What is adrenoleukodystrophy
Adrenoleukodystrophy also known as X-linked adrenoleukodystrophy (X-ALD) or ALD disease, is an X-linked recessive genetic disorder caused by an abnormality in the ABCD1 gene on the X chromosome 1. The faulty ABCD1 gene means those affected are unable to process very long chain fatty acids (VLCFAs). Very long chain fatty acids (VLCFAs) are mainly esterified with cholesterol and glycerophospholipids, resulting in pathology 2. Studies show that other lipids such as free cholesterol, the oxysterols, gangliosides and the plasmalogens may also contribute to the pathophysiology of adrenoleukodystrophy 3. It is thought that these very long chain fatty acids (VLCFAs) accumulate and destroy the myelin sheath that covers the nerves in the body and brain. The myelin acts like the coating around an electric cable, and allows messages to be transmitted along nerve cells. In X-linked adrenoleukodystrophy (X-ALD), lipids containing very long chain fatty acids (VLCFAs) accumulate in all tissues; however, the brain, spinal cord, adrenal cortex and the Leydig cells of the testis have the greatest increase of VLCFA 4. Some of those affected experience serious neurological problems that can affect mental function and lead to disability and reduced life span. X-linked adrenoleukodystrophy (X-ALD) should not be confused with neonatal adrenoleukodsystrophy, which is a disease of newborns and young infants and belongs to the group of peroxisomal biogenesis disorders.
X-linked adrenoleukodystrophy (X-ALD) mainly affects the nervous system and the adrenal glands, which are small glands located on top of each kidney and in some cases testicular insufficiency 5. People with X-linked adrenoleukodystrophy (X-ALD) accumulate high levels of saturated, very long chain fatty acids (VLCFA) in the brain and adrenal cortex. The loss of myelin and the progressive dysfunction of the adrenal gland are the primary characteristics of adrenoleukodystrophy. While nearly all patients with adrenoleukodystrophy suffer from adrenal insufficiency, also known as Addison’s disease, the neurological symptoms can begin either in childhood or in adulthood. The childhood cerebral form is the most severe, with onset between ages 4 and 10. The most common symptoms are usually behavioral changes such as abnormal withdrawal or aggression, poor memory, and poor school performance. Other symptoms include visual loss, learning disabilities, seizures, poorly articulated speech, difficulty swallowing, deafness, disturbances of gait and coordination, fatigue, intermittent vomiting, increased skin pigmentation, and progressive dementia. The milder adult-onset form is also known as adrenomyeloneuropathy (AMN), which typically begins between ages 21 and 35. Symptoms may include progressive stiffness, weakness or paralysis of the lower limbs, and ataxia. Although adult-onset adrenoleukodystrophy progresses more slowly than the classic childhood form, it can also result in deterioration of brain function.
X-linked adrenoleukodystrophy is a genetic disorder that occurs primarily in males. Because females have two X chromosomes and are the carriers of the disease, but since men only have one X chromosome and lack the protective effect of the extra X chromosome, they are more severely affected. Almost half the women who are carriers of adrenoleukodystrophy will develop a milder form of adrenomyeloneuropathy (AMN); such carriers almost never develop symptoms that are seen in boys the X-linked adrenoleukodystrophy (X-ALD). Most men and women with adrenoleukodystrophy have a slowly progressive spinal cord disease, adrenomyeloneuropathy (AMN), men, typically beginning in their 30s, and women beginning postmenopausal 6. However, 35%–40%of adrenoleukodystrophy males may develop a rapidly progressive inflammatory cerebral demyelination peaking in the ages 3–10 years of age. About 20% of adult males with adrenomyeloneuropathy (AMN) also develop cerebral disease that rapidly progresses to disability and death 7. Additionally, the adrenal glands are commonly affected with a lifetime risk of adrenal insufficiency of ~80% in adrenoleukodystrophy males 8.
In cerebral adrenoleukodystrophy the damage to myelin happens in the brain. When the myelin is damaged the nerves in the brain cannot work properly, and the person’s physical and mental abilities begin to deteriorate. Functions such as reasoning, speech and mobility are lost. Eventually, they become completely dependent.
In most males with adrenoleukodystrophy the adrenal glands are also affected. Damage to the outer layer of the adrenal glands (adrenal cortex) causes a shortage of certain hormones such as adrenaline and cortisol (adrenocortical insufficiency), leading to abnormalities in blood pressure, heart rate, sexual development and reproduction. Adrenocortical insufficiency may cause weakness, weight loss, tiredness, skin changes, muscle pains, vomiting, and coma.
It is not possible to predict how the adrenoleukodystrophy gene will affect any one person: its effects can vary. Once diagnosis has been confirmed, the affected person needs to be closely monitored.
Adrenoleukodystrophy or ALD, is the most common leukodystrophy, accounting for about half of all leukodystrophies. The prevalence is approximately 1/20,000-1/50,000 births and most of those affected are boys. Approximately half of all females who carry the abnormal ABCD1 gene will develop some symptoms of ALD. Adrenoleukodystrophy occurs in all ethnic groups. The presentation of symptoms occurs somewhere between the ages of 4 and 10, and affects the brain with demyelination 9. Demyelination is the stripping away of the fatty coating that keeps nerve pulses confined and maintains the integrity of nerve signals. This process inhibits the nerves ability to conduct properly, thereby causing neurological deficits, including visual disturbances, auditory discrimination, impaired coordination, dementia, and seizures. Demyelination is an inflammatory response and nerve cells throughout the brain are destroyed.
Boys develop normally until the onset of symptoms occurs. Symptoms typically rival those of attention deficit disorder before serious neurological involvement becomes apparent. The symptoms progress rapidly and lead to vegetative state within two years, and death anytime thereafter.
Adrenocortical insufficiency, Addison’s Disease, is seen in 90 percent of the cases of ALD.
There are a wide range of clinical severities of X-linked adrenoleukodystrophy (X-ALD), and these have been classified into six broad categories: childhood cerebral ALD, adolescent cerebral ALD, adult cerebral, adrenomyeloneuropathy, adrenal insufficiency-only, and symptomatic heterozygotes. The clinical phenotypes of each are described below.
Adrenoleukodystrophy has been categorized into six types based on symptoms and age of onset:
- Childhood cerebral adrenoleukodystrophy (48% of cases): Childhood cerebral ALD is one of the most common forms of X-linked ALD, comprising approximately 30% of all patients with X-linked adrenoleukodystrophy. Onset of childhood cerebral adrenoleukodystrophy occurs between the ages of 2 and 10. Up to the point of onset, development is normal. The most common initial symptoms are difficulty in school, behavioral disturbance, impaired vision, or impaired hearing. After initial neurological symptoms appear, the health of the patients deteriorates rapidly. Further symptoms may include dementia, poor coordination, seizures, hyperactivity, difficulty with speech, and headaches. The average time between the initial symptoms and a vegetative state (where the patient is bedridden) or death is approximately 2 years, although it can range anywhere from 6 months to 20 years.
- Adolescent cerebral adrenoleukodystrophy (5% of cases): A small number of patients with X-linked adrenoleukodystrophy will present between the ages of 11 and 21 years. The symptoms are similar to those of childhood cerebral adrenoleukodystrophy, though progression of the disease may be somewhat slower.
- Adrenomyeloneuropathy (AMN) (26% of cases): Adrenomyeloneuropathy (AMN) is the most common form of adrenoleukodystrophy disease, and comprises approximately 40% of all X-linked adrenoleukodystrophy patients. The first symptoms of adrenomyeloneuropathy (AMN) usually occur in the twenties. Generally, initial symptoms noted are stiffness/clumsiness in the legs, weight loss, attacks of nausea, and generalized weakness. Adrenal impairment occurs, and other major manifestations may include difficulty with walking, urinary disturbance and impotence, cognitive defects, emotional disturbances, and depression. 70% of adrenomyelonueropathy (AMN) patients experience Addison’s disease. Adrenomyeloneuropathy (AMN) disease progresses slowly, and within 5 to 15 years the patient will generally need the aid of a cane or wheelchair.
- Adult cerebral adrenoleukodystrophy (3% of cases): Adult cerebral ALD is relatively rare, only representing approximately 3% of all ALD cases. Age of onset varies from the twenties to the fifties. The symptoms are similar to those of schizophrenia with dementia, and the progression of the disorder is rapid. The average time from the initial symptoms to vegetative state or death is approximately 3-4 years.
- Adrenoleukodystrophy that occurs in females: Women have two copies of the X chromosome, which is where the gene responsible for X-adrenoleukodystrophy resides. As a result, most women who carry a defective copy of a ABCD1 gene on the X-chromosome also carry one good copy of the ABCD1 gene, so they often won’t have any symptoms of the disease. However, some women who carry one good copy and one bad copy of the X-adrenoleukodystrophy gene (heterozygotes) do show some symptoms of adrenoleukodystrophy (symptomatic heterozygotes). The symptoms can range from very mild to very severe. They resemble those of other adrenoleukodystrophy patients, with the exception that heterozygote women rarely have impaired adrenal function.
- Adrenal insufficiency or Addison’s disease (10% of cases) only. Addison’s disease occurs when the adrenal glands do not produce enough of the hormone cortisol and, in some cases, the hormone aldosterone. Addison’s disease is also hypocortisolism.
Children with the cerebral form of X-linked adrenoleukodystrophy experience learning and behavioral problems that usually begin between the ages of 4 and 10. Over time the symptoms worsen, and these children may have difficulty reading, writing, understanding speech, and comprehending written material. Additional signs and symptoms of the cerebral form include aggressive behavior, vision problems, difficulty swallowing, poor coordination, and impaired adrenal gland function. The rate at which this disorder progresses is variable but can be extremely rapid, often leading to total disability within a few years. The life expectancy of individuals with this type depends on the severity of the signs and symptoms and how quickly the disorder progresses. Individuals with the cerebral form of X-linked adrenoleukodystrophy usually survive only a few years after symptoms begin but may survive longer with intensive medical support.
Signs and symptoms of the adrenomyeloneuropathy (AMN) type appear between early adulthood and middle age. Affected individuals develop progressive stiffness and weakness in their legs (paraparesis), experience urinary and genital tract disorders, and often show changes in behavior and thinking ability. Most people with the adrenomyeloneuropathy (AMN) type also have adrenocortical insufficiency. In some severely affected individuals, damage to the brain and nervous system can lead to early death.
People with X-linked adrenoleukodystrophy whose only symptom is adrenocortical insufficiency are said to have the Addison disease only form. In these individuals, adrenocortical insufficiency can begin anytime between childhood and adulthood. However, most affected individuals develop the additional features of the adrenomyeloneuropathy (AMN) type by the time they reach middle age. The life expectancy of individuals with this form depends on the severity of the signs and symptoms, but typically this is the mildest of the three types.
Rarely, individuals with X-linked adrenoleukodystrophy develop multiple features of the disorder in adolescence or early adulthood. In addition to adrenocortical insufficiency, these individuals usually have psychiatric disorders and a loss of intellectual function (dementia). It is unclear whether these individuals have a distinct form of the condition or a variation of one of the previously described types.
For reasons that are unclear, different forms of X-linked adrenoleukodystrophy can be seen in affected individuals within the same family.
Can female carriers display symptoms of adrenoleukodystrophy?
Women carriers of a defective adrenoleukodystrophy gene can display symptoms, though many remain asymptomatic for their entire lives. The symptoms vary dramatically, and tend to be displayed in later life. Symptoms may be similar to those of adrenomyeloneuropathy (AMN) (described earlier), though they are generally milder. Roughly half of all women who are carriers of adrenoleukodystrophy will eventually develop some sort of adrenomyeloneuropathy (AMN)-like symptoms in later life.
Can adrenoleukodystrophy be cured?
Sadly, once symptoms of cerebral adrenoleukodystrophy have appeared, there is currently no cure. However, it may be possible to prevent the disorder in people who carry the ABCD1 gene but have so far developed none or minor symptoms.
Why is genetic testing of the family important?
Once one member of a family has been diagnosed, it is very important to get the rest of your family checked genetically, especially if there are brothers, who could also have the disease. If the ABCD1 gene is discovered early, before symptoms appear, there is a chance of preventing adrenoleukodystrophy from developing. Sisters could be adrenoleukodystrophy carriers. When a boy is diagnosed with adrenoleukodystrophy it will most likely be inherited from his mother. You should be offered genetic counseling as soon as the diagnosis has been confirmed. Genetic tests are advised for all siblings and parents. The genetic counselor will talk you through the implications of the tests. If siblings are found to have adrenoleukodystrophy they should be considered for a preventative programme
Adrenoleukodystrophy causes
Mutations in the ABCD1 gene cause X-linked adrenoleukodystrophy. The ABCD1 gene provides instructions for producing the adrenoleukodystrophy protein (ALDP), which is involved in transporting certain fat molecules called very long-chain fatty acids into peroxisomes. Peroxisomes are small sacs within cells that process many types of molecules, including very long-chain fatty acids.
ABCD1 gene mutations result in a shortage (deficiency) of ALDP. When this protein is lacking, the transport and subsequent breakdown of very long-chain fatty acids is disrupted, causing abnormally high levels of these fats in the body. The accumulation of very long chain fatty acids (VLCFAs) may be toxic to the adrenal cortex and myelin. Research suggests that the accumulation of very long-chain fatty acids triggers an inflammatory response in the brain, which could lead to the breakdown of myelin. The destruction of these tissues leads to the signs and symptoms of X-linked adrenoleukodystrophy.
The faulty ABCD1 gene can also cause several related but different conditions, such as Asymptomatic ALD (for males affected by the gene who do not yet have symptoms). The other condition caused by the faulty ABCD1 gene is called adrenomyelonueropathy (AMN), which affects nerves in the spinal cord and body. Men that experience adrenomyelonueropathy (AMN) also endure demyelination, however, it most commonly restricts itself to the long tracts of the spinal column causing increasing difficulty with walking, as well as bladder and bowel disturbances over a period of decades. 70% of adrenomyelonueropathy (AMN) patients experience Addison’s disease. There is also an Addison’s-only form of the disease that appears to spare the nervous system. Many, but not all of the Addison’s-only patients develop adrenomyelonueropathy (AMN) characteristics later in life..
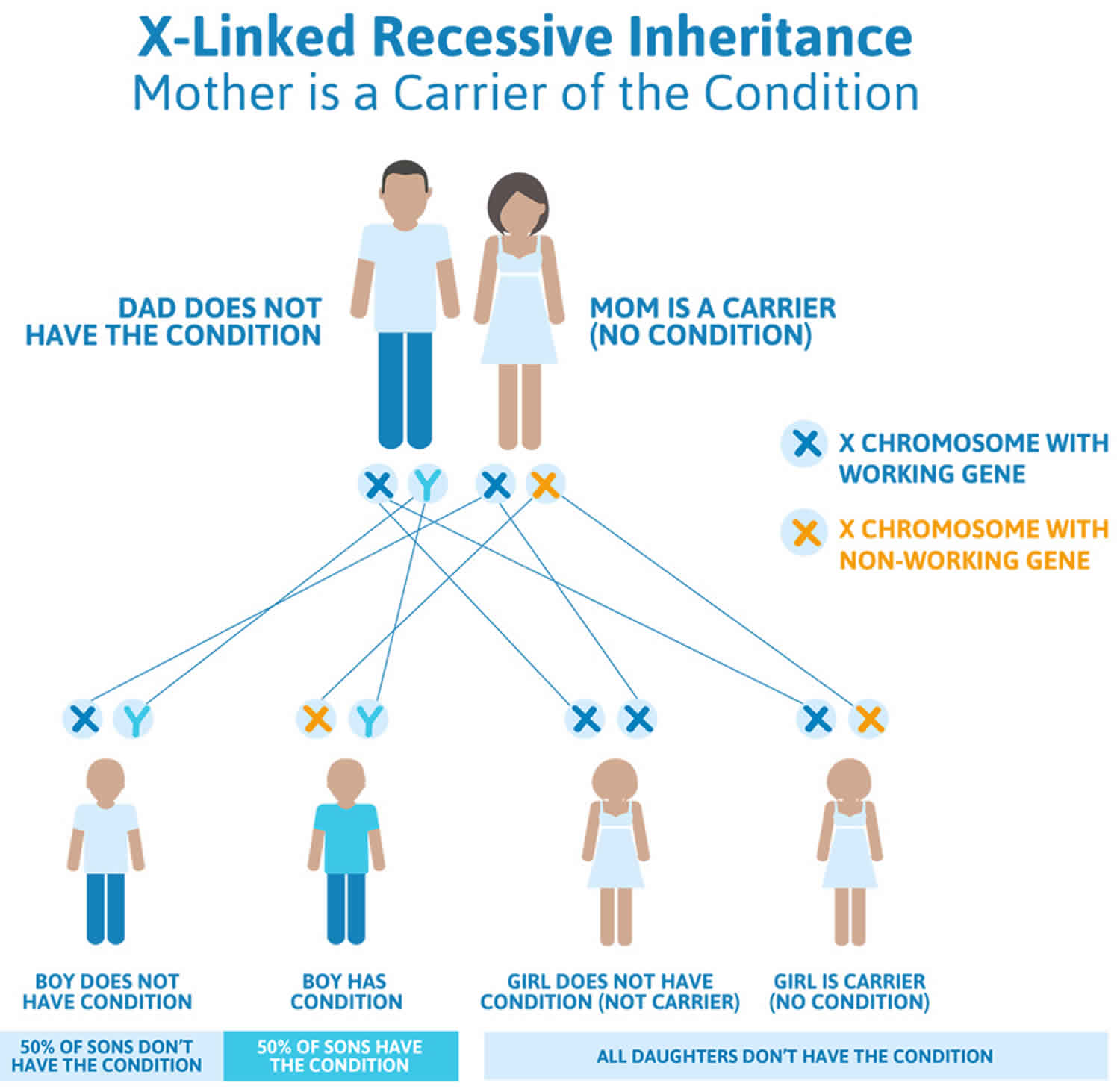
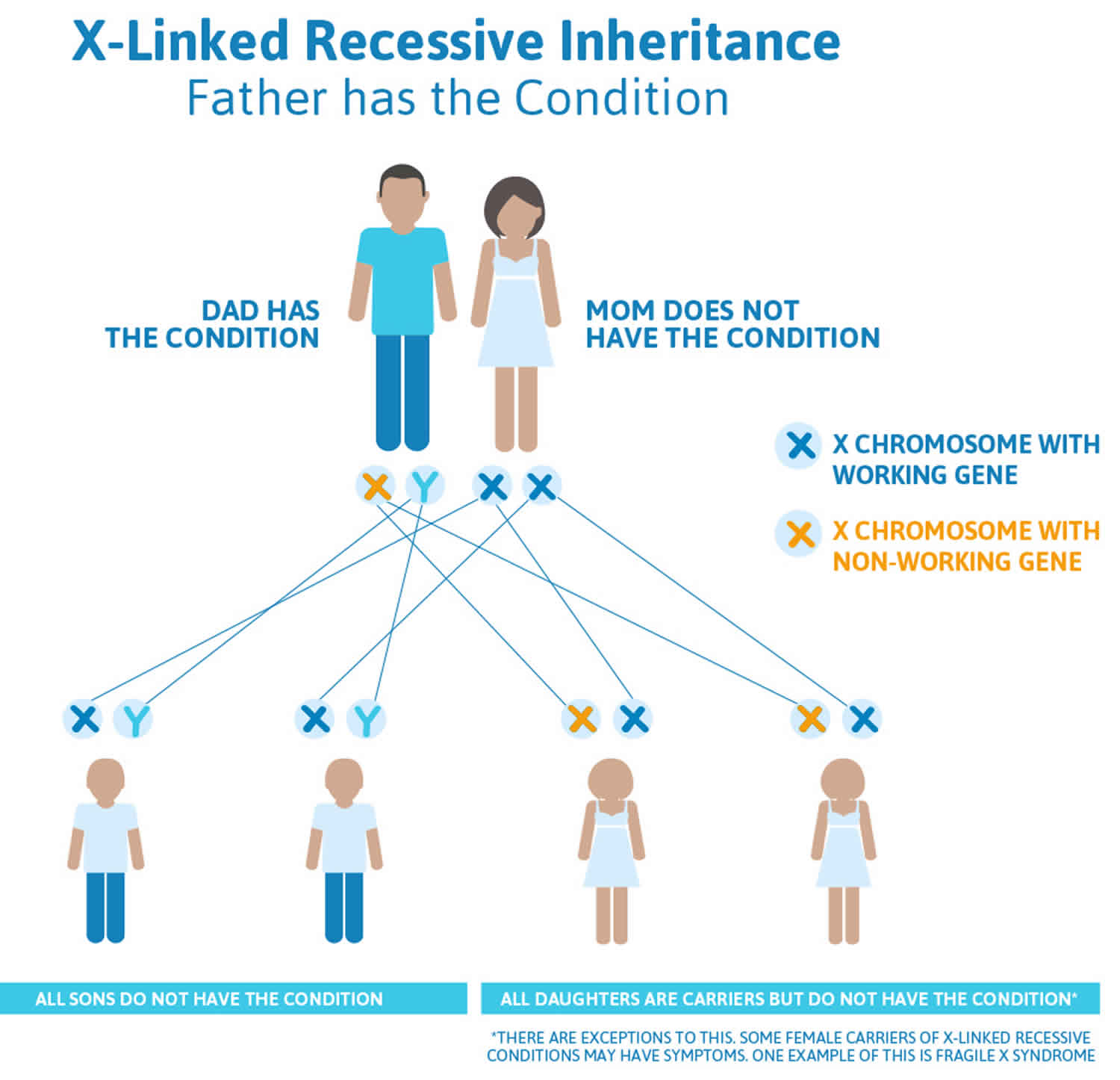
Adrenoleukodystrophy inheritance pattern
Adrenoleukodystrophy (ALD) is inherited as an X-linked recessive genetic disease. X-linked recessive genetic disorders are conditions caused by an abnormal gene on the X chromosome, one of the two sex chromosomes in each cell. Females have two X chromosomes but one of the X chromosomes is “turned off” and all of the genes on that chromosome are inactivated. Females who have a disease gene (ABCD1 gene) present on one of their X chromosomes are carriers for adrenoleukodystrophy (ALD). Carrier females often do not display symptoms of adrenoleukodystrophy (ALD) because it is usually the X chromosome with the abnormal gene that is turned off. Because females have two copies of the X chromosome, one altered copy of the ABCD1 gene in each cell usually does not cause any features of X-linked adrenoleukodystrophy; however, some females with one altered copy of ABCD1 gene have health problems associated with adrenoleukodystrophy (ALD). The signs and symptoms of X-linked adrenoleukodystrophy tend to appear at a later age in females than in males. Affected women usually develop features of the adrenomyeloneuropathy type.
A male has only one X chromosome and if he inherits an X chromosome that contains a disease gene (ABCD1 gene), he will develop adrenoleukodystrophy (ALD). Males with X-linked disorders pass the disease gene to all of their daughters, who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring. Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease, and a 25% chance to have an unaffected son.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counsellors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
- The National Cancer Institute provides a Cancer Genetics Services Directory (https://www.cancer.gov/about-cancer/causes-prevention/genetics/directory), which lists professionals who provide services related to cancer genetics. You can search by type of cancer or syndrome, location, and/or provider name.
If you have a health condition that has not been diagnosed, you may be interested in the Undiagnosed Diseases Network (https://undiagnosed.hms.harvard.edu/). They have information about how to apply for this multicenter research study.
Figure 1. Adrenoleukodystrophy X-linked inheritance pattern
Adrenoleukodystrophy phenotypes
Clinical presentation of X‐linked adrenoleukodystrophy phenotypes are summarized in Table 1. These phenotypes are commonly used to describe cerebral, adrenal and spinal cords and peripheral nerve involvement. While no genotype–phenotype correlation is known, phenotype shift to deadly cerebral disease is typically only seen in homozygous individuals 10. Further details regarding clinical have been comprehensively published in multiple reviews 11.
Table 1. Adrenoleukodystrophy Phenotypes
| Adrenoleukodystrophy Phenotypes | ||
|---|---|---|
| Presentation & pathology | Reported cumulative frequency and age of onset | |
| Phenotypes in MALES | ||
| Childhood cerebral adrenoleukodystrophy (CCALD) | Progressive behavioral, cognitive and neurologic deficit often leading to total disability and death within 4 years of diagnosis. Pathologic hallmark is inflammatory cerebral demyelination | 31%–35% Onset at 3–11 years of age |
| Adolescent cerebral adrenoleukodystrophy | Presentation & pathology as in childhood cerebral adrenoleukodystrophy (CCALD). Onset 11–21 years with somewhat slower progression than childhood cerebral adrenoleukodystrophy (CCALD) | 4%–7% Onset 11–21 years of age |
| Adrenomyeloneuropathy (AMN) | Characterized by weakness, spasticity, pain, bladder & bowel dysfunction and impaired movement often resulting in assistive device or wheelchair use. Pathology includes slow progressive distal axonopathy with atrophy of the spinal cord, and peripheral neuropathy | Most adult males will develop adrenomyeloneuropathy (AMN) Onset typically starting in third‐fourth decade of life |
| Adult cerebral adrenoleukodystrophy | Dementia, behavioral disturbances and focal neurologic deficits. Symptom progression may parallel childhood cerebral adrenoleukodystrophy (CCALD), however, rate of progression is variable with rare self‐limiting cerebral demyelination termed “arrested‐cerebral disease.” | 20% 7 |
| Addison’s disease only adrenoleukodystrophy | Primary adrenal involvement without apparent neurologic involvement. Most will continue to develop adrenomyeloneuropathy (AMN) | Common in childhood |
| Asymptomatic adrenoleukodystrophy | Biochemical and gene abnormality without demonstrable adrenal or neurologic deficit. Detailed studies often show adrenal hypofunction or subtle signs of adrenomyeloneuropathy (AMN) on examination in adulthood | Common in childhood. 50% of asymptomatic develop adrenomyeloneuropathy (AMN) within 10 years |
| Phenotypes in FEMALES | ||
| Asymptomatic | No evidence of adrenal or neurologic involvement | |
| Adrenomyeloneuropathy. Mild, moderate and severe | Symptomatology resembles adrenomyeloneuropathy (AMN) in men, albeit with later onset and a slower rate of progression | Increases with age. Estimates of 50% >40 and ca. 65% by 65 |
| Cerebral involvement | Rare, reported in cases with confirmed and suspected X chromosomal inactivation | Few cases reported 12 |
| Addison’s disease | Rare in females and does not precede adrenomyeloneuropathy (AMN) phenotype as seen in males | 1.00% |
Adrenoleukodystrophy symptoms
Adrenoleukodystrophy symptoms can vary depending on age, gender, and the body tissues affected. The tissues that are most severely affected in adrenoleukodystrophy are myelin, blood, and the adrenal glands. Not all tissues are affected at the same time in all patients. In the world of genetic disorders, doctors group collections of symptoms into “phenotypes” based on the cells and tissues that are most severely affected by a gene abnormality. Individuals with the adrenoleukodystrophy gene may have different phenotypes. In adrenoleukodystrophy, the phenotypes are not mutually exclusive. In fact, it is common for individuals to have more than one phenotype at any given time.
The childhood cerebral form of adrenoleukodystrophy usually begins between four and eight years of age and symptoms include attention deficit disorder, progressive loss of intellectual function, and vision, hearing and motor deterioration. Adolescent cerebral adrenoleukodystrophy begins between 11 and 21 years of age and the symptoms are similar to the childhood cerebral type but the disease progresses more slowly. The adrenomyeloneuropathy (AMN) type of adrenoleukodystrophy usually begins in the late twenties and is characterized by difficulty walking, a progressive weakness and stiffness in the legs (paraparesis), a loss in ability to coordinate muscle movements, excessive muscle tone (hypertonia), vision loss, difficulty speaking dysarthria), seizures and adrenal insufficiency. Adult cerebral adrenoleukodystrophy can begin between the twenties and fifties with symptoms similar to schizophrenia with dementia. Individuals with adrenal insufficiency only do not initially have neurological problems, but symptoms such as those seen in adrenomyeloneuropathy usually develop later. Adrenoleukodystrophy in females usually begins later in life and symptoms can vary greatly from mild to severe, but usually do not include adrenal insufficiency.
There are 4 primary phenotypes that can occur in MALES with the adrenoleukodystrophy gene:
1. Asymptomatic phenotype:
All individuals with the adrenoleukodystrophy gene are free of clinical symptoms for at least the first three years of life. And some may continue to have no symptoms. But the percentage of asymptomatic men and women decreases with age.
2. Adrenomyeloneuropathy (AMN) phenotype symptoms:
Walking and balance problems. General leg weakness and stiffness progresses into walking difficulty and reduced balance. With the weakening of leg muscles, changes in gait, or how a person walks, becomes noticeable. The use of mobility devices, such as canes, walkers, and wheelchairs may become necessary.
Pain, numbness, or tingling in the legs. Mild to moderate weakness of the arms/hands Urinary problems or incontinence and bowel urgency or incontinence Sexual dysfunction, or the inability to obtain or maintain an erection.
Adrenomyelopathy other symptoms include:
- Difficulty controlling urination
- Possible worsening muscle weakness or leg stiffness
- Problems with thinking speed and visual memory
Adrenomyeloneuropathy (AMN) is a specific form of adrenoleukodystrophy characterized by onset in the late 20s to middle ages in affected men. Adrenomyeloneuropathy (AMN) eventually affects almost all males who do not present in childhood. Initial symptoms are usually progressive stiffness and weakness in the legs (spastic paraparesis). Affected men may develop problems walking or walk in an unusual manner (abnormal gait). Numbness and pain from polyneuropathy are also common symptoms. Polyneuropathy is a general term for degeneration of the peripheral nerves, which are the nerves outside of the brain and spinal cord (i.e. central nervous system).
Affected men may also exhibit erectile dysfunction and problems with bowel and bladder control due to sphincter dysfunction. Sphincters are muscles which control the narrowing or widening certain passageways in the body. The urinary sphincters are two muscles that control the passage of urine from the bladder through the tiny tube that carries urine out of the body (urethra). Poor control of the urinary sphincter leads to urinary dysfunction. In addition, many men also develop premature balding and thinning of hair.
3. Adrenal insufficiency (Addison’s disease) phenotype symptoms:
Addison’s disease also known as primary adrenal insufficiency or hypoadrenalism, is a rare endocrine disorder that occurs as a result of permanent injury to the adrenal glands resulting in your adrenal glands not producing enough of their hormones – cortisol (a “stress” hormone) and aldosterone and androgens (the other hormones made by the adrenal glands) 13. Most men with adrenoleukodystrophy will eventually develop adrenal insufficiency over their lifespan. Women develop adrenal insufficiency much less commonly.
Your adrenal glands are composed of two sections. The interior (the adrenal medulla) produces adrenaline-like hormones. The outer layer (the adrenal cortex) produces a group of hormones called corticosteroids, which include glucocorticoids, mineralocorticoids and male sex hormones (androgens).
Some of the hormones the adrenal glands cortex produces are essential for life — the glucocorticoids (Cortisol) and the mineralocorticoids (Aldosterone).
Although it is easily treatable, adrenal insufficiency can be life-threatening if it is not recognized promptly. Addison’s disease treatment involves taking hormones to replace those that are missing.
Addison’s disease (primary adrenal insufficiency) symptoms are often non-specific and can include weakness/fatigue, nausea, abdominal pain, and low blood pressure. Darkening of the skin is also common.
Early symptoms of Addison’s disease may include 14:
- lack of energy or motivation (fatigue)
- muscle weakness
- low mood
- loss of appetite and unintentional weight loss
- increased thirst
Over time, these problems may become more severe and you may experience further symptoms, such as dizziness, fainting, cramps and exhaustion.
You may also develop small areas of darkened skin, or darkened lips or gums (hyperpigmentation). Hyperpigmentation is the physical finding most characteristic of Addison disease, arising from continual stimulation of the corticotrophs in the anterior pituitary. Specifically, hyperpigmentation results from cross-reactivity between the adrenocorticotropic hormone (ACTH) produced by the corticotrophs and the melanocortin 1 receptor on keratinocytes. Hyperpigmentation is usually generalized over the entire body and can be found in palmar creases, buccal mucosa, vermilion border of the lips, and around scars and nipples. It is not a feature of secondary adrenal insufficiency because of the lack of increased adrenocorticotropic hormone (ACTH) in these patients.
These symptoms relate to the degree of cortisol, mineralocorticoid, and adrenal androgen deficiency 15.
Adrenal insufficiency is sometimes referred to as Addison’s disease (based on the doctor, Thomas Addison MD, who first described it). There are many causes of adrenal insufficiency in the general population. adrenoleukodystrophy is the cause of approximately 33% of all cases of adrenal insufficiency. This means that not all patients diagnosed with adrenal insufficiency have adrenoleukodystrophy. Nonetheless, all patients with adrenal insufficiency should be tested for adrenoleukodystrophy (and vice versa).
4. Cerebral adrenoleukodystrophy (childhood cerebral adrenoleukodystrophy [CCALD]) symptoms:
35% of affected males develop neurological symptoms between three and ten years of age. It almost never occurs before approximately two and a half to three years of age. Affected boys’ symptoms may include “spacing out” in school: inattention, deterioration in handwriting skills, and decreased school performance; difficulty in understanding speech (though sound perception is normal); difficulty in reading and understanding written material; clumsiness; visual disturbances and occasionally double-vision; and aggressive or unexplained inappropriate behavior. In some boys, seizures may be the first symptom. Symptom severity varies from patient to patient and is not determined by phenotype. Even identical twins may have different experiences with symptom onset and severity. Other symptoms may include:
- Behavioral problems
- Hyperactivity
- Eye pain/Childhood onset migraines
- Recurring viral infections
- Lethargy, tires easily, clumsiness
- Hypoglycemia
- Tanning or bronzing of the skin
- Adrenal insufficiency
- Attention deficit and hyperactivity disorder (ADHD)
- Changes in muscle tone, especially muscle spasms and uncontrolled movements
- Crossed eyes
- Handwriting that gets worse
- Difficulty at school
- Difficulty understanding what people are saying
- Hearing loss
- Worsening nervous system damage, including coma, decreased fine motor control, and paralysis
- Seizures
- Swallowing difficulties
- Visual impairment or blindness
Affected males will develop normally and then start to show a loss (regression) of previously acquired skills. Before the loss of skills, affected males may exhibit behavioral problems including attention deficit and hyperactivity disorder (ADHD) and learning disabilities. Affected individuals usually develop cognitive deficits which means that they may have impairment of their mental processes and have difficulty acquiring information and knowledge. This means affected children may show a decline in performance at school. They may “space out” in school or at various times, have difficulty understanding speech, difficulty reading or understanding written words, difficulty with spatial references, and show a deterioration in handwriting skills.
Later on, they will develop additional symptoms including diminished clarity of vision (diminished visual acuity), hearing loss, gait difficulty, and eventually weakness and stiffness of limbs, convulsions or seizures. Eventually, affected children loose most neurological function and become totally disabled with blindness, deafness and inability to move voluntarily. The disorder will further progress to result in a vegetative state and death typically within 2-3 years from onset of neurological symptoms.
Adrenoleukodystrophy diagnosis
X-adrenoleukodystrophy is diagnosed by a simple blood test that analyzes the amount of very long chain fatty acids (VLCFAs); the levels of these very long chain fatty acids (VLCFAs) are elevated in in 99% of males with adrenoleukodystrophy and in approximately 85% of female carriers of the abnormal ABCD1 gene. While the test is accurate in males, in about 15-20% of women who are proven carriers, the test shows normal results and thus gives a “false negative” result. A DNA-based blood test is available. This test permits accurate identification of carriers by genetic testing, and if it is normal can assure a woman that she is not a carrier.
Molecular genetic testing for the ABCD1 gene is available and is used primarily to confirm a diagnosis if other testing is not conclusive, to provide genetic counseling to family members and for prenatal diagnosis.
Additionally, the patient will be evaluated for adrenal insufficiency (by another blood test), as this is a common symptom of the disease that can be corrected. Adrenal function tests are abnormal in 90% of boys with adrenoleukodystrophy who have neurologic symptoms and in approximately 70% of men with adrenomyeloneuropathy.
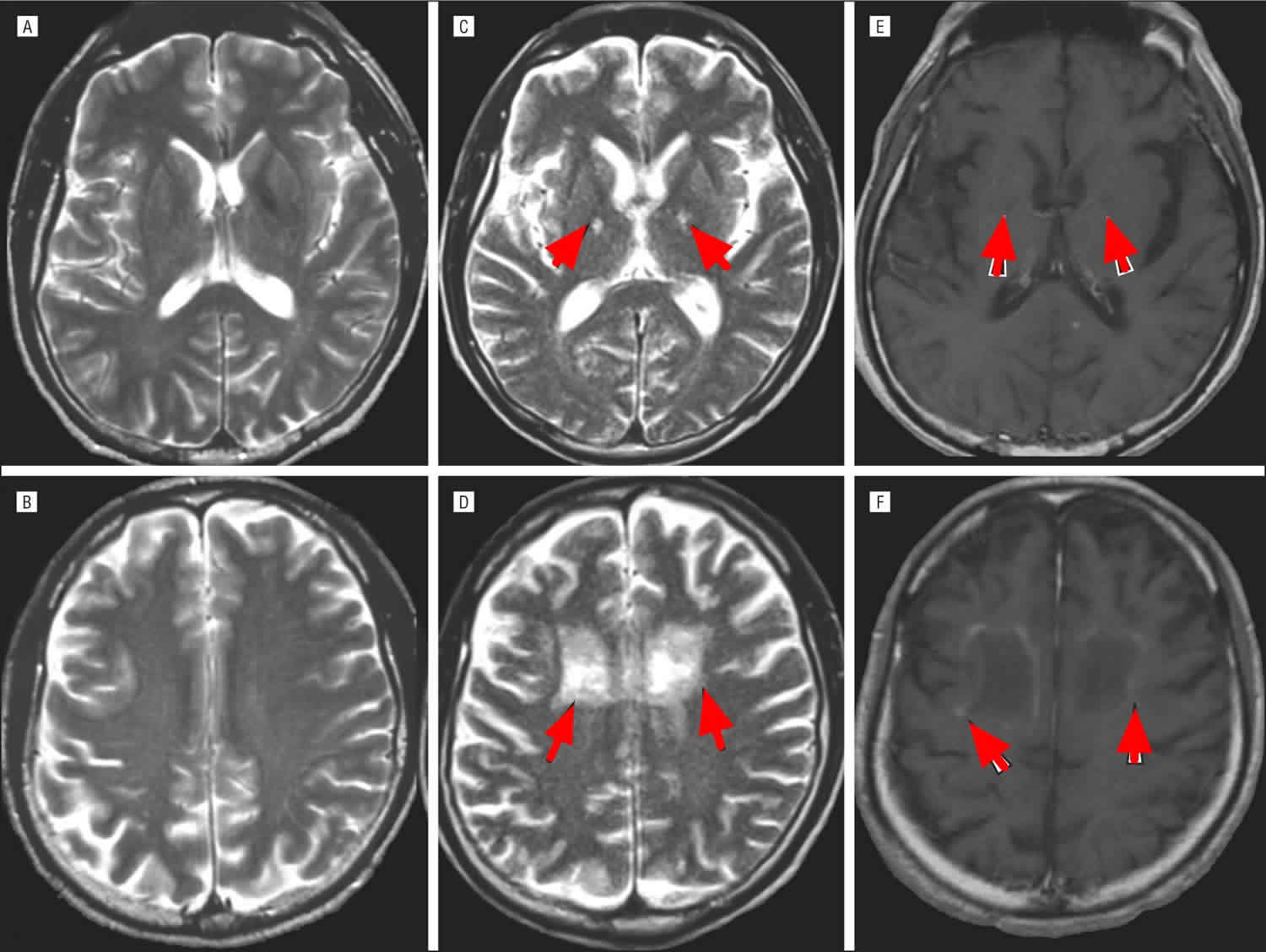
If the blood test suggests X-adrenoleukodystrophy, then generally an magnetic resonance imaging (MRI) will be performed in order to assess cerebral involvement. An MRI uses a magnetic field and radio waves to produce cross-sectional images of particular organs and bodily tissues including the brain. This allows physicians to see whether the brain has been damaged, including the loss of myelin in the cerebral white matter. It is important to identify the brain MRI changes as early as possible, since individuals with early MRI changes prior to neurological symptoms have the best outcome when undergoing therapy.
In 2016, adrenoleukodystrophy was added to the Recommended Uniform Newborn Screening Panel (RUSP) in the United States. However, each state determines what specific disorders are included in its newborn screening program within that state. As of October 2019, only a dozen of states test for adrenoleukodystrophy through newborn screening, although more states are planning on adding the disorder to testing programs.
Adrenoleukodystrophy prognosis
Prognosis for patients with childhood cerebral X-adrenoleukodystrophy is generally poor due to progressive neurological deterioration unless bone marrow transplantation is performed early. It leads to a long-term coma (vegetative state) about 2 years after nervous system symptoms develop. The child can live in this condition for as long as 10 years until death occurs. Death usually occurs within 1 to 10 years after the onset of symptoms.
X-linked cerebral adrenoleukodystrophy – left untreated, all but 10% of patients are bedridden, blind, lacking speech and require fulltime care, dying within 2-5 years 16. Arrested X-linked cerebral adrenoleukodystrophy may enter a phase of rapid neurological deterioration at any time.
Adult-onset adrenomyeloneuropathy (AMN) will progress over decades.
The deterioration rate is different for each patient. Some will continue to walk and talk several years before succumbing to the effects of the disease, and will then suddenly deteriorate very quickly into a dependent state. Others will deteriorate into the dependent state within a few months of diagnosis. There is no way to predict how quickly or slowly the process will happen.
Adrenoleukodystrophy life expectancy
Medical guidelines say that on average, boys with symptomatic cerebral adrenoleukodystrophy live for 2-4 years. But many boys have gone on to live for longer periods. There are boys who have symptomatic adrenoleukodystrophy that are living in their teens and early 20s with quality of life 17.
Adrenoleukodystrophy treatment
Adrenoleukodystrophy treatment may require the coordinated efforts of a team of specialists. Pediatricians, general internists, physicians who specialize in diagnosing and treating disorders of the brain and nervous system (pediatric neurologists), adult neurologists, physicians who specialize in diagnosing and treating disorders of the urinary system (urologists), physicians who specialize in diagnosing and treating disorders of the endocrine system (endocrinologists), psychiatrists, physical therapists, and other healthcare professionals may need to systematically and comprehensively plan treatment.
Genetic counseling is recommended for affected individuals and their families. Psychosocial support for the entire family is essential as well.
Boys without symptoms (asymptomatic) should be closely monitored for signs of cerebral disease. This group tends to be boys identified through newborn screening or were diagnosed early because of a previous affected family member. Treatment such as hematopoietic stem cell transplantation should only be considered in boys with abnormal MRI changes who do not yet have neurological symptoms.
Periodic reassessments and adjustment of services should be provided with all children and adults. Additional medical, social, and/or vocational services including specialized learning programs may be necessary.
1. The first treatment deals with the replacement of the faulty adrenal function often present in adrenoleukodystrophy. The adrenal glands are next to the kidney, and produce certain important hormones. If the adrenal functions are not properly functioning, these hormones need to be replaced.
Addison’s disease treatment usually involves corticosteroid (steroid) replacement therapy for life. Corticosteroid medicine is used to replace the hormones cortisol and aldosterone that your body no longer produces. A medicine called hydrocortisone is usually used to replace the cortisol. Other possible medicines are prednisolone or dexamethasone, although these are less commonly used.
Aldosterone is replaced with a medicine called fludrocortisone. Your doctor may also ask you to add extra salt to your daily diet, although if you’re taking enough fludrocortisone medicine this may not be necessary. Unlike most people, if you feel the urge to eat something salty, then you should eat it.
Management of spinal cord disease, urinary complications, polyneuropathy tend to follow routine or standard guidelines.
2. Lorenzo’s oil. This is a mixture of two oils (glyceryl trieucate, or GTE, and glyceryl trioleate, or GTO). It is thought to aid in the normalization of the fatty acid levels. However, this oil does not seem to alter the progression of the disease once the brain is involved. It is not yet clear if Lorenzo’s oil could prevent progression prior to symptoms. Lorenzo’s Oil has been successfully used as a therapy for boys with ALD. If started early, it can help lessen the risk of developing the childhood cerebral form of ALD. This oil, along with a low-fat diet, can help to reduce the very long chain fatty acids that accumulate. Lorenzo’s Oil is not approved by the FDA. Lorenzo’s oil therapy is not FDA approved as a double‐blind placebo‐controlled trial has not seen successful completion. In open‐label studies, Lorenzo’s oil has not shown to halt or slow disease progression in adrenomyeloneuropathy (AMN) nor in cerebral adrenoleukodystrophy (CALD) 18. To learn more about obtaining the Lorenzo’s Oil, please visit (https://www.myelin.org/lorenzos-oil/). Financial assistance through the Myelin Project is also available. Eating a diet low in very-long-chain fatty acids and taking special oils can lower the blood levels of very-long-chain fatty acids, such as Lorenzo’s Oil, which may be help slow the progression of ALD disease.
Lorenzo’s oil together with a low‐fat diet was given to 89 asymptomatic adrenoleukodystrophy boys in an open, non‐placebo‐controlled, trial. After a mean follow‐up of 6.9 years, 24% developed cerebral adrenoleukodystrophy (CALD); however, compared to historical data, the percentage of boys that would develop cerebral adrenoleukodystrophy (CALD) was 37% suggesting a protective effect 19. Studies in postmortem brain of cerebral adrenoleukodystrophy (CALD) who were on Lorenzo’s oil showed no evidence of erucic acid in brain lipids 20; however, later studies in rats showed that erucic acid entered the brain and was either degraded or chain elongated to nervonic acid 21. The mechanism for Lorenzo’s oil was thought to be by competitive inhibition of the chain elongation of saturated fatty acids by providing an excess of monounsaturated fatty acid precursors. The in vitro study (test tube study) of Lorenzo’s oil in HeLa cells expressing high levels of ELOVL1, the enzyme that catalyzes the chain elongation of C22 to C26 fatty acids in the endoplasmic reticulum, showed that Lorenzo’s oil strongly inhibits ELOVL1 22.
3. Bone marrow transplantation (allogenic hematopoietic stem cell transplantation [HSCT]). Hematopoietic stem cells are special cells found in bone marrow that grow or mature into different types of cells. Allo means other. In allogenic stem cell transplantation, the stem cells are from a person other than the patient, either a matched related or unrelated donor. This is the most successful treatment for adrenoleukodystrophy so far identified. Bone marrow transplantation has been successful in individuals who are in the early stages of adrenoleukodystrophy. A series of studies conducted over the last two decades have shown that a allogenic hematopoietic stem cell transplantation (HSCT) stops the progression of neurological disease in adrenoleukodystrophy, although it does not improve adrenal insufficiency. Allogenic hematopoietic stem cell transplantation (HSCT) is a major medical procedure that carries significant risk. Since allogenic hematopoietic stem cell transplantation (HSCT) is only effective in early stages of the disease, it is pertinent that newborn screening is expanded to all states and that all boys diagnosed with adrenoleukodystrophy undergo repeated brain MRI studies and follow up routinely with a neurologist.
Ideal candidates for allogenic hematopoietic stem cell transplantation (HSCT) are individuals with a Loes score of 9 or lower, without any neurologic deficits, who receive HLA‐matched sibling or related donor hematopoietic stem cell transplantation (HSCT) 4. However, this intervention has a high‐morbidity and long‐term complications related to immunosuppression and graft versus host disease. It is important to note that disease progression continues for some 6–9 months following allogenic hematopoietic stem cell transplantation (HSCT) 23. Importantly, adrenal dysfunction is not corrected following allogeneic stem cell transplant for cerebral disease 24. Moreover, it must be noted that bone marrow transplantation has not been successful in patients with advanced neurological involvement, but only in patients in earlier stages of the disease. Although this procedure is considered risky, successful transplants are possible with early intervention. Outcomes depend on the child’s clinical status at the time of the evaluation, along with interpretation of the brain MRI. Only about roughly 30% of boys who undergo allogenic hematopoietic stem cell transplantation (HSCT) will go on to develop childhood cerebral disease. Currently, doctors typically will not perform stem cell transplantation on adults with adrenoleukodystrophy, generally because the risks of the treatment are considered to outweigh the potential benefits. But as transplantation technology improves and becomes safer, it is possible that stem cell transplantation will be available for men with adrenomyelonueropathy (AMN).
4. Medications. Your doctor may prescribe medications to help relieve symptoms, including stiffness and seizures.
5. Exercise and physical therapy. Patients who experience walking difficulties may benefit from seeing a physical therapist, who can provide exercises to strengthen muscles and improve walking ability.
Affected individuals can benefit from supportive care from psychologists, educators, physical therapists, urologists, and family and vocational counselors. Genetic counseling is recommended for affected individuals and their family members.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counsellors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
- The National Cancer Institute provides a Cancer Genetics Services Directory (https://www.cancer.gov/about-cancer/causes-prevention/genetics/directory), which lists professionals who provide services related to cancer genetics. You can search by type of cancer or syndrome, location, and/or provider name.
If you have a health condition that has not been diagnosed, you may be interested in the Undiagnosed Diseases Network (https://undiagnosed.hms.harvard.edu/). They have information about how to apply for this multicenter research study.
How does blood or marrow transplant work for adrenoleukodystrophy?
Bone marrow transplant or blood stem cell transplant, can stop the adrenoleukodystrophy from causing more damage. Bone marrow transplant replaces the blood-forming cells (stem cells) that are missing the important protein with healthy ones. With healthy blood-forming cells, the body is able to break down fat-based substances normally. This keeps the brain, spinal cord and nervous system from more damage. But, bone marrow transplant can’t fix any damage that has already happened.
Allogeneic transplant is used for adrenoleukodystrophy. This type of transplant uses healthy, blood-forming cells donated by someone else to replace the unhealthy blood-forming cells. These healthy cells can come from a family member, unrelated donor or umbilical cord blood. First, you get chemotherapy (chemo), with or without radiation, to kill the unhealthy cells. Then, the healthy, donated cells are given to you through an intravenous (IV) catheter. The new cells travel to the inside of the bones and begin to make healthy cells.
The entire transplant process, from the start of chemo or radiation, until hospital discharge, can last weeks to months. This is followed by many months of recovery near the transplant center and at home. The transplant team will closely care for you to prevent and treat any complications.
When should I see a transplant doctor?
Most people have a better chance of a cure if they have a bone marrow transplant soon after diagnosis. You or your child should see a transplant doctor as soon as you’re diagnosed.
Your first appointment with a transplant doctor
At the first appointment, the transplant doctor will:
- Review your or your child’s medical history
- Talk with you about treatment options
- Discuss the risks and benefits of transplant
- Recommend the best time for you or your child to get a transplant and prepare for treatment
- Start a donor search
- Schedule appointments with other doctors, like a neurologist.
Questions to ask your doctor
Ask questions so you understand your treatment options and can make decisions that are best for you and your child. Questions you may want to ask include:
- What are the chances transplant will stop the adrenoleukodystrophy from causing more damage?
- What are the possible side effects of transplant? How can they be reduced?
- How might my or my child’s quality of life change over time, with or without transplant?
Experimental therapeutic trials and investigational therapies
Metabolic modulators
One inhibitor of ELOVL1 is bezafibrate that showed reduced VLCFA in adrenoleukodystrophy fibroblasts 25. However, in a clinical trial of administering bezafibrate to adrenomyeloneuropathy (AMN) men and women VLCFA in plasma, lymphocytes and dried whole blood spots were not lowered 26.
Statins lower LDL (low-density lipoprotein) cholesterol. In 1998 Singh et al. 27 reported that adrenoleukodystrophy patients given lovastatin normalized their plasma VLCFA. Another randomized double‐blind crossover trial comparing lovastatin to placebo showed no normalization of C26:0 in adrenoleukodystrophy patients 28.
Various studies have shown an upregulation of peroxisomal beta‐oxidation in cells from adrenoleukodystrophy subjects and the ABCD1 mutant mouse model by upregulation of the ABCD2 protein and peroxisome proliferation. Normalization of C24:0 levels in brain and C26:0 levels were lowered by 80% in Abcd1 mice after 6 weeks of feeding 4‐phenyl‐ butyrate, 4PBA 29.
Overexpression of Abcd2 in an Abcd1 knockout mouse normalizes VLCFA in spinal cord, sciatic nerve and adrenal gland 30. However, to date there have not been clinical trials of 4PBA in adrenoleukodystrophy.
It was shown that thyroid hormone receptor agonist sobetirome increased Abcd2 mRNA levels in brain and liver of wild‐type mice. Adult Abcd1 KO mice were treated with sobetirome for 12 weeks resulting in a lowering of C26:0‐LPC levels in plasma, brain, testes and adrenal tissue by ~20% 31. Other thyroid hormone agonists are currently being pursued as potential therapeutic candidates in adrenoleukodystrophy.
Morato et al. demonstrated that pioglitazone, a PPARγ agonist, halts axonal degeneration in the Abcd1 mouse by restoring mitochondria 32. Recently, a derivative of pioglitazone has been developed and is currently in being assessed in a multinational, placebo‐controlled, randomized trial in adrenomyeloneuropathy (AMN).
Anti‐inflammatory drugs
Anti‐inflammatory drugs such as immunoglobulin, cyclosporine, cyclophosphamide and interferon‐beta were tried to halt or reverse the cerebral inflammation 10. These drugs were not effective in halting the cerebral inflammation.
Antioxidant therapy
A combination of multiple high‐dose antioxidants was recently demonstrated to normalize biomarkers for oxidative damage and inflammation in a small open‐label trial of adult patients with adrenomyeloneuropathy (AMN) 33. There appeared to be a potential effect on the 6‐min walk test, justifying larger placebo‐controlled trials in future.
Several centers have utilized N‐Acetyl cysteine (NAC) adjunct therapy for individuals undergoing allogeneic hematopoietic stem cell transplantation (HSCT). One study showed individuals with more advanced MRI undergoing allogeneic hematopoietic stem cell transplantation (HSCT) had improved survival with N‐Acetyl cysteine (NAC) therapy, not however demonstrating an improvement in residual neurologic deficit 34.
As with most neurologic disease, main pharmacodynamic challenges are in passing the blood–brain barrier and in the targeted delivery of a therapeutic compound. Recently, a nanoparticle polyamidoamine, PAMAM, dendrimer drug delivery platform conjugated to NAC (D‐NAC), having shown rescue of motor impairments and inflammatory status in a neonatal rabbit model of cerebral palsy 35, demonstrated targeted drug delivery into Abcd1 mouse spinal cord microglia, and in ex vivo cerebral adrenoleukodystrophy peripheral blood monocytic patient cells stimulated by VLCFA (C26:0), normalization of antioxidant and inflammatory status 36.
Gene therapy
In order to decrease the burden of morbidity of allogenic hematopoietic stem cell transplantation (HSCT), a trial of autologous hematopoietic stem cell transplantation (HSCT) with ex vivo lentiviral gene correction of CD34‐positive stem cells is ongoing 37. Auto means self. The stem cells in autologous hematopoietic stem cell transplantation (HSCT) come from the same person who will get the transplant, so the patient is their own donor. Interim findings of this trial in 17 adrenoleukodystrophy boys with early stage brain disease who received the Lenti‐D ABCD1 gene therapy have been reported. No treatment death or graft versus host disease was seen. Fifteen of the boys survived; however, 1 died of rapid neurologic deterioration and the other, who had evidence of rapid disease progression on MRI, withdrew from the study to undergo allogenic stem cell transplantation and died of complications. All 15 boys who survived remained free of major functional disabilities at the 24‐month follow‐up. A longer follow‐up and larger sample size is needed to confirm the efficacy and safety of ABCD1 gene therapy with the Lenti‐D lentiviral vector.
In addition to ex vivo lentiviral gene correction, in vivo adeno‐associated virus 9 (AAV9)‐based gene therapy is being pursued. Gong et al. 38 have showed intrathecal delivery of an AAV9 carrying ABCD1 in mice corrected VLCFA metabolism and behavioral outcomes. This therapeutic strategy may show promise for adrenomyeloneuropathy (AMN) due to the intrathecal delivery.
References- Adrenoleukodystrophy. https://rarediseases.org/rare-diseases/adrenoleukodystrophy/
- Johnson AB, Schaumburg HH, Powers JM. Histochemical characteristics of the striated inclusions of adrenoleukodystrophy. J Histochem Cytochem. 1976 Jun;24(6):725-30. doi: 10.1177/24.6.59773
- Nury, T. , Zarrouk, A. , Ragot, K. , Debbabi, M. , Riedinger, J. M. , Vejux, A. , … Lizard, G. (2017). 7‐Ketocholesterol is increased in the plasma of X‐ALD patients and induces peroxisomal modifications in microglial cells: Potential roles of 7‐ketocholesterol in the pathophysiology of X‐ALD. Journal of Steroid Biochemistry and Molecular Biology, 169, 123–136. 10.1016/j.jsbmb.2016.03.037
- Turk, B. R., Theda, C., Fatemi, A., & Moser, A. B. (2020). X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. International journal of developmental neuroscience : the official journal of the International Society for Developmental Neuroscience, 80(1), 52–72. https://doi.org/10.1002/jdn.10003
- X-linked adrenoleukodystrophy. https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=761&Disease_Disease_Search_diseaseGroup=adrenoleukodystrophy&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=X-linked-adrenoleukodystrophy&title=X-linked%20adrenoleukodystrophy&search=Disease_Search_Simple
- Huffnagel, I. C., Dijkgraaf, M., Janssens, G. E., van Weeghel, M., van Geel, B. M., Poll-The, B. T., Kemp, S., & Engelen, M. (2019). Disease progression in women with X-linked adrenoleukodystrophy is slow. Orphanet journal of rare diseases, 14(1), 30. https://doi.org/10.1186/s13023-019-1008-6
- van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol. 2001 Feb;49(2):186-94. doi: 10.1002/1531-8249(20010201)49:2<186::aid-ana38>3.0.co;2-r
- Huffnagel IC, Laheji FK, Aziz-Bose R, Tritos NA, Marino R, Linthorst GE, Kemp S, Engelen M, Eichler F. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration. J Clin Endocrinol Metab. 2019 Jan 1;104(1):118-126. doi: 10.1210/jc.2018-01307
- What is ALD? http://www.aldfoundation.org/ald.php
- Moser, H. W. , Smith, K. D. , Watkins, P. A. , Powers, J. , & Moser, A. B. (2001). X‐linked adrenoleukodystrophy In Scriver C. R., Beaudet A. L., Sly W. S., & Valle D. (Eds.), The metabolic and molecular basis of inherited disease (8th ed., pp. 3257–3301). New York, NY: McGraw‐Hill Book Co.
- Huffnagel IC, van Ballegoij WJC, van Geel BM, Vos JMBW, Kemp S, Engelen M. Progression of myelopathy in males with adrenoleukodystrophy: towards clinical trial readiness. Brain. 2019 Feb 1;142(2):334-343. doi: 10.1093/brain/awy299
- Fatemi A, Barker PB, Uluğ AM, Nagae-Poetscher LM, Beauchamp NJ, Moser AB, Raymond GV, Moser HW, Naidu S. MRI and proton MRSI in women heterozygous for X-linked adrenoleukodystrophy. Neurology. 2003 Apr 22;60(8):1301-7. doi: 10.1212/01.wnl.0000059546.15529.cb
- Adrenal Insufficiency. https://www.hormone.org/diseases-and-conditions/adrenal/adrenal-insufficiency
- Burke CW. Adrenocortical insufficiency. Clin Endocrinol Metab. 1985;14(4):947–976.
- Addison Disease: Early Detection and Treatment Principles. Am Fam Physician. 2014 Apr 1;89(7):563-568. https://www.aafp.org/afp/2014/0401/p563.html
- X-linked cerebral adrenoleukodystrophy. https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=16884&Disease_Disease_Search_diseaseGroup=adrenoleukodystrophy&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=X-linked-cerebral-adrenoleukodystrophy&title=X-linked%20cerebral%20adrenoleukodystrophy&search=Disease_Search_Simple
- PRACTICAL INFORMATION FOR MALES WITH SYMPTOMATIC ALD. http://www.aldlife.org/wp-content/uploads/2015/04/Practical-Information-for-Males-with-Symptomatic-ALD.pdf
- van Geel, B. M., Assies, J., Haverkort, E. B., Koelman, J. H., Verbeeten, B., Jr, Wanders, R. J., & Barth, P. G. (1999). Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with “Lorenzo’s oil”. Journal of neurology, neurosurgery, and psychiatry, 67(3), 290–299. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1736534/pdf/v067p00290.pdf
- Moser HW, Raymond GV, Lu SE, Muenz LR, Moser AB, Xu J, Jones RO, Loes DJ, Melhem ER, Dubey P, Bezman L, Brereton NH, Odone A. Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo’s oil. Arch Neurol. 2005 Jul;62(7):1073-80. https://jamanetwork.com/journals/jamaneurology/fullarticle/788970
- Poulos A, Gibson R, Sharp P, Beckman K, Grattan-Smith P. Very long chain fatty acids in X-linked adrenoleukodystrophy brain after treatment with Lorenzo’s oil. Ann Neurol. 1994 Nov;36(5):741-6. doi: 10.1002/ana.410360509
- Golovko MY, Murphy EJ. Uptake and metabolism of plasma-derived erucic acid by rat brain. J Lipid Res. 2006 Jun;47(6):1289-97. doi: 10.1194/jlr.M600029-JLR200
- Sassa, T., Wakashima, T., Ohno, Y., & Kihara, A. (2014). Lorenzo’s oil inhibits ELOVL1 and lowers the level of sphingomyelin with a saturated very long-chain fatty acid. Journal of lipid research, 55(3), 524–530. https://doi.org/10.1194/jlr.M044586
- Raymond GV, Aubourg P, Paker A, Escolar M, Fischer A, Blanche S, Baruchel A, Dalle JH, Michel G, Prasad V, Miller W, Paadre S, Balser J, Kurtzberg J, Nascene DR, Orchard PJ, Lund T. Survival and Functional Outcomes in Boys with Cerebral Adrenoleukodystrophy with and without Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2019 Mar;25(3):538-548. doi: 10.1016/j.bbmt.2018.09.036
- Burtman E, Regelmann MO. Endocrine Dysfunction in X-Linked Adrenoleukodystrophy. Endocrinol Metab Clin North Am. 2016 Jun;45(2):295-309. doi: 10.1016/j.ecl.2016.01.003
- Engelen, M., Schackmann, M. J., Ofman, R., Sanders, R. J., Dijkstra, I. M., Houten, S. M., Fourcade, S., Pujol, A., Poll-The, B. T., Wanders, R. J., & Kemp, S. (2012). Bezafibrate lowers very long-chain fatty acids in X-linked adrenoleukodystrophy fibroblasts by inhibiting fatty acid elongation. Journal of inherited metabolic disease, 35(6), 1137–1145. https://doi.org/10.1007/s10545-012-9471-4
- Engelen, M., Tran, L., Ofman, R., Brennecke, J., Moser, A. B., Dijkstra, I. M., Wanders, R. J., Poll-The, B. T., & Kemp, S. (2012). Bezafibrate for X-linked adrenoleukodystrophy. PloS one, 7(7), e41013. https://doi.org/10.1371/journal.pone.0041013
- Singh I, Khan M, Key L, Pai S. Lovastatin for X-linked adrenoleukodystrophy. N Engl J Med. 1998 Sep 3;339(10):702-3. doi: 10.1056/NEJM199809033391012
- Engelen M, Ofman R, Dijkgraaf MG, Hijzen M, van der Wardt LA, van Geel BM, de Visser M, Wanders RJ, Poll-The BT, Kemp S. Lovastatin in X-linked adrenoleukodystrophy. N Engl J Med. 2010 Jan 21;362(3):276-7. doi: 10.1056/NEJMc0907735
- Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, Moser AB, Watkins PA, Smith KD. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med. 1998 Nov;4(11):1261-8. doi: 10.1038/3242
- Pujol A, Ferrer I, Camps C, Metzger E, Hindelang C, Callizot N, Ruiz M, Pàmpols T, Giròs M, Mandel JL. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum Mol Genet. 2004 Dec 1;13(23):2997-3006. doi: 10.1093/hmg/ddh323
- Hartley, M. D., Kirkemo, L. L., Banerji, T., & Scanlan, T. S. (2017). A Thyroid Hormone-Based Strategy for Correcting the Biochemical Abnormality in X-Linked Adrenoleukodystrophy. Endocrinology, 158(5), 1328–1338. https://doi.org/10.1210/en.2016-1842
- Morató, L., Galino, J., Ruiz, M., Calingasan, N. Y., Starkov, A. A., Dumont, M., Naudí, A., Martínez, J. J., Aubourg, P., Portero-Otín, M., Pamplona, R., Galea, E., Beal, M. F., Ferrer, I., Fourcade, S., & Pujol, A. (2013). Pioglitazone halts axonal degeneration in a mouse model of X-linked adrenoleukodystrophy. Brain : a journal of neurology, 136(Pt 8), 2432–2443. https://doi.org/10.1093/brain/awt143
- Casasnovas, C., Ruiz, M., Schlüter, A., Naudí, A., Fourcade, S., Veciana, M., Castañer, S., Albertí, A., Bargalló, N., Johnson, M., Raymond, G. V., Fatemi, A., Moser, A. B., Villarroya, F., Portero-Otín, M., Artuch, R., Pamplona, R., & Pujol, A. (2019). Biomarker Identification, Safety, and Efficacy of High-Dose Antioxidants for Adrenomyeloneuropathy: a Phase II Pilot Study. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics, 16(4), 1167–1182. https://doi.org/10.1007/s13311-019-00735-2
- Miller WP, Rothman SM, Nascene D, Kivisto T, DeFor TE, Ziegler RS, Eisengart J, Leiser K, Raymond G, Lund TC, Tolar J, Orchard PJ. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011 Aug 18;118(7):1971-8. doi: 10.1182/blood-2011-01-329235
- Kannan, S., Dai, H., Navath, R. S., Balakrishnan, B., Jyoti, A., Janisse, J., Romero, R., & Kannan, R. M. (2012). Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Science translational medicine, 4(130), 130ra46. https://doi.org/10.1126/scitranslmed.3003162
- Turk, B. R., Nemeth, C. L., Marx, J. S., Tiffany, C., Jones, R., Theisen, B., Kambhampati, S., Ramireddy, R., Singh, S., Rosen, M., Kaufman, M. L., Murray, C. F., Watkins, P. A., Kannan, S., Kannan, R., & Fatemi, A. (2018). Dendrimer-N-acetyl-L-cysteine modulates monophagocytic response in adrenoleukodystrophy. Annals of neurology, 84(3), 452–462. https://doi.org/10.1002/ana.25303
- Eichler, F., Duncan, C., Musolino, P. L., Orchard, P. J., De Oliveira, S., Thrasher, A. J., Armant, M., Dansereau, C., Lund, T. C., Miller, W. P., Raymond, G. V., Sankar, R., Shah, A. J., Sevin, C., Gaspar, H. B., Gissen, P., Amartino, H., Bratkovic, D., Smith, N., Paker, A. M., … Williams, D. A. (2017). Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. The New England journal of medicine, 377(17), 1630–1638. https://doi.org/10.1056/NEJMoa1700554
- Gong, Y., Mu, D., Prabhakar, S., Moser, A., Musolino, P., Ren, J., Breakefield, X. O., Maguire, C. A., & Eichler, F. S. (2015). Adenoassociated virus serotype 9-mediated gene therapy for x-linked adrenoleukodystrophy. Molecular therapy : the journal of the American Society of Gene Therapy, 23(5), 824–834. https://doi.org/10.1038/mt.2015.6





{kind=link}