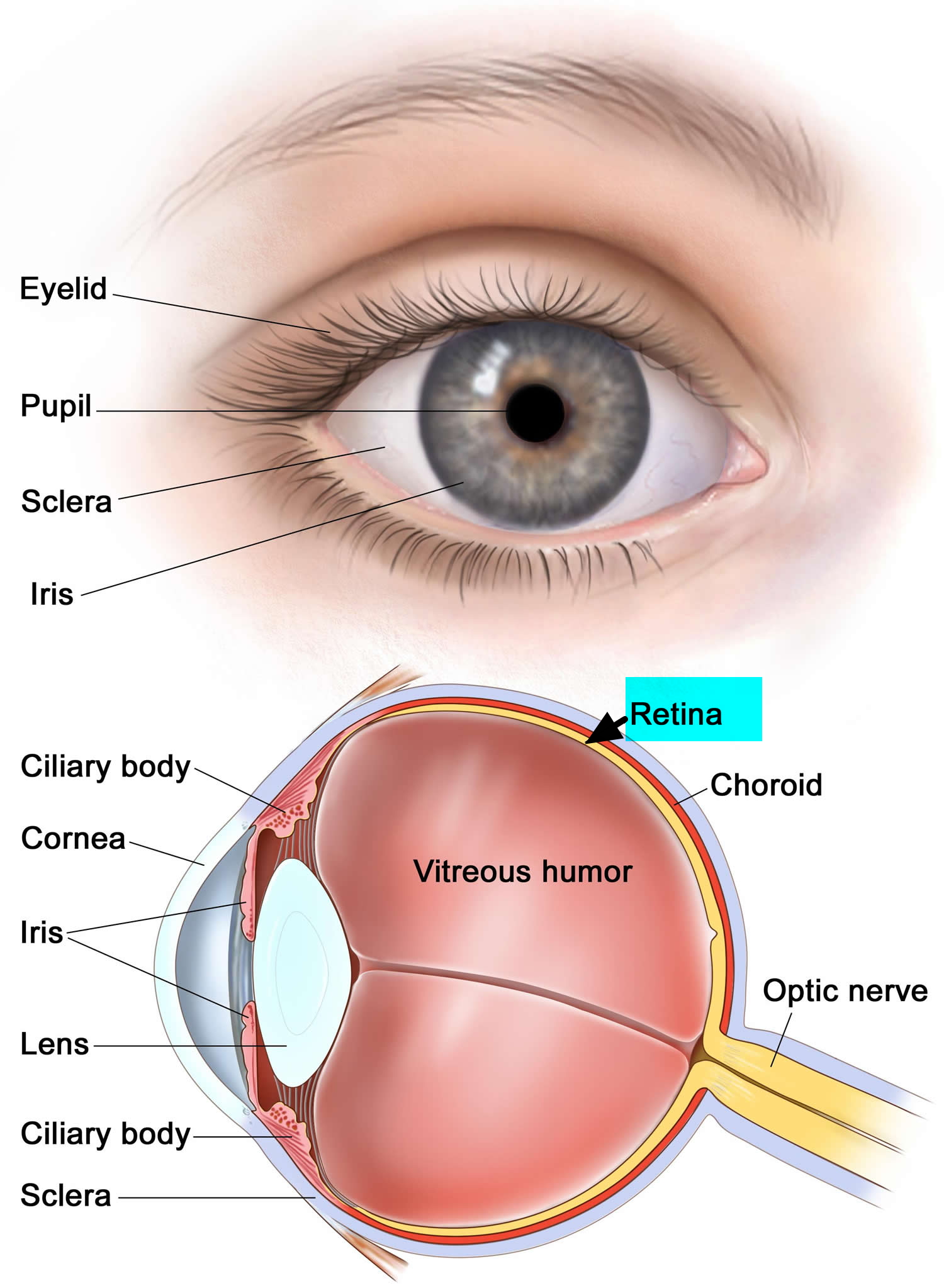
The human eye
The main part of the eye is the eyeball (also known as the globe), which is filled with a jelly-like material called vitreous humor. The front of the eyeball has a clear lens with an iris (the colored part of the eye that acts like a camera shutter), which allows light to enter the eye and focuses it on the retina.
The retina is the inner layer of cells in the back of the eye. It is made up of special nerve cells that are sensitive to light. These light-sensing cells are connected to the brain by the optic nerve, which runs out the back of the eyeball. The pattern of light (image) that reaches the retina is sent through the optic nerve to an area of the brain called the visual cortex, allowing us to see.
Figure 1. Human eye and retina
How does retinoblastoma develop?
The eyes develop very early as babies grow in the womb. During the early stages of development, the eyes have cells called retinoblasts that divide into new cells and fill the retina. At a certain point, these cells stop dividing and develop into mature retinal cells.
Rarely, something goes wrong with this process. Instead of maturing into special cells that detect light, some retinoblasts continue to divide and grow out of control, forming a cancer known as retinoblastoma.
The chain of events inside cells that leads to retinoblastoma is complex, but it almost always starts with a change (mutation) in a gene called the retinoblastoma (RB1) gene. The normal RB1 tumor suppressor gene helps keep cells from growing out of control, but the change in the gene stops it from working like it should. Depending on when and where the change in the RB1 gene occurs, 2 different types of retinoblastoma can result.
Congenital (hereditary) retinoblastoma
In about 1 out of 3 children with retinoblastoma, the abnormality in the RB1 gene is congenital (present at birth) and is in all the cells of the body, including all of the cells of both retinas. This is known as a germline mutation.
In most of these children, there is no family history of this cancer. Only about 25% of the children born with this gene change inherit it from a parent. In about 75% of children the gene change first occurs during early development in the womb. The reasons for this are not clear.
Children born with a mutation in the RB1 gene usually develop retinoblastoma in both eyes (known as bilateral retinoblastoma), and there are often several tumors within the eye (known as multifocal retinoblastoma).
Because all of the cells in the body have the changed RB1 gene, these children also have a higher risk of developing cancers in other areas as well.
A small number of children with this form of retinoblastoma will develop another tumor in the brain, usually in the pineal gland at the base of the brain (a pineoblastoma). This is also known as trilateral retinoblastoma.
For survivors of hereditary retinoblastoma, the risk of developing other cancers later in life is also higher than average.
Sporadic (non-hereditary) retinoblastoma
In about 2 out of 3 children with retinoblastoma, the abnormality in the RB1 gene develops on its own in only one cell in one eye. It is not known what causes this change. A child who has sporadic (non-hereditary) retinoblastoma develops only one tumor in one eye. This type of retinoblastoma is often found at a later age than the hereditary form.
Children with this type of retinoblastoma do not have the same increased risk of other cancers as children with congenital retinoblastoma.
Intraocular medulloepithelioma
Medulloepithelioma is another type of tumor that can start in the eye. It is not a type of retinoblastoma, but it is mentioned here because it also usually occurs in young children. These tumors are very rare.
Medulloepitheliomas start in the ciliary body, which is near the front of the eye (see image above). Most of these tumors are malignant (cancerous), but they rarely spread outside the eye. They usually cause eye pain and loss of vision.
The diagnosis is made when a doctor finds a tumor mass in the eye by using an ophthalmoscope (an instrument that helps doctors to look inside the eye). Like retinoblastoma, the diagnosis is usually made based on where the tumor is inside the eye and how it looks. A biopsy (removing cells from the tumor to be looked at under a microscope) to confirm the diagnosis is almost never done because it might harm the eye or risk spreading the cancer outside of the eye.
Treatment for medulloepithelioma is almost always surgery to remove the eye. This usually gets rid of all of the cancer, as long as it was still only in the eye.
How does retinoblastoma grow and spread?
If retinoblastoma tumors are not treated, they can grow and fill much of the eyeball. Cells might break away from the main tumor on the retina and float through the vitreous to reach other parts of the eye, where they can form more tumors. If these tumors block the channels that let fluid circulate within the eye, the pressure inside the eye can rise. This can cause glaucoma, which can lead to pain and loss of vision in the affected eye.
Most retinoblastomas are found and treated before they have spread outside the eyeball. But retinoblastoma cells can occasionally spread to other parts of the body. The cells sometimes grow along the optic nerve and reach the brain. Retinoblastoma cells can also grow through the covering layers of the eyeball and into the eye socket, eyelids, and nearby tissues. Once the cancer reaches tissues outside the eyeball, it can then spread to lymph nodes (small bean-shaped collections of immune system cells) and to other organs such as the liver, bones, and bone marrow (the soft, inner part of many bones).
Retinoblastoma complications
Children treated for retinoblastoma have a risk of cancer returning in and around the treated eye. For this reason, your child’s doctor will schedule follow-up exams to check for recurrent retinoblastoma. The doctor may design a personalized follow-up exam schedule for your child. In most cases, this will likely involve eye exams every few months for the first few years after retinoblastoma treatment ends.
Additionally, children with the inherited form of retinoblastoma have an increased risk of developing other types of cancers in any part of the body in the years after treatment. For this reason, children with inherited retinoblastoma may have regular exams to screen for other cancers.
Retinoblastoma causes
Retinoblastoma is caused by mutations (changes) in certain genes. Over the past few decades, scientists have learned how certain changes in a person’s DNA can cause cells of the retina to become cancerous. The DNA in each of your cells makes up your genes, which are the instructions for how your cells function. You usually look like your parents because they are the source of your DNA. But DNA affects much more than how you look.
Some genes control when your cells grow, divide into new cells, and die at the right time. Certain genes that help cells grow, divide, or stay alive are called oncogenes. Others that slow down cell division or cause cells to die at the right time are called tumor suppressor genes. Cancers can be caused by DNA changes that turn on oncogenes or turn off tumor suppressor genes.
The most important gene in retinoblastoma is the RB1 tumor suppressor gene. This gene makes a protein (pRb) that helps stop cells from growing too quickly. Each cell normally has 2 RB1 genes. As long as a retinal cell has at least one RB1 gene that works as it should, it will not form a retinoblastoma. But when both of the RB1 genes are mutated or missing, a cell can grow unchecked. This can lead to further gene changes, which in turn may cause cells to become cancerous.
Hereditary or bilateral retinoblastoma
About 1 out of 3 children with retinoblastoma have a germline mutation in one RB1 gene; that is, all the cells in the body have a defective RB1 gene. In most of these children (75%), this mutation developed after conception while in the womb. The other 25% of children have inherited it from one of their parents.
About 9 of 10 children who are born with this RB1 germline mutation develop retinoblastoma. This happens when the second RB1 gene is lost or mutated. Most often the retinoblastoma is bilateral (in both eyes), but sometimes it is found early enough that it is still only in one eye.
These children have hereditary retinoblastoma. Hereditary retinoblastoma also tends to occur in both eyes, as opposed to just one eye, although not all hereditary retinoblastomas are bilateral when they are found. Children with the inherited form of retinoblastoma tend to develop the disease at an earlier age.
Hereditary retinoblastoma is passed from parents to children in an autosomal dominant pattern, which means only one parent needs a single copy of the mutated gene to pass the increased risk of retinoblastoma on to the children. If one parent carries a mutated gene, each child has a 50 percent chance of inheriting that gene. Children with the inherited form of retinoblastoma tend to develop the disease at an earlier age.
Every person has 2 RB1 genes but passes only 1 on to each of their children. (The child gets the other RB1 gene from the other parent.) Therefore there is a 1 in 2 chance that a parent who had hereditary retinoblastoma will pass the mutated gene on to his or her child.
However, most children with hereditary retinoblastoma don’t have an affected parent. But these children can still pass their RB1 gene mutation on to their children. This is why we call this form of retinoblastoma “hereditary” (even though neither of the child’s parents may have been affected).
Although a genetic mutation increases a child’s risk of retinoblastoma, it doesn’t mean that cancer is inevitable.
Figure 2. Hereditary retinoblastoma – autosomal dominant inheritance pattern
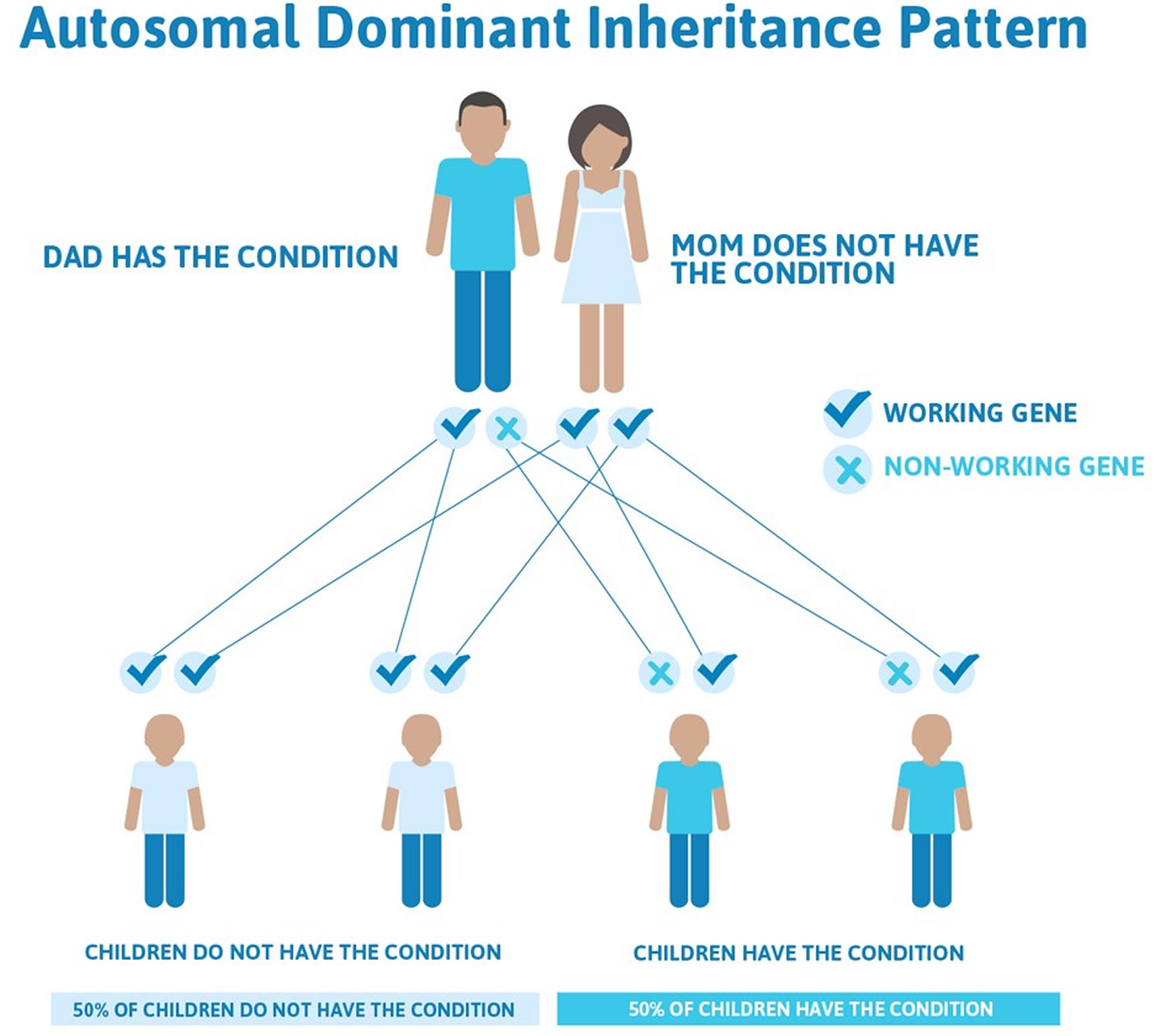
Note: In an autosomal dominant disorder, the mutated gene is a dominant gene located on one of the nonsex chromosomes (autosomes). You need only one mutated gene to be affected by this type of disorder. A person with an autosomal dominant disorder — in this case, the father — has a 50 percent chance of having an affected child with one mutated gene (dominant gene) and a 50 percent chance of having an unaffected child with two normal genes (recessive genes).
Non-hereditary (sporadic) retinoblastoma
Most of the remaining 2 out of 3 children with retinoblastoma do not have the RB1 gene mutation in all the cells of their body. Instead, the RB1 mutation happens early in life and first occurs only in one cell in one eye.
Whether the changes in the RB1 gene are hereditary or sporadic, it’s not clear what causes these changes. They may result from random gene errors that sometimes occur when cells divide to make new cells. There are no known lifestyle-related or environmental causes of retinoblastoma, so it’s important to remember that there is nothing these children or their parents could have done to prevent these cancers.
Risk factors for retinoblastoma
A risk factor is anything that affects a person’s chance of getting a disease such as cancer. Different cancers have different risk factors.
Lifestyle-related risk factors such as body weight, physical activity, diet, and tobacco use play a major role in many adult cancers. But these factors usually take many years to influence cancer risk, and they are not thought to play much of a role in childhood cancers, including retinoblastomas.
There are very few known risk factors for retinoblastoma.
Age
Most children diagnosed with retinoblastoma are younger than 3 years old. Most congenital or hereditary retinoblastomas are found during the first year of life, while non-inherited retinoblastomas tend to be diagnosed in 1- and 2-year-olds. Retinoblastomas are rare in older children and in adults.
Heredity
About 1 out of 3 retinoblastomas is caused by a mutation (change) in the RB1 gene that is present in all the cells of the child’s body. But of these cases, only about 1 in 4 is inherited from one of the child’s parents. In the rest, the gene mutation is not inherited, but occurs during early development in the womb. Children born with a mutation in the RB1 gene usually develop retinoblastoma in both eyes. Regardless of whether the mutated RB1 gene was inherited from a parent or not, because these children have the mutated gene in all of their cells, they have a 1 in 2 chance of eventually passing it on to their children.
Most of the remaining 2 out of 3 retinoblastomas occur as a result of a random RB1 gene mutation that occurs only in one cell of one eye. These mutations are not inherited from a parent, and children who have them do not pass on a greatly increased risk of retinoblastoma to their children. Non-hereditary retinoblastomas always affect one eye only.
Retinoblastoma prevention
In adults, the risk for many cancers can be reduced by avoiding certain risk factors, such as smoking. But there are no known avoidable risk factors for retinoblastoma. If your child does develop retinoblastoma, it’s important to realize that you or your child did nothing to cause it.
Some gene changes that put a child at high risk of retinoblastoma can be passed on from a parent. Children born to a parent with a history of retinoblastoma should be screened for this cancer starting shortly after birth because early detection of this cancer greatly improves the chance for successful treatment.
Genetic testing enables families to know which children have an increased risk of retinoblastoma, so eye exams can begin at an early age. That way, retinoblastoma may be diagnosed very early — when the tumor is small and a chance for a cure and preservation of vision is still possible.
If your doctor determines that your child’s retinoblastoma was caused by an inherited genetic mutation, your family may be referred to a genetic counselor.
Genetic testing can be used to determine whether:
- Your child with retinoblastoma is at risk of other related cancers
- Your other children are at risk of retinoblastoma and other related cancers, so they can start eye exams at an early age
- You and your partner have the possibility of passing the genetic mutation on to future children
The genetic counselor can discuss the risks and benefits of genetic testing and help you decide whether you, your partner or your other children will be tested for the genetic mutation.
Children with a family history of retinoblastoma should have eye exams to check for retinoblastoma.
A child with a family history of retinoblastoma should have regular eye exams beginning early in life to check for retinoblastoma, unless it is known that the child does not have the RB1 gene change. Early diagnosis of retinoblastoma may mean the child will need less intense treatment.
Brothers or sisters of a child with retinoblastoma should have regular eye exams by an ophthalmologist until age 3 to 5 years, unless it is known that the brother or sister does not have the RB1 gene change.
A child who has heritable retinoblastoma has an increased risk of trilateral retinoblastoma and other cancers.
A child with heritable retinoblastoma has an increased risk of a pineal tumor in the brain. When retinoblastoma and a brain tumor occur at the same time, it is called trilateral retinoblastoma. The brain tumor is usually diagnosed between 20 and 36 months of age. Regular screening using MRI (magnetic resonance imaging) may be done for a child thought to have heritable retinoblastoma or for a child with retinoblastoma in one eye and a family history of the disease. CT (computerized tomography) scans are usually not used for routine screening in order to avoid exposing the child to ionizing radiation.
Heritable retinoblastoma also increases the child’s risk of other types of cancer such as lung cancer, bladder cancer, or melanoma in later years. Regular follow-up exams are important.
Can Retinoblastoma Be Found Early?
Retinoblastoma is a rare cancer, and there are no widely recommended screening tests to look for retinoblastoma in children without symptoms. Still, many retinoblastomas are found early by parents, relatives, or a child’s doctor.
During children’s regular physical exams, doctors routinely check their eyes. Some of the things doctors look for include changes in how the eyes look (inside or outside), changes in how the eyes move, and changes in the child’s vision. Any of these might be a sign of retinoblastoma, although they are more often caused by something else.
Sometimes, a parent or relative may notice that a young child’s eye doesn’t look normal, prompting a visit to the doctor. It’s important for parents to be aware of the possible signs and symptoms of retinoblastoma, and to report anything unusual to the doctor as soon as possible. Most often the cause is something other than retinoblastoma, but it’s important to have it checked so that the cause can be found and treated right away, if needed.
For children in families known to carry an abnormal RB1 gene, or in families with a history of retinoblastoma who have not had genetic testing for the RB1 gene, doctors recommend regular eye exams during the first years of life to detect any tumors at an early stage. These children often have an eye exam a few days after birth, again at about 6 weeks of age, then every few months until at least age 5. The RB1 gene defect can be found by a special blood test, so most doctors now advise that children with parents or siblings with a history of retinoblastoma have this genetic test done during the first few weeks after birth. The results of the test then help define how often eye exams should be done.
Most hereditary retinoblastomas develop and are diagnosed in infants only a few months old. Usually, if tumors develop in both eyes, it happens at the same time. But in some children, tumors develop in one eye first, then a few months (or even years) later in the other eye. So even if retinoblastoma is diagnosed in only one eye, these children will still need regular exams of the other eye for several years after treatment.
If a child has retinoblastoma that is thought to be hereditary, many doctors also recommend magnetic resonance imaging (MRI) scans of the brain at regular intervals for up to 5 years to check for a trilateral retinoblastoma (a brain tumor such as a pineoblastoma).
Retinoblastoma signs and symptoms
Retinoblastomas nearly always occur in young children. They are often found when a parent or doctor notices a child’s eye looks unusual.
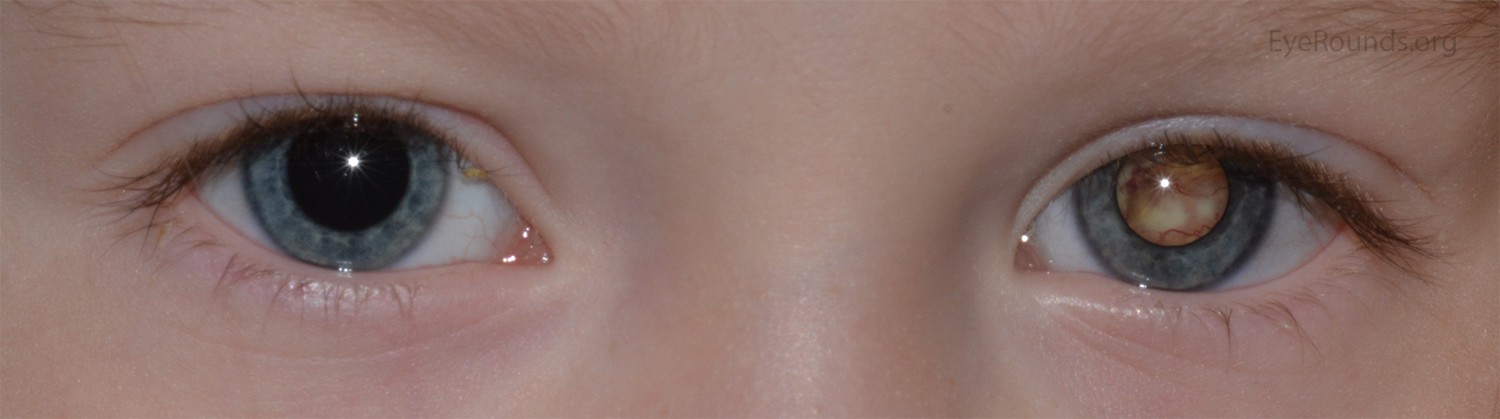
White pupillary reflex
This is the most common early sign of retinoblastoma. Normally when you shine a light in the eye, the pupil (the dark spot in the center of the eye) looks red because of the blood vessels in the back of the eye. In an eye with retinoblastoma, the pupil often appears white or pink instead, which is known as a white pupillary reflex (or leukocoria).
This white glare of the eye may be noticed by a parent after a flash photograph is taken, especially if the pupils are different colors. It also might be noted by the child’s doctor during a routine eye exam.
Lazy eye
Sometimes the eyes don’t appear to look in the same direction, a condition often called lazy eye. (Doctors call this strabismus.) There are many possible causes of this in children. Most of the time lazy eye is caused by a mild weakness of the muscles that control the eyes, but it can also be caused by retinoblastoma.
Other possible signs and symptoms
Less common signs and symptoms of retinoblastoma include:
- Vision problems
- Eye pain
- Redness of the white part of the eye
- Bleeding in the front part of the eye
- Bulging of the eye
- A pupil that doesn’t get smaller when exposed to bright light
- A different color in each iris (the colored part of the eye)
Many of these signs and symptoms are more likely to be caused by something other than retinoblastoma. Still, if your child has any of these, check with your child’s doctor so the cause can be found and treated, if needed.
Retinoblastoma Diagnosis
Retinoblastomas are usually found when a child is brought to a doctor because he or she has certain signs or symptoms.
For most types of cancer, the diagnosis is made with a biopsy. During a biopsy, the doctor removes a sample from the tumor and sends it to a lab to be looked at under a microscope.
But biopsies are not usually done to diagnose retinoblastoma for 2 reasons. First, taking a biopsy specimen from a tumor in the eye can’t be done easily without harming the eye and risking spreading cancer cells outside the eye. Second, retinoblastoma can usually be diagnosed accurately without a biopsy by doctors who have experience with this disease, and it’s unlikely to be confused with other eye problems in children.
Medical history and physical exam
If your child has signs or symptoms of retinoblastoma, the doctor will examine your child’s eyes and get a complete medical history. The doctor will ask about the child’s symptoms and may ask about any family history of retinoblastoma or other cancers. This information is important when deciding if more tests and exams are needed. Your family history is also useful for determining whether other relatives could possibly pass the retinoblastoma (RB1) gene change on to their children or develop this cancer themselves (if they are young children) and might benefit from genetic counseling.
If a retinoblastoma is suspected, the doctor will refer you to an ophthalmologist (a doctor who specializes in eye diseases), who will examine the eye closely to be more certain about the diagnosis. The ophthalmologist will use special lights and magnifying lenses to look inside the eye. Usually, the child needs to be under general anesthesia (asleep) during the exam so that the doctor can take a careful and detailed look.
If a diagnosis of retinoblastoma seems likely based on the eye exam, imaging tests will be done to help confirm it and to find out how far it may have spread within the eye and possibly to other parts of the body. Usually an ophthalmologist who specializes in treating cancers of the eye (called an ocular oncologist) will make the final determination. This doctor should also be part of the team of doctors treating the cancer.
Imaging tests
Imaging tests use x-rays, sound waves, magnetic fields, or radioactive substances to create pictures of the inside of the body. Imaging tests may be done for a number of reasons, including:
- To help tell if a tumor in the eye is likely to be a retinoblastoma
- To determine how large the tumor is and how far it has spread
- To help determine if treatment is working
Children thought to have retinoblastoma may have one or more of these tests.
Ultrasound
Ultrasound uses sound waves to create images of tissues inside the body, such as the inner parts of the eye. For this test, a small ultrasound probe is placed up against the eyelid or eyeball. The probe gives off sound waves and detects the echoes that bounce off the tissues inside and around the eye. The echoes are converted by a computer into an image on a computer screen.
Ultrasound is one of the most common imaging tests for confirming the diagnosis of retinoblastoma. It is painless and does not expose the child to radiation, but the child may need to be given medicine to help keep them calm or even asleep so the doctor can get a good look at the eye. This test can be very useful when tumors in the eye are so large they prevent doctors from seeing inside the whole eye.
Optical coherence tomography (OCT) is a similar type of test that uses light waves instead of sound waves to create very detailed images of the back of the eye.
Magnetic resonance imaging (MRI) scan
MRI scans are often used for retinoblastomas because they provide very detailed images of the eye and surrounding structures without using radiation. This test is especially good at looking at the brain and spinal cord. Most children with retinoblastoma will have at least one MRI scan. For children with bilateral retinoblastomas (tumors in both eyes), many doctors continue to do MRI scans of the brain for several years after treatment to look for tumors of the pineal gland (sometimes called trilateral retinoblastoma).
Unlike CT scans, MRI scans use radio waves and strong magnets to create images instead of x-rays. A contrast material called gadolinium may be injected into a vein before the scan to show details better.
MRI scans can take up to an hour. Your child may have to lie inside a narrow tube, which is confining and can be upsetting. Newer, more open MRI machines can help with this, but the test still requires staying still for long periods of time. The machines also make buzzing and clicking noises that may be disturbing. Young children may be given medicine to help keep them calm or even asleep during the test.
Computed tomography (CT) scan
CT scans use x-rays to make detailed images of parts of the body. CT scans can help determine the size of a retinoblastoma tumor and how much it has spread within the eye and to nearby areas.
Normally, either a CT or an MRI scan is needed, but usually not both. Because CT scans give off radiation, which might raise a child’s risk for other cancers in the future, most doctors prefer to use MRI. However, a CT scan can show deposits of calcium in the tumor much better than an MRI, which can be very helpful when the diagnosis of retinoblastoma is not clear.
Instead of taking one picture, like a regular x-ray, a CT scanner takes many pictures as it rotates around your child while he or she lies on a table. A computer then combines these pictures into images of slices of the part of the body being studied.
Before the scan, your child may get an IV (intravenous) injection of a contrast dye that helps better outline structures in the body. The dye can cause some flushing (a feeling of warmth, especially in the face). Some people are allergic and get hives. Rarely, more serious reactions like trouble breathing or low blood pressure can occur. Be sure to tell the doctor if your child has any allergies (especially to iodine or shellfish) or has ever had a reaction to any contrast material used for x-rays.
CT scans take longer than regular x-rays, but not as long as MRI scans. A CT scanner has been described as a large donut, with a narrow table that slides in and out of the middle opening. Your child will need to lie still on the table while the scan is being done. Your child may be given medicine to help them stay calm or even go to sleep during the test to help make sure the pictures come out well.
Bone scan
A bone scan can help show if the retinoblastoma has spread to the skull or other bones. Most children with retinoblastoma don’t need to have a bone scan. It is normally used only when there is a strong reason to think retinoblastoma might have spread beyond the eye.
For this test, a small amount of low-level radioactive material is injected into a vein (intravenously, or IV). (The amount of radioactivity used is very low and will pass out of the body within a day or so.) The material settles in abnormal areas of bone throughout the body over the course of a couple of hours. Your child then lies on a table for about 30 minutes while a special camera detects the radioactivity and creates a picture of the skeleton. Younger children may be given medicine to help them stay calm or even go to sleep during the test.
Areas of active bone changes appear as “hot spots” on the scan. These areas may suggest cancer is in an area, but other bone diseases can also cause the same pattern. To help tell these apart, other tests such as plain x-rays or MRI scans of the bone might be needed.
How Is Retinoblastoma Staged?
The stage of cancer describes how far it has spread. The outlook (prognosis) for children with retinoblastoma depends, to some extent, on the cancer’s stage. The stage is also an important factor in choosing treatment.
Retinoblastoma is staged based on the results of eye exams, imaging tests, and any biopsies that were done.
A staging system is a standard way for your child’s cancer care team to sum up how far a cancer has spread. Doctors use staging systems to predict the outlook for saving the child’s vision, as well as for survival and the likelihood that certain treatments will be effective.
Several detailed systems can be used to stage retinoblastoma. But for practical purposes, when determining the best treatment options, doctors often divide retinoblastomas into 2 main groups:
- Intraocular retinoblastoma: The cancer is still within the eye.
- Extraocular retinoblastoma: The cancer has spread beyond the eye. Extraocular cancers can be divided further into orbital retinoblastomas, which have spread only to the eye socket, and metastatic retinoblastomas, which have spread to distant parts of the body, such as the brain or bone marrow.
In the United States, most retinoblastomas are diagnosed before they have spread outside the eye, so staging systems that apply only to intraocular retinoblastoma are used most often in this country. There are 2 staging systems for intraocular retinoblastomas.
It’s important to know that regardless of the stage, almost all children with intraocular retinoblastoma can be cured if they are properly treated. But the stage has a bigger impact on whether the affected eye (or the vision in the eye) can be saved.
International Classification for Intraocular Retinoblastoma
The International Classification for Intraocular Retinoblastoma is the newer staging system, which takes into account what has been learned about the disease in recent decades. Most doctors now use this system. It divides intraocular retinoblastomas into 5 groups, labeled A through E, based on the chances that the eye can be saved using current treatment options.
Group A
Small tumors (3 millimeters [mm] across or less) that are only in the retina and are not near important structures such as the optic disc (where the optic nerve enters the retina) or the foveola (the center of vision).
Group B
All other tumors (either larger than 3 mm or small but close to the optic disc or foveola) that are still only in the retina.
Group C
Well-defined tumors with small amounts of spread under the retina (subretinal seeding) or into the jelly-like material that fills the eye (vitreous seeding).
Group D
Large or poorly defined tumors with widespread vitreous or subretinal seeding. The retina may have become detached from the back of the eye.
Group E
The tumor is very large, extends near the front of the eye, is bleeding or causing glaucoma (high pressure inside the eye), or has other features that mean there is almost no chance the eye can be saved.
The Reese-Ellsworth staging system
The Reese-Ellsworth system was developed in the 1960s, when most children were being treated with external beam radiation therapy. While this is no longer a common treatment, some doctors may still use this system to classify retinoblastomas that have not spread beyond the eye. This system can help determine the likelihood of preserving vision while still treating the tumor.
Terms such as favorable, doubtful, and unfavorable used in this staging system refer to the likelihood that the cancer can be treated while preserving the affected eye. These terms do not refer to the likelihood of the child’s survival. Indeed, more than 9 in 10 children with intraocular retinoblastomas are cured. The major challenge is saving the vision in the affected eye.
To explain the groupings below, it helps to define a few terms. The optic disc is the end of the optic nerve where it is attached to the retina. Retinoblastomas are diagnosed by looking at the retina through an ophthalmoscope, so doctors can’t measure their size directly using a ruler. Instead they compare the size of the tumor with the size of the optic disc, which is usually about 1.5 millimeters (1/16 inch) across. For example, a tumor estimated to be 3 times the size of the disc (3 disc diameters or 3 DD) would be about 4.5 millimeters (3/16 inch) across.
The equator is an imaginary line that divides the front and back halves of the eyeball.
The Reese-Ellsworth staging system divides intraocular retinoblastoma into 5 groups. The higher the group number, from 1 to 5, the lower the chance of controlling the retinoblastoma or of saving the eye or any useful vision.
Group 1 (very favorable for saving [or preserving] the eye)
- 1A: one tumor, smaller than 4 disc diameters (DD), at or behind the equator
- 1B: multiple tumors smaller than 4 DD, all at or behind the equator
Group 2 (favorable for saving [or preserving] the eye)
- 2A: one tumor, 4 to 10 DD, at or behind the equator
- 2B: multiple tumors, with at least one 4 to 10 DD, and all at or behind the equator
Group 3 (doubtful for saving [or preserving] the eye)
- 3A: any tumor in front of the equator
- 3B: one tumor, larger than 10 DD, behind the equator
Group 4 (unfavorable for saving [or preserving] the eye)
- 4A: multiple tumors, some larger than 10 DD
- 4B: any tumor extending toward the front of the eye to the ora serrata (front edge of the retina)
Group 5 (very unfavorable for saving [or preserving] the eye)
- 5A: tumors involving more than half of the retina
- 5B: vitreous seeding (spread of tumors into the jelly-like material that fills the eye)
Other staging systems
Other staging systems that include both intraocular retinoblastomas and those that have spread beyond the eye (extraocular retinoblastomas) may be used by some doctors. These can be especially useful in countries where these cancers are more likely to have spread by the time they are found. For example, the American Joint Commission on Cancer (AJCC) staging system takes into account 3 key pieces of information:
- T: The size of the main (primary) tumor and how far it has grown within and outside of the eye
- N: Whether or not the cancer has reached the lymph nodes (small, bean shaped collections of immune cells, to which cancers sometimes spread)
- M: Whether or not the cancer has spread (metastasized) to distant parts of the body, such as the bone marrow, brain, skull, or long bones
This system can be used to describe the extent of retinoblastomas in detail, particularly for those that have spread outside the eye, which rarely happens in the United States.
Be sure to ask your child’s doctor if you have any questions about the stage of your child’s cancer.
Retinoblastoma treatment
Retinoblastoma is rare, so not many doctors other than those in specialty eye hospitals and major children’s cancer centers have much experience treating it. Children with retinoblastoma and their families have special needs that can best be met by these children’s cancer centers. These centers have teams of specialists who know about retinoblastoma and the unique needs of children with cancer. This gives the child the best chance for recovery and, if possible, keeping their sight.
If your child has retinoblastoma, be sure he or she is treated at a children’s cancer center that has expertise in treating children with this rare form of cancer. Ask about the services offered at your treatment center. Your child’s doctor or nurse can tell you what is available to help with any problems you or your child might have.
Children with retinoblastoma are treated by a team of doctors that often includes:
- A pediatric ophthalmologist: a doctor who treats eye diseases in children
- An ocular oncologist: a doctor (usually an ophthalmologist) who treats cancers of the eye
- A pediatric oncologist: a doctor who treats children with cancer
- A radiation oncologist: a doctor who treats cancer with radiation therapy
The team might also include other doctors, physician assistants, nurse practitioners, nurses, therapists, child psychologists, social workers, genetic counselors, and other professionals. Having a child go through cancer treatment often means meeting lots of specialists and learning about parts of the medical system you probably haven’t been exposed to before.
Retinoblastoma treatment principles
The goals of treatment for retinoblastoma are:
- To get rid of the cancer and save the child’s life
- To save the eye if possible
- To preserve as much vision as possible
- To limit the risk of second cancers later in life, which can be caused by treatment, particularly in children with hereditary retinoblastoma
The most important factors that help determine treatment are:
- Whether the tumor is just in one eye or both
- How good the vision in the eye is
- Whether the tumor has extended outside the eye
Overall, more than 9 in 10 children with retinoblastoma are cured. The chances of long-term survival are much better if the tumor has not spread outside the eye.
The main types of treatment for retinoblastoma are:
- Surgery
- Radiation therapy
- Photocoagulation (using lasers to kill small tumors or the blood vessels that feed them)
- Cryotherapy (using cold to freeze and kill small tumors)
- Thermotherapy (using a type of laser to apply heat to kill small tumors)
- Chemotherapy
- High-dose chemotherapy and stem cell transplant
Sometimes more than one type of treatment may be used. The treatment options are based on the extent of the cancer and other factors.
Retinoblastoma treatment options
The best treatments for your child’s retinoblastoma depend on the size and location of the tumor, whether cancer has spread to areas other than the eye, your child’s overall health and your own preferences. When possible, your child’s doctor will work to preserve your child’s vision.
Chemotherapy
Chemotherapy is a drug treatment that uses chemicals to kill cancer cells. Chemotherapy can be taken in pill form, or it can be given through a blood vessel. Chemotherapy drugs travel throughout the body to kill cancer cells.
In children with retinoblastoma, chemotherapy may help shrink a tumor so that another treatment, such as radiation therapy, cryotherapy, thermotherapy or laser therapy, may be used to treat the remaining cancer cells. This may improve the chances that your child won’t need surgery.
Chemotherapy may also be used to treat retinoblastoma that has spread to tissues outside the eyeball or to other areas of the body.
Chemo can be given in different ways.
- Systemic chemotherapy: In most cases, chemo drugs are injected into a vein (IV) or given by mouth. These drugs enter the bloodstream and reach throughout the body. This is known as systemic chemotherapy.
- Periocular (subtenon) chemotherapy: For some advanced intraocular cancers, higher doses of chemo are needed inside the eye. Along with systemic chemotherapy, one of the drugs (carboplatin) may be injected in the tissues around the eye, where it slowly diffuses into the eyeball. This is called periocular or subtenon chemotherapy. These injections are done while the child is under anesthesia (asleep). This can cause redness and swelling around the eye.
- Intra-arterial chemotherapy: A newer approach sometimes used instead of systemic chemotherapy is to inject chemo directly into the ophthalmic artery, the main artery that supplies blood to the eye. In this technique, a very thin catheter (a long, hollow, flexible tube) is inserted into a large artery on the inner thigh and slowly threaded through the blood vessels all the way up into the ophthalmic artery. (This is done with the child asleep under general anesthesia.) The chemo is then infused into the artery. The drug used most often is melphalan, but other drugs such as carboplatin and topotecan can also be used. This process may then be repeated every few weeks, depending on how much the tumor shrinks. Because the chemo is put directly into the artery feeding the eye, doctors can use much smaller doses of chemo drugs (less than 10% of the doses used for systemic chemo). Therefore, there are fewer side effects from the chemo. Early results with this technique in eyes with advanced tumors have been promising, generally with good tumor control and few side effects. In most cases it has allowed doctors to save an eye that otherwise would have needed to be removed.
- Intravitreal chemotherapy: In this newer approach, chemotherapy is given directly into the vitreous humor, the jelly-like substance inside the eye.
Uses of chemotherapy
Chemotherapy may be used in different situations:
- Chemo can be used as the first treatment to shrink some tumors that have not spread outside the eye. This is called chemoreduction. These tumors can then be treated more effectively with focal therapies such as laser therapy, cryotherapy, thermotherapy, or brachytherapy.
- Systemic (IV) chemo may be given to children whose tumors don’t seem to have spread beyond the eye, but might be likely to spread because of the tumor’s size and/or location.
- Chemo is sometimes used when the eye has already been removed, but the tumor was found to extend into some areas in the eye that make it more likely to have spread. This type of treatment is called adjuvant chemotherapy.
- Systemic chemo is also used to treat children whose retinoblastoma has spread beyond the eye, a much more critical situation. If the cancer has spread to the brain, chemo may also be given directly into the cerebrospinal fluid that surrounds it (known as intrathecal chemotherapy). Tumors outside the eye may shrink for a time with standard doses of chemo, but they will usually start growing again. For this reason, doctors often prefer to give more intense chemo, usually along with a stem cell transplant.
Doctors give systemic chemo in cycles, with each period of treatment followed by a rest period to give the body time to recover. Each chemo cycle typically lasts for a few weeks, and the total length of treatment is often several months.
Some of the drugs used to treat children with retinoblastoma include:
- Carboplatin
- Cisplatin
- Vincristine
- Etoposide
- Cyclophosphamide
- Topotecan
- Doxorubicin
Most often, 2 or 3 drugs are given at the same time. A standard combination used to shrink intraocular retinoblastomas is carboplatin, vincristine, and etoposide, although for very small tumors, only carboplatin and vincristine may be enough. Other drugs might be used if these are not effective.
Possible side effects
Chemo drugs attack cells that are dividing quickly, which is why they work against cancer cells. But other cells in the body, such as those in the bone marrow (where new blood cells are made), the lining of the mouth and intestines, and the hair follicles, also divide quickly. These cells are also likely to be affected by chemo, which can lead to side effects.
Children tend to have less severe side effects from chemo and to recover from side effects more quickly than adults do. One benefit of this is that doctors can give them the high doses of chemo needed to kill the tumor.
The side effects of chemo depend on the type of drugs, the doses used, and how long they are given. Possible short-term side effects include:
- Hair loss
- Mouth sores
- Loss of appetite
- Nausea and vomiting
- Diarrhea or constipation
- Increased chance of infections (from having too few white blood cells)
- Easy bruising or bleeding (from having too few blood platelets)
- Fatigue (from having too few red blood cells)
Most of these side effects go away after treatment is finished. There are often ways to lessen these side effects. For example, drugs can be given to help prevent or reduce nausea and vomiting. Be sure to discuss any questions about side effects with your child’s cancer care team, and let them know if your child has side effects so they can be managed.
Along with those listed above, certain chemo drugs can cause specific side effects. For example:
- Cisplatin and carboplatin can affect the kidneys. Giving the child plenty of fluids during treatment can help reduce this risk. These drugs can also cause hearing loss in young children, especially in babies younger than 6 months. Your child’s doctor may check your child’s hearing with tests during or after treatment. When carboplatin is injected directly into the tissues near the eye (periocular chemotherapy), it can cause redness and swelling in the area.
- Vincristine can damage nerves. Some children may feel tingling and numbness, particularly in their hands and feet.
Some drugs, such as etoposide, doxorubicin, and cyclophosphamide, can increase the risk of developing a cancer of white blood cells known as acute myeloid leukemia (AML) later in life. Fortunately, this is not common. - Doxorubicin can damage the heart. The risk of this happening goes up with the total amount of the drug given. Doctors try to limit this risk as much as possible by not giving more than the recommended doses and by checking the heart with an echocardiogram (an ultrasound of the heart) during treatment.
- Cyclophosphamide can damage the bladder, which can cause blood in the urine. This risk can be lowered by giving this drug along with plenty of fluids and with a drug called mesna, which helps protect the bladder.
Radiation therapy
Radiation therapy uses high-energy beams, such as X-rays, to kill cancer cells. Two types of radiation therapy used in treating retinoblastoma include:
- Internal radiation (brachytherapy) or plaque radiotherapy. During internal radiation, the treatment device is temporarily placed in or near the tumor. Internal radiation for retinoblastoma uses a small disk made of radioactive material. The disk is stitched in place and left for a few days while it slowly gives off radiation to the tumor. Placing radiation near the tumor reduces the chance that treatment will affect healthy eye tissue.
- External beam radiation. External beam radiation delivers high-powered beams to the tumor from a large machine outside of the body. As your child lies on a table, the machine moves around your child, delivering the radiation. External beam radiation can cause side effects when radiation beams reach the delicate areas around the eye, such as the brain. For this reason, external beam radiation is typically reserved for children with advanced retinoblastoma and those for whom other treatments haven’t worked.
Figure 4. Internal radiation (plaque radiotherapy)
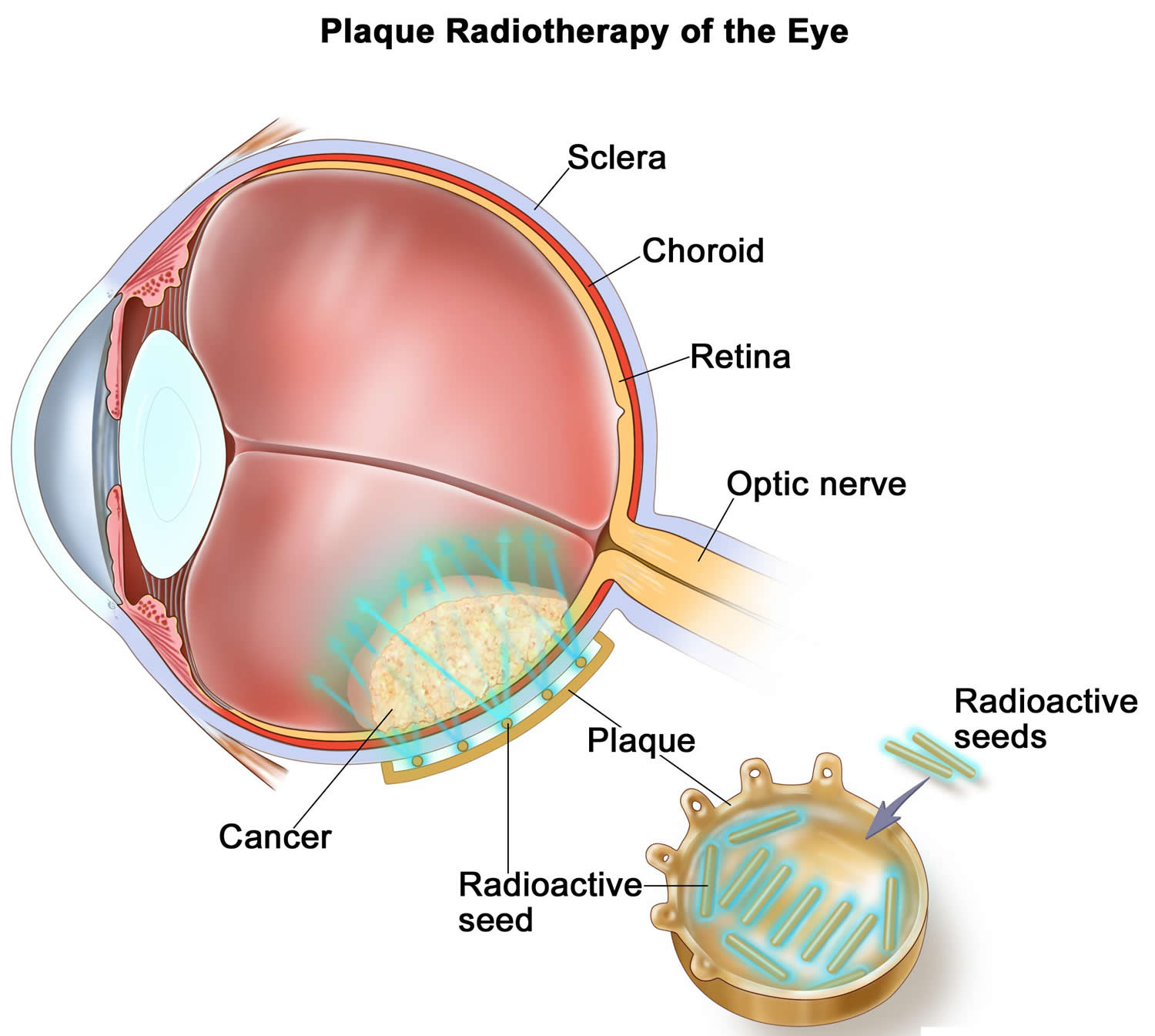 Note: Radioactive seeds are placed on one side of a thin piece of metal (usually gold) called a plaque. The plaque is sewn onto the outside wall of the eye. The seeds give off radiation which kills the cancer. The plaque is removed at the end of treatment, which usually lasts for several days.
Note: Radioactive seeds are placed on one side of a thin piece of metal (usually gold) called a plaque. The plaque is sewn onto the outside wall of the eye. The seeds give off radiation which kills the cancer. The plaque is removed at the end of treatment, which usually lasts for several days.
Laser therapy (laser photocoagulation)
During laser therapy, a laser is used to destroy blood vessels that supply oxygen and nutrients to the tumor. Without a source for fuel, cancer cells may die.
Cold treatments (cryotherapy)
Cryotherapy uses extreme cold to kill cancer cells.
During cryotherapy, a very cold substance, such as liquid nitrogen, is placed in or near the cancer cells. Once the cells freeze, the cold substance is removed and the cells thaw. This process of freezing and thawing, repeated a few times in each cryotherapy session, causes the cancerous cells to die.
Heat treatments (thermotherapy)
Thermotherapy uses extreme heat to kill cancer cells. During thermotherapy, heat is directed at the cancer cells using ultrasound, microwaves or lasers.
Surgery
When the tumor is too large to be treated by other methods, surgery may be used to treat retinoblastoma. In these situations, surgery to remove the eye may help prevent the spread of cancer to other parts of the body. Surgery for retinoblastoma includes:
- Surgery to remove the affected eye (enucleation). During surgery to remove the eye, surgeons disconnect the muscles and tissue around the eye and remove the eyeball. A portion of the optic nerve, which extends from the back of the eye into the brain, also is removed.
- Surgery to place an eye implant. Immediately after the eyeball is removed, the surgeon places a special ball — made of plastic or other materials — in the eye socket. The muscles that control eye movement are attached to the implant. After your child heals, the eye muscles will adapt to the implanted eyeball, so it may move just as the natural eye did. However, the implanted eyeball cannot see.
- Fitting an artificial eye. Several weeks after surgery, a custom-made artificial eye can be placed over the eye implant. The artificial eye can be made to match your child’s healthy eye. The artificial eye sits behind the eyelids and clips onto the eye implant. As your child’s eye muscles move the eye implant, it will appear that your child is moving the artificial eye.
Side effects of surgery include infection and bleeding. Removing an eye will affect your child’s vision, though most children will adapt to the loss of an eye over time.
High-Dose Chemotherapy and Stem Cell Transplant for Retinoblastoma
Doctors are studying the use of this type of treatment in children with retinoblastoma that has spread outside the eye and who are unlikely to be cured with other treatments.
A stem cell transplant lets doctors give higher doses of chemotherapy (chemo) than could safely be given otherwise). In the past, this type of treatment was commonly referred to as a bone marrow transplant.
The bone marrow is the soft, inner part of some bones where new blood cells are made. Chemo drugs can affect quickly dividing cells like those in the bone marrow. Even though higher doses of these drugs might be more effective in treating tumors, they can’t be given because they would cause severe damage to bone marrow cells, leading to life-threatening shortages of blood cells.
To try to get around this problem, the doctor may treat the child with high-dose chemo (sometimes along with radiation therapy) and then use a stem cell transplant to “rescue” the bone marrow.
How is it done?
The first step in a stem cell transplant is to collect, or harvest, the child’s own blood-making stem cells to use later. (These are the cells that make the different types of blood cells.)
In the past, the stem cells were often taken from the child’s bone marrow, which required a minor operation. But doctors have found that these cells can be taken from the bloodstream during a procedure similar to donating blood. Instead of going into a collecting bag, the blood goes into a special machine that filters out the stem cells and returns the rest of the blood to the child’s body. The stem cells are then frozen until the transplant. This process may need to be done more than once.
Once the stem cells have been stored, the child gets high-dose chemotherapy in the hospital, sometimes along with radiation therapy. When the treatment is finished (a few days later), the stem cells are thawed and returned to the body in a process similar to a normal blood transfusion. The stem cells travel through the blood and settle in the bone marrow.
Over the next few weeks, the stem cells start to make new blood cells. Until this happens, the child is at high risk of infection because of a low white blood cell count, as well as bleeding because of a low platelet count. To avoid infection, protective measures are taken, such as using special air filters in the hospital room and having visitors wear protective clothing. Blood and platelet transfusions and treatment with IV antibiotics may also be used to prevent or treat infections or bleeding problems.
The child can usually leave the hospital once their blood cell counts return to a safe level. They may then need to make regular visits to the outpatient clinic for about 6 months, after which time their care may be continued by their regular doctors.
Practical points
A stem cell transplant is a complex treatment that can cause life-threatening side effects. If the doctors think your child can benefit from a transplant, the best place to have it done is at a nationally recognized cancer center where the staff has experience with the procedure and with managing the recovery period.
A stem cell transplant often requires a long hospital stay and can be very expensive, often costing well over $100,000. Be sure to get a written approval from your insurer before treatment if it is recommended for your child. Even if the transplant is covered by your insurance, your co-pays or other costs could easily amount to many thousands of dollars. It’s important to find out what your insurer will cover before the transplant to get an idea of what you might have to pay.
Possible side effects
The possible side effects from stem cell transplant are generally divided into early and long-term effects.
Short-term, early side effects: The early complications and side effects are basically the same as those listed in the chemotherapy for retinoblastoma section, but they can be more severe because the drug doses are higher. Side effects can include:
- Low blood cell counts (with fatigue and an increased risk of infection and bleeding)
- Nausea and vomiting
- Loss of appetite
- Mouth sores
- Diarrhea
- Hair loss
One of the most common and serious short-term effects is an increased risk of serious infections. Antibiotics are often given to try to keep this from happening. Other side effects, like low red blood cell and platelet counts, may require blood product transfusions or other treatments.
Long-term and late side effects: Some complications and side effects can last for a long time or might not occur until months or years after the transplant. These can include:
- Radiation damage to the lungs
- Problems with the thyroid or other hormone-making glands
- Problems with fertility
- Damage to bones or problems with bone growth
- Development of another cancer (including leukemia) years later
Be sure to talk to your child’s doctor before the transplant to learn about possible long-term effects your child might have.
Clinical trials
Clinical trials are studies to test new treatments and new ways of using existing treatments. While clinical trials give your child a chance to try the latest in retinoblastoma treatments, they can’t guarantee a cure.
Ask your child’s doctor whether your child is eligible to participate in clinical trials. Your child’s doctor can discuss the benefits and risks of enrolling in a clinical trial.
New types of treatment are being tested in clinical trials.
- Targeted therapy
Targeted therapy is a type of treatment that uses drugs or other substances to attack cancer cells. Targeted therapies usually cause less harm to normal cells than chemotherapy or radiation therapy do.
Targeted therapy is being studied for the treatment of retinoblastoma that has recurred (come back).
Coping and support
When your child is diagnosed with cancer, it’s common to feel a range of emotions — from shock and disbelief to guilt and anger. Everyone finds his or her own way of coping with stressful situations, but if you’re feeling lost, you might try to:
- Gather all the information you need. Find out enough about retinoblastoma to feel comfortable making decisions about your child’s care. Talk with your child’s health care team. Keep a list of questions to ask at the next appointment. Visit your local library and ask for help searching for information. Consult the websites of the National Cancer Institute and the American Cancer Society for more information.
- Organize a support network. Find friends and family who can help support you as a caregiver. Loved ones can accompany your child to doctor visits or sit by his or her bedside in the hospital when you can’t be there. When you’re with your child, your friends and family can help out by spending time with your other children or helping around your house.
- Take advantage of resources for kids with cancer. Seek out special resources for families of kids with cancer. Ask your clinic’s social workers about what’s available. Support groups for parents and siblings put you in touch with people who understand what you’re feeling. Your family may be eligible for summer camps, temporary housing and other support.
- Maintain normalcy as much as possible. Small children can’t understand what’s happening to them as they undergo cancer treatment. To help your child cope, try to maintain a normal routine as much as possible. Try to arrange appointments so that your child can have a set nap time each day. Have routine mealtimes. Allow time for play when your child feels up to it. If your child must spend time in the hospital, bring items from home that help him or her feel more comfortable.
Ask your health care team about other ways to comfort your child through his or her treatment. Some hospitals have recreation therapists or child-life workers who can give you more specific ways to help your child cope.




{kind=link}