Adult onset still’s disease
Adult-onset Still’s disease also known as AOSD or Wissler-Fanconi syndrome, is a rare type of inflammatory arthritis of unknown cause usually affecting young adults that is thought to be autoummune or autoinflammatory in origin 1. The inflammation can destroy affected joints, particularly the wrists. Adult-onset Still’s disease (AOSD) is a rare systemic autoinflammatory disease that is characterized by four main symptoms such as arthralgia or arthritis, spiking fever (≥39 °C), skin rash and hyperleukocytosis (≥10,000 cells/mm3) with elevated ferritin levels 2, 3. However, many other clinical features are possible and adult-onset Still’s disease can appear in all age groups with potentially severe inflammatory onset accompanied by a broad spectrum of disease manifestation and complications. Hence, it remains a diagnostic challenge 3. The reason behind the nomenclature of this condition is that adult-onset Still’s disease (AOSD) shares certain symptoms with Still’s disease in children, currently named systemic-onset juvenile idiopathic arthritis (SJIA) sometimes also known as Still’s disease. Adult Still’s disease is a severe version of juvenile idiopathic arthritis (JIA), which occurs in children.
By definition, adult-onset Still’s disease affects people older than 16, either de novo or those with a history of systemic-onset juvenile idiopathic arthritis (SJIA) 4. In the systemic-onset juvenile idiopathic arthritis (SJIA), a disease-free interval between the childhood episode and the adulthood recurrence differentiates Adult-onset Still’s disease (AOSD) from persistent systemic-onset juvenile idiopathic arthritis (SJIA) 3. Adult Still’s disease may lead to long-term (chronic) arthritis.
Adult-onset Still’s disease has similar symptoms to systemic-onset juvenile idiopathic arthritis (SJIA) sometimes referred to as Still’s disease– with fever, rash and joint pain. Adult Still’s disease symptoms usually begin with a high fever that spikes once or twice a day and a salmon-pink rash on the trunk, arms or legs (see Figure 1). Other symptoms include sore throat and swollen lymph nodes in your neck. A few weeks after these initial symptoms, your joints and muscles begin aching. These aches last at least two weeks. The most commonly affected joints are the knee and wrist. The ankles, shoulders, elbows and finger joints may also be involved.
Other frequently observed clinical features include sore throat, hepatomegaly (enlarged liver), splenomegaly (enlarged spleen), lymphadenopathy (enlarged lymph glands) and serositis (inflammation of the serous tissues of your body that protect your organs and allow them to move smoothly within your body such as the tissues lining your lungs [pleura], heart [pericardium], and the inner lining of your abdomen [peritoneum] and organs within the body). Furthermore, adult Still’s disease patients may experience different life-threatening complications. Macrophage activation syndrome (MAS) has been reported up to 15% of Adult onset Still’s disease patients and it is considered to be the most severe complication of the disease being characterized by high mortality rate 1.
Adult onset Still’s disease begins in adulthood, so it is compared to rheumatoid arthritis (RA). Inflammation may affect a few joints at first. Over time, more joints may be involved. Some people may have only one bout of the illness followed by lasting remission (no visible symptoms), while others develop chronic arthritis. Fewer than 1 out of 100,000 people develop adult Still’s disease each year. Adult-onset Still’s disease affects men and women equally, usually young adults between the ages of 16 and 35 5.
There are 3 different patterns in the clinical evolution of adult-onset Still’s disease. The monocyclic pattern is self-limited and characterized by one flare of variable duration followed by complete remission. The polycyclic pattern is marked by 2 or more episodes with intervening symptom-free periods. The chronic articular pattern is characterized by severe joint manifestations that lead to joint-space narrowing and destruction. Adult-onset Still’s disease can also have serious systemic complications including serositis, chronic arthropathy, and macrophage activation syndrome (MAS). Late carpal ankylosis occurs in approximately 25% of patients and represents a distinct clinical feature differentiating AOSD from rheumatoid arthritis.
No single test identifies adult Still’s disease. Imaging tests can reveal damage caused by the disease, while blood tests can help rule out other conditions that have similar symptoms.
Doctors use several medications that help control inflammation to treat adult Still’s disease. Over-the-counter or prescription nonsteroidal anti-inflammatory drugs (NSAIDs) help to reduce mild pain and inflammation. Corticosteroids, such as prednisone, are needed if the disease is severe or doesn’t respond to prescription NSAIDs. Disease-modifying drugs (DMARDs), such as methotrexate, and biologic response modifiers, are needed in more severe cases or if the arthritis becomes chronic. It may be necessary to take more than one medication at a time to control symptoms.
With adult Still’s disease, the medications may need to be taken even after symptoms go away. This is called maintenance therapy. It’s important to keep inflammation under control to prevent damage to joints and organs.
Figure 1. Adult onset Still’s disease rash (salmon-pink maculopapular rash)
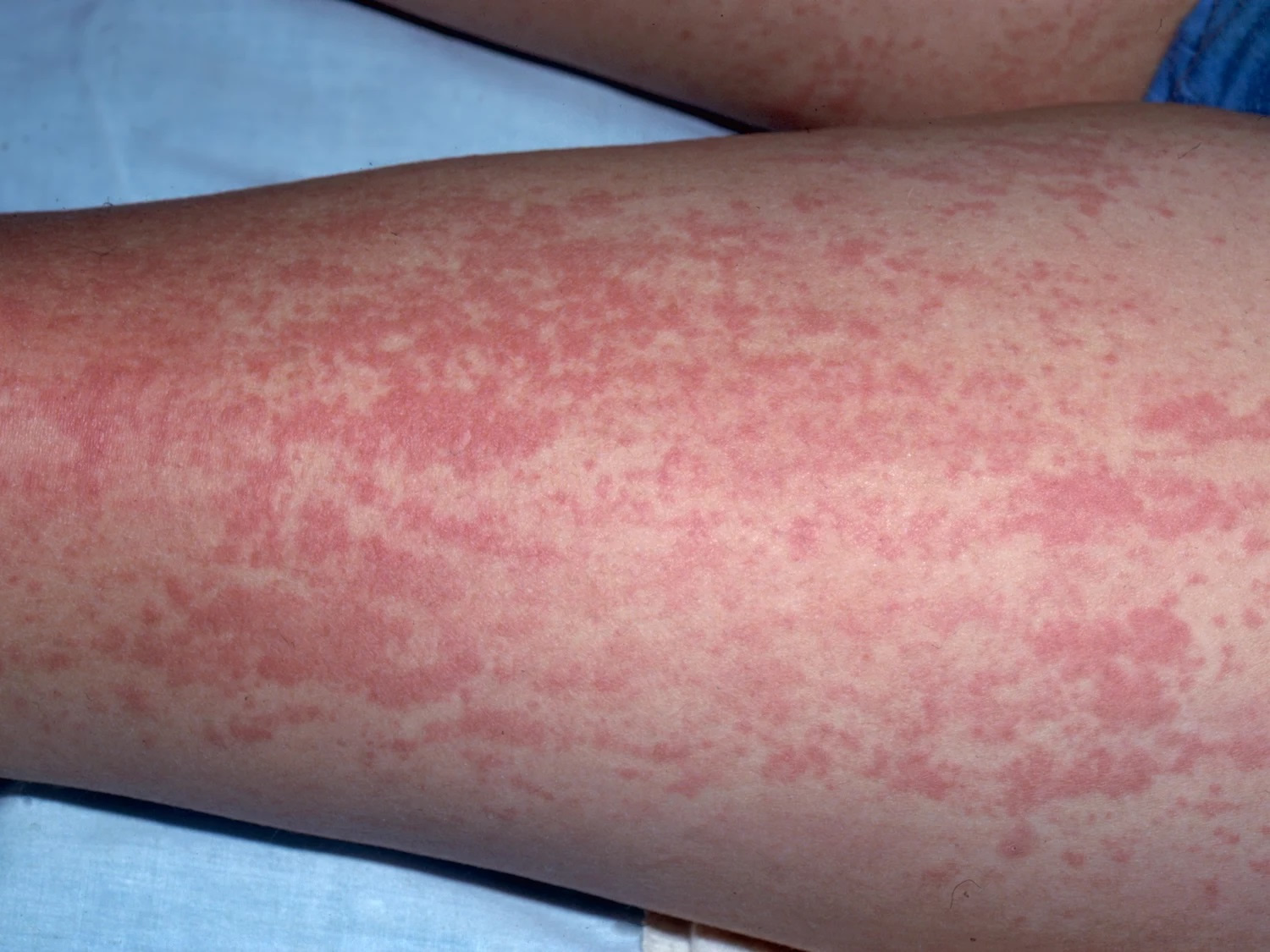
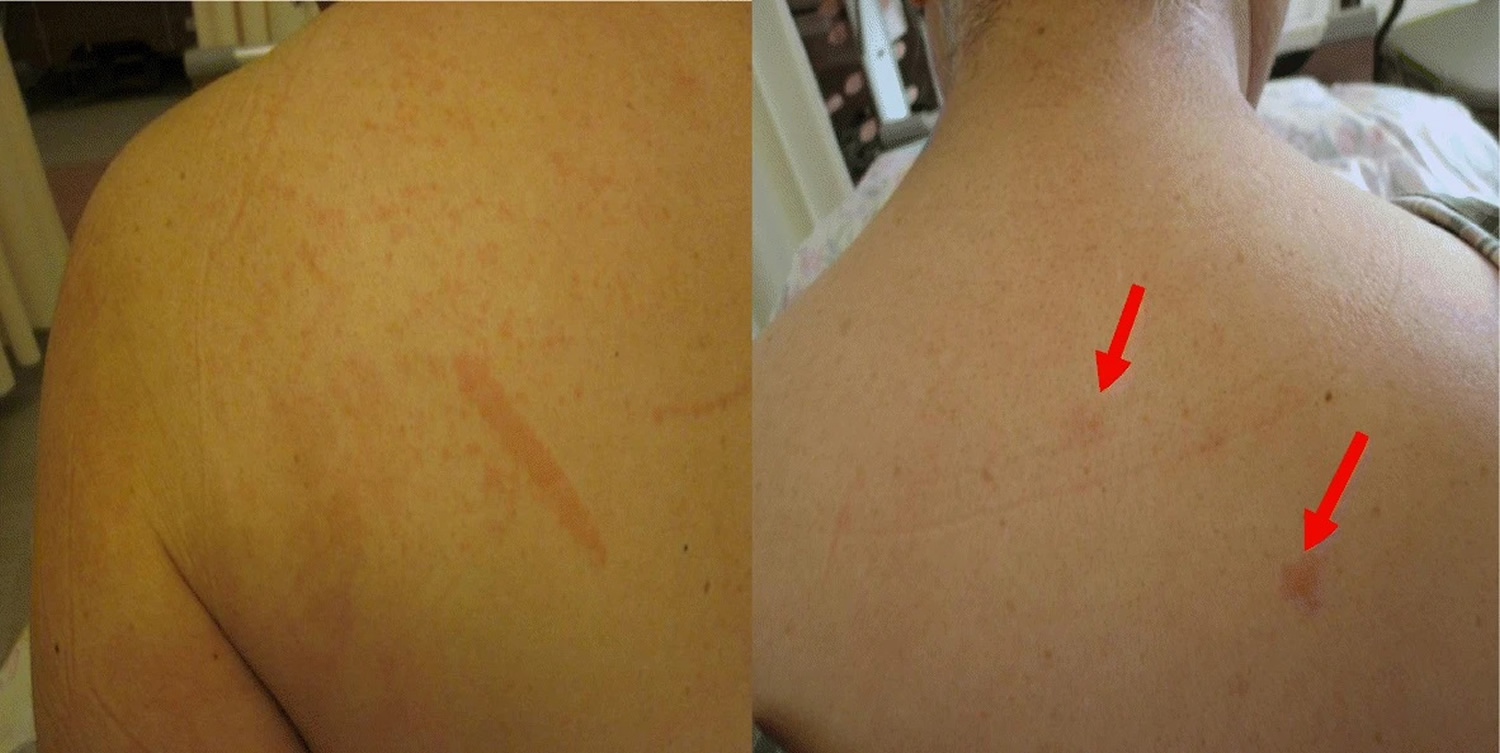
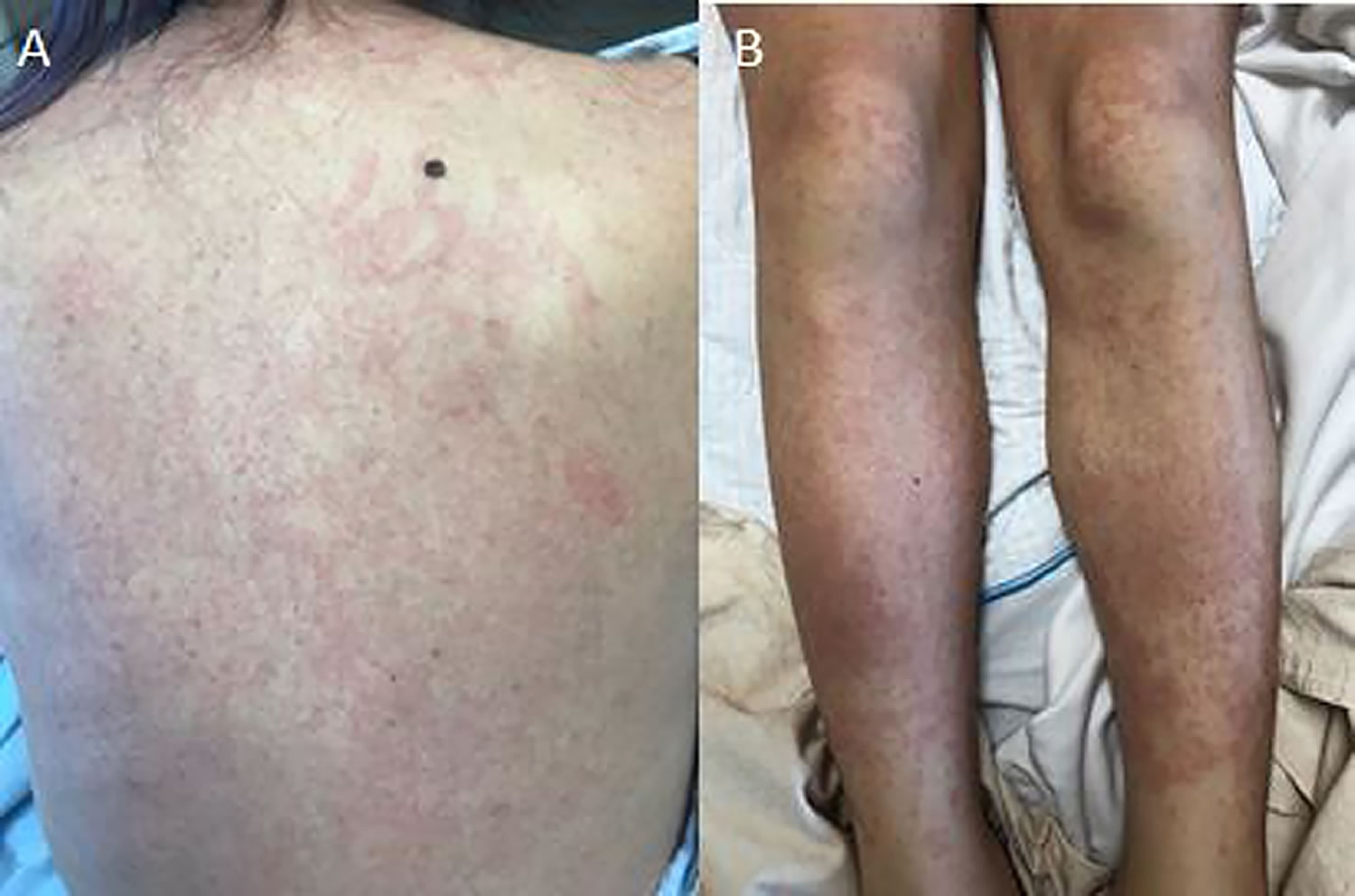
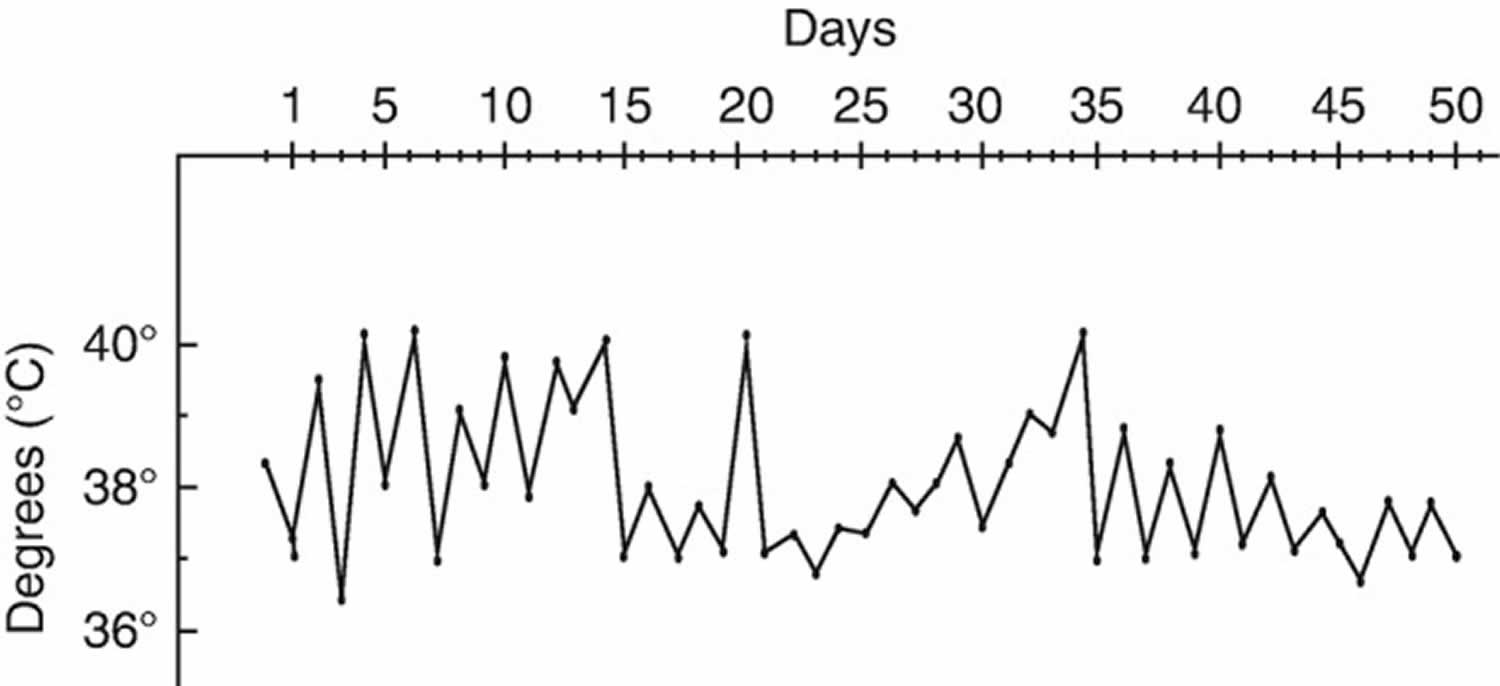
Figure 2. Adult Still’s disease fever (typical temperature curve during an Adult Still’s disease flare)
What is Still’s disease?
Still’s disease is a type of juvenile chronic arthritis previously called juvenile rheumatoid arthritis that is characterized by high fever and signs of systemic illness that can exist for weeks or months before the onset of arthritis.
How many people have adult Still’s disease?
To date, epidemiologic data about adult onset Still’s disease have been scarce and imprecise, because of the heterogeneous clinical presentation of the disease and the complex assessment of diagnosis 3. Adult Still’s disease occurs worldwide, and no specific familial aggregation has been reported. Adult onset Still’s disease satisfies the definition of an orphan disease because historically its prevalence was estimated between one case per million in Europe 6 and ten cases per million in Japan 7. In the Japanese study, the incidence was estimated at about one case per million in the mid-1990s. However, because of the substantial advances in adult onset Still’s disease diagnosis as well as differential diagnoses during the last two decades, these prevalence and incidence estimates lack robustness, and new studies are clearly needed to update these figures 3.
Initially, adult onset Still’s disease was characterized as affecting exclusively young adults (i.e., 16–35 years of age) 8; however, more recent series identified cases in adults older than 35 and even elderly people 9. The sex ratio is almost balanced, with only a slight female predominance 10.
Could patients with adult onset Still’s disease develop intestinal pseudo-obstruction as a complication?
Pseudo-obstruction develops secondarily to various acute diseases, and it can also develop in patients with immunological diseases. Intestinal pseudo-obstruction can occur in patients with systemic lupus erythematosus, scleroderma, and chronic inflammation from autoimmune diseases 11. On the other hand, intestinal pseudo-obstruction in patients with adult onset Still’s disease is extremely rare, and to our knowledge, only three cases like this have been reported 12, 13. Amyloidosis should also be excluded in cases of intestinal pseudo-obstruction in patients with adult onset Still’s disease 14. Adult onset Still’s disease may cause intestinal pseudo-obstruction in patients with acute onset and chronic stages complicated with amyloidosis 12.
What is Macrophage activation syndrome (MAS)?
Macrophage activation syndrome (MAS) is the term used to describe a potentially life-threatening complication of systemic inflammatory disorders especially rheumatic disease, for unknown reasons, occurs much more frequently in individuals with systemic-onset juvenile idiopathic arthritis (SJIA) sometimes known as Still’s disease and in its adult equivalent, adult-onset
Still’s disease (AOSD) 15. Macrophage activation syndrome (MAS) also occurs in patients with other autoimmune or autoinflammatory conditions, such as adult- and childhood-onset systemic lupus erythematosus 16, Kawasaki disease 17, and periodic fever syndromes 18, is being reported with increased frequency.
Macrophage activation syndrome (MAS) is characterized by pancytopenia, liver insufficiency, coagulopathy, and neurologic symptoms and is thought to be caused by the activation and uncontrolled proliferation of T lymphocytes and well-differentiated macrophages, leading to widespread hemophagocytosis and cytokine overproduction 19.
Adult Still’s disease symptoms
Almost all people with adult Still’s disease will have a combination of the following signs and symptoms – fever, joint pain, sore throat, and a rash.
- Fever. You might have a daily fever of at least 102 °F (38.9 °C) for a week or longer. The fever usually peaks in the late afternoon or early evening. You might have two fever spikes daily, with your temperature returning to normal in between. Typically, the fever starts suddenly once per day, most commonly in the afternoon or evening and temperature rapidly reaches 39 °C or more, associated with shivers. The fever evolves with daily evening spikes for more than a week (Figure 2). Patients usually experience rapid deterioration of general health as well as significant weight loss. Of importance, the fever may be the only clinical symptom of adult Still’s disease as a potential diagnosis in patients with fever of unknown origin 20.
- Achy and swollen joints (arthralgia or arthritis). Joint pain or arthritis is the second most common symptom, with synovitis, usually with concomitant fever spikes, occurring in more than two-thirds of patients. Your joints — especially your knees and wrists — might be stiff, painful, swollen and inflamed. Most often, several joints are involved at the same time with your ankles, elbows, hands, shoulders, sacroiliac and distal interphalangeal joints might also ache. Often, people with adult-onset Still’s disease have morning stiffness of joints that lasts for several hours. The joint discomfort usually lasts at least two weeks. In some patients, the presentation is that of a bilateral symmetrical rheumatoid arthritis (RA)-like polyarthritis 21. During the evolution, arthritis becomes erosive in one-third of patients; in these patients, isolated bilateral carpal ankylosis (i.e., without structural damage of metacarpophalangeal or proximal interphalangeal joints, in contrast to rheumatoid arthritis) is very suggestive of adult Still’s disease 21.
- Skin rash. A salmon-pink colored macular or maculopapular skin rash might come and go with the fever. The rash usually appears on your trunk, arms or legs. The rash is transient, mainly visible during fever spikes on the proximal limbs or trunk, and rarely involves the face, palms, or soles of the feet 21, 22. No specific histological feature has been described. Misdiagnosis with drug allergy is frequent and usually attributed to nonsteroidal anti-inflammatory drugs (NSAIDs) prescribed for joint symptoms or fever. Complete regression without scars is the rule.
- Atypical patterns also reported are urticarial or pruritic with dermographism 22. Presence of purpuric lesions should lead to urgent coagulation workup because these lesions are suggestive of the adult onset Still’s disease hematologic complications of hemophagocytic syndrome or reactive hemophagocytic lymphoproliferation, disseminated intravascular coagulation (DIC), or thrombotic microangiopathy, also called thrombocytic thrombocytopenic purpura or Moschcowitz syndrome 21.
- Sore throat (odynophagia) and sometimes pharyngitis. This is one of the first symptoms of adult Still’s disease. The lymph nodes in your neck might be swollen and tender.
- Muscle pain. Muscular pain usually ebbs and flows with the fever, but the pain can be severe enough to disrupt your daily activities.
Additional symptoms include:
- Abdominal pain and swelling
- Pain when taking a deep breath (pleurisy)
- Swollen lymph nodes (lymphadenopathy)
- Weight loss
The spleen or liver may become swollen. Lung and heart inflammation may also occur.
The signs and symptoms of adult Still’s disease can mimic those of other conditions, including lupus and a type of cancer called lymphoma.
Adult onset Still’s disease complications
Adult onset Still’s disease can be life-threatening because of the predominant involvement of one organ or of systemic complications with multiple organ failure from the outset, while the diagnosis isn’t clearly confirmed. Most complications from adult Still’s disease arise from chronic inflammation of organs and joints. A rare form of adult onset Still’s disease, called macrophage activation syndrome (MAS), can be very severe with high fevers, severe illness and low blood cell counts. The bone marrow is involved and biopsy is needed to make the diagnosis.
Other complications of adult onset Still’s disease may include:
- Arthritis in several joints. Chronic inflammation can damage your joints. The most commonly involved joints are your knees and wrists. Your neck, foot, finger and hip joints also may be affected, but much less frequently.
- Liver disease
- Inflammation of your heart. Adult Still’s disease can lead to an inflammation of the saclike covering of your heart (pericarditis) or of the muscular portion of your heart (myocarditis).
- Pleural effusion. Inflammation may cause fluid to build up around your lungs, which can make it hard to breathe deeply.
- Spleen enlargement
Many other symptoms attributed to adult onset Still’s disease have appeared in the literature. As these are almost exclusively in case reports, drawing conclusions is difficult:
- Ophthalmologic involvement related to sicca syndrome, conjunctivitis, uveitis, or episcleritis 23.
- Neurological manifestations, such as ischemic stroke, aseptic meningitis, or encephalitis 4. Such symptoms might reveal hepatic failure or coagulation dysfunction, as in disseminated intravascular coagulopathy (DIC), reactive hemophagocytic lymphohistiocytosis (RHL) or thrombotic microangiopathy (TMA).
- Renal involvement limited to isolated proteinuria or related to glomerular or interstitial nephritis [102, 103]. Acute renal failure is possible in the context of severe myositis, hepatic insufficiency, or hematological complications 24.
Table 1. Main life-threatening complications of adult onset Still’s disease
| Complications | Signs and symptoms | Diagnosis | Treatment |
|---|---|---|---|
| Reactive hemophagocytic lymphohistiocytosis (RHL) | • High (>38.5 °C), persistent fever a • Peripheral lymphadenopathy, hepatomegaly, splenomegaly • Polymorphous skin rashes b: rashes, edema, petechiae, urticaria, or purpura • Multiple organ involvement: pulmonary, neurological, gastrointestinal, or renal involvement, bleeding • Rapid change on hemogram: resolution of high leukocyte and neutrophil counts, occurrence of anemia and thrombocytopenia | • Cytopeniac: anemia, thrombocytopenia, and/or leukopenia • Decreased ESR d • Increased liver function tests: transaminases and alkaline phosphatases • Elevated LDH level • Hypertriglyceridemia • Hyperferritinemia • High soluble CD25 or CD163 level • Hemophagocytosis in bone marrow smear (or reticuloendothelial organs) • Increased CD163 staining in bone marrow Or • HScore f | • Supportive care in ICU • Exclusion of an additional trigger: mainly infections g • Immunomodulatory agents: – High-dose corticosteroids – ±IL-1 or IL-6 inhibitors after a few days – If refractory, etoposide or cyclosporine A |
| Disseminated intravascular coagulopathy (DIC) | • Hematoma, bleeding • Thrombotic event • Multi-organ involvement or failure: ARDS, pleural effusion, myocarditis, pulmonary embolism, gastrointestinal bleeding, CNS involvement | • Thrombocytopenia • Prolongation of prothrombin time and activated thromboplastin time • Decreased fibrinogen • Increased levels of fibrin degradation products | • Supportive measures in ICU • Immunomodulatory agents: – High-dose corticosteroids – ±IL-1 or IL-6 inhibitors – If refractory, cyclosporine A |
| Thrombotic microangiopathy (TMA) or Moschcowitz syndrome | • Acute vision impairment (early sign) • Weakness • Confusion, seizures, or coma • Abdominal pain, nausea, vomiting, diarrhea • Cutaneous gangrene • Arrhythmias caused by myocardial damage • Multi-organ failure | • Mechanical hemolytic anemia (schistocytes on the peripheral blood smear with a negative Coomb’s test) • Thrombocytopenia • Multiple organ failure of varying severity (mainly the kidneys and the CNS) | • Supportive measures in ICU • Specific treatment: – High-dose corticosteroids and plasma exchange – ±Hemodialysis – If insufficient, multidisciplinary round ±IL-1 or IL-6 inhibitors or cyclosporine or IVIG |
| Fulminant hepatitis | • Physical status deterioration: appetite loss, fatigue • Jaundice • Hepatomegaly • Right abdominal pain • Rarely, bleeding • Elevated liver function test findings | • Abnormal and rapidly increasing liver function test findings • High activity on multi-biomarker FibroTest • Liver biopsy (if performed): nonspecific portal infiltrates of lymphocytes, plasma cells, and neutrophils, hepatocytic lesions or massive necrosis | • Supportive measures in ICU • Discontinuation of all potentially hepatotoxic drugsh: mainly acetaminophen, aspirin, NSAIDs, or methotrexate • Rule out RHL, DIC, and/or TMA • Rule out viral reactivation • Specific treatment: – High-dose corticosteroids – ±IL-1 or IL-6 inhibitorsi or cyclosporine • Liver transplantation in extreme cases |
| Cardiac complications | • Pericarditis, sometimes recurrent • Tamponade • Myocarditis • Endocarditis (exceptional cases) | • Electrocardiography • Echocardiography • Troponin and creatinine kinase level increase if myocarditis | • Supportive measures in ICU • Specific treatment: – High-dose corticosteroids usually – ±IL-1 or IL-6 inhibitors |
| Pulmonary arterial hypertension | • Dyspnea (key symptom) • Fatigue, dizziness • Syncope • ARDS | • Electrocardiography: right atrium hypertrophy • Echocardiography: systolic PAP >35 mmHg • Right heart catheterization (gold standard): – Mean PAP ≥25 mmHg at rest – End-expiratory PAWP ≤15 mmHg – Pulmonary vascular resistance >3 wood units | • Close monitoring and refer to a pulmonary hypertension reference center (multidisciplinary rounds) • Vasodilatation therapy: calcium channel blockers, endothelin receptor antagonists, phosphodiesterase-5 inhibitors, and/or prostacyclin analogues • Immunosuppressive therapy: – High-dose steroids – ±IL-1 or IL-6 inhibitors or cyclosporine |
| Pulmonary complications | • Pleuritis • Interstitial lung disease with or without ARDS • Aseptic empyema • Diffuse alveolar hemorrhage | • High-resolution computed tomography • Bronchoalveolar lavage (for differential diagnosis: in AoSD, nonspecific neutrophilic profile) • Pulmonary function tests | • Supportive measures in ICU if ARDS • Rule out differential diagnoses: infections, cardiogenic causes (brain natriuretic peptide dosage, echocardiography), drug-related or iatrogenic causes, cancer • Specific treatment: – High-dose corticosteroids usually – ±IL-1 or IL-6 inhibitors |
| Amyloid A amyloidosis | • Exceptional these days • Renal failure, proteinuria, edema, hydrops • Digestive involvement • Orthostatic hypotension, other neuropathies | • Biopsy of minor salivary gland, or abdominal fat pad, or the kidney | • Inflammation control with IL-1 or IL-6 inhibitors |
Footnotes:
a In adult onset Still’s disease, fever is often remitting, with spikes (hectic)
b In adult onset Still’s disease, skin rash is classically maculopapular and evanescent
c In noncomplicated adult onset Still’s disease, white blood count is often ≥10,000/mm³, with mainly neutrophils, and thrombocytosis is frequent
d In noncomplicated adult onset Still’s disease, ESR is high
e Hyperferritinemia is also seen in adult onset Still’s disease, but, in case of very high ferritin levels or sudden increase, a reactive hemophagocytic lymphohistiocytosis (RHL) or another systemic complication should be suspected
f This scoring system is a set of nine weighted criteria (known underlying immunosuppression, temperature, organomegaly, number of cytopenia, ferritin, triglyceride, fibrinogen, serum glutamic oxaloacetic transaminase, hemophagocytosis features on bone marrow aspirate) that have been elaborated and validated for the diagnosis of reactive hemophagocytic lymphohistiocytosis (RHL) in adults 25
g Mainly infections, in particular viral reactivation (Epstein-Barr virus, cytomegalovirus), which may trigger adult onset Still’s disease or reactivated by immunosuppressive treatments
h Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, potentially induced by interleukin (IL)-1 or IL-6 or another immunosuppressive agent, is a differential diagnosis that should always been ruled out because it can be responsible for a fulminant hepatitis and mimic adult onset Still’s disease systemic manifestations
i Keeping in mind that tocilizumab-induced hepatic injury has also been reported 26
Abbreviations: AoSD = adult-onset Still’s disease; ARDS = acute respiratory distress syndrome; BAL = bronchoalveolar lavage; CNS = central nervous system; DIC = disseminated intravascular coagulopathy; DRESS = drug reaction with eosinophilia and systemic symptoms; ESR = erythrocyte sedimentation rate; ICU = intensive care unit; IL = interleukin; IVIG = intravenous immunoglobulins; LDH = lactate dehydrogenase; NSAIDs = nonsteroidal anti-inflammatory drugs; PAP = pulmonary arterial pressure; PAWP = pulmonary artery wedge pressure; RHL = reactive hemophagocytic lymphohistiocytosis; TMA = thrombotic microangiopathy
[Source 3 ]Reactive Hemophagocytic Lymphohistiocytosis (RHL)
Reactive Hemophagocytic Lymphohistiocytosis (RHL) also known as reactive hemophagocytic syndrome (RHS), is a common complication of adult onset Still’s disease at the time of diagnosis, right after treatment introduction or during the course of the disease 27. In systemic-onset juvenile idiopathic arthritis (SJIA), reactive hemophagocytic lymphohistiocytosis (RHL) is also called macrophage activation syndrome (MAS). This serious complication should be suspected in a patient with persisting fever (in contrast to evening spiking fever) and decrease in initially elevated leukocyte and neutrophil counts 28. In adult-onset Still disease (AOSD), reactive hemophagocytic lymphohistiocytosis (RHL) can occur with either a disease flare or an active infection from a complication of immunosuppressive treatment 29. The key issue with reactive hemophagocytic lymphohistiocytosis (RHL) is to determine whether its occurrence is related to adult onset Still’s disease intense inflammation or to concomitant infection, potentially favored by immunomodulators introduced because of adult onset Still’s disease.
Reactive Hemophagocytic Lymphohistiocytosis (RHL) and adult onset Still’s disease share several clinical characteristics and laboratory findings including fever, lymphadenopathy, hepatosplenomegaly, elevated liver enzyme levels, and hyperferritinemia, and these conditions cannot be easily distinguished from each other 30. However, most of the previous studies on adult onset Still’s disease related to reactive hemophagocytic lymphohistiocytosis (RHL) have been case reports 31. The list of possible infections is quite long, with viral reactivation at first place. Therefore, the inducing factors, course, treatment, and prognosis of reactive hemophagocytic lymphohistiocytosis (RHL) in adult onset Still’s disease are not yet clearly known. It is suggested that reactive hemophagocytic lymphohistiocytosis (RHL) in adult onset Still’s disease is more common than has been previously recognized in the literature and has probably been underdiagnosed in several cases 32.
Coagulation disorders
Adult onset Still’s disease can be complicated by two serious coagulation disorders, mainly at the acute phase of the disease.
The first disorder, disseminated intravascular coagulation (DIC), is not rare and may occur at a frequency of 1–5% 33. This diagnosis should be suspected with the combination of thrombotic events and cutaneous or mucosal bleeding and sometimes specific organ involvements, such as fulminant hepatitis, cardiac or respiratory failure, or stroke. Hemostasis workups reveal platelet and coagulation factor consumption, increased thromboplastin time, and high D-dimer levels.
The other rare but quite severe coagulation disorder is thrombotic microangiopathy (TMA). It must be suspected with unexplained multiorgan failure or stroke, related to multiple small thrombi leading to tissue ischemia and mechanical hemolytic anemia 34. Acute vision impairment, such as blurred vision related to Purtscher-like retinopathy, is frequently the first symptom 35. Key diagnostic tests display thrombocytopenia by platelet consumption, anemia, and schistocytes (fragmented red blood cells), which are specific to this diagnosis. Organ imaging may reveal multiple infarctions. In addition, ADAMTS13 enzymatic activity needs to be tested because acquired deficiency has been found to predispose to thrombotic microangiopathy (TMA) 36. TMA has been mainly described during adult onset Still’s disease flare, related to intense inflammation or to concomitant infection with Shiga toxin-producing microorganisms 35.
Cardiac and Pulmonary Involvements
Although pleural effusion or pericarditis is frequent, other serious cardiac or pulmonary manifestations have been described.
Specific attention should be dedicated to a rare and very severe adult onset Still’s disease manifestation, pulmonary arterial hypertension (PAH), with several cases recently reported [77–88]. PAH, either idiopathic or occurring with adult onset Still’s disease or other connective tissue diseases, is thought to be related at least in part to immune-related alteration of the endothelium with overproduction of IL-1 , IL-6, IL-18, and TNF [79, 80, 82, 83, 99]. PAH may occur at adult onset Still’s disease onset or later and seems to predominate in women. The diagnosis must be suspected with unexplained and often rapidly progressing dyspnea [59, 79, 81, 100, 101]. Electrocardiography is rarely contributive, eventually disclosing signs of right atrium hypertrophy. Echocardiography is more useful for detection by measuring systolic pulmonary artery pressure, which is >35 mmHg, and the exclusion of left ventricle dysfunction. The formal PAH diagnosis remains the right heart catheterization [100, 101].
Hepatitis
While liver abnormalities are most often limited to mild to moderate increases in aminotransferase activity which is frequent (up to 60% of cases), fulminant and fatal hepatitis have been reported 28. The potential role of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), or methotrexate has been mentioned by some authors 23. Thus, clinicians should closely monitor liver function from the onset of the disease and particularly after prescription of potentially hepatotoxic drugs. Self-medication should be avoided.
Liver biopsy, if performed, reveals nonspecific portal infiltrates of lymphocytes, plasma cells, and polymorphonuclears 28. Hepatocytic lesions or massive necrosis has been described in fulminant hepatitis with rapidly progressive hepatic insufficiency. Liver transplantation was necessary in a few exceptional cases 37.
Amyloid A amyloidosis
Amyloid A amyloidosis is becoming extremely rare owing to the better ability of adult onset Still’s disease treatments to control inflammation 38. It could be observed in refractory patients or in patients who did not have access to adequate treatments for long periods of time. Multiple organs can be involved. Secondary amyloid A amyloidosis (AA amyloidosis) is nowadays exceptional and is related to sustained uncontrolled inflammation 39.
Macrophage activation syndrome (MAS)
The most severe complication of the spectrum of Still’s disease and adult onset Still’s disease is secondary hemophagocytic lymphohistiocytosis (HLH), better known as macrophage activation syndrome (MAS). The term cytokine storm best describes excessive cytokinaemia during macrophage activation syndrome (MAS). The prevalence varies from 10 to 15% and is associated with high mortality 40. Macrophage activation syndrome (MAS) is characterized by pancytopenia, liver insufficiency, coagulopathy, and neurologic symptoms and is thought to be caused by the activation and uncontrolled proliferation of T lymphocytes and well-differentiated macrophages, leading to widespread hemophagocytosis and cytokine overproduction 41. Possible triggers such as infections or medications in combination with uncontrolled and prolonged inflammation in patients with genetic predisposition may lead to this life-threatening condition 42.
The clinical presentation of macrophage activation syndrome (MAS) is generally acute and occasionally dramatic. Typically, patients become acutely ill with the sudden onset of nonremitting high fever, profound depression in all 3 blood cell lines (ie, leukopenia, anemia, and thrombocytopenia), hepatosplenomegaly, lymphadenopathy, and elevated serum liver enzyme levels 43. High levels of triglycerides and lactic dehydrogenase and low sodium levels are consistently observed.
The coagulation profile is often abnormal, with prolongation of prothrombin time (PT) and partial thromboplastin time (aPTT), hypofibrinogenemia, and detectable fibrin degradation products. In children with systemic juvenile idiopathic arthritis, the clinical picture may mimic sepsis or an exacerbation of the underlying disease. However, the pattern of nonremitting fever is different from the remitting high-spiking fever seen in systemic juvenile idiopathic arthritis. Moreover, patients may show a paradoxical improvement in the underlying inflammatory disease at the onset of macrophage activation syndrome, with disappearance of signs and symptoms of arthritis and a precipitous fall in the erythrocyte sedimentation rate. The latter phenomenon probably reflects the degree of hypofibrinogenemia secondary to fibrinogen consumption and liver dysfunction 44.
Researchers from France developed diagnostic criteria for macrophage activation syndrome (MAS) to shorten the critical process of reaching an accurate diagnosis. In this multicentre retrospective cohort study of 312 patients, the diagnosis relied on a set of nine variables: known underlying immunosuppression, high temperature, organomegaly, triglyceride, ferritin, serum aspartate transaminase, fibrinogen levels, cytopenia and haemophagocytosis features on bone marrow aspirate (Table 2). Based on a scoring system, physicians can then calculate the “HScore” and assess the probability of the patient having macrophage activation syndrome (MAS). MAS can be ruled out with an HScore of ≤90 MAS, whereas an HScore ≥ 250 has a diagnostic accuracy of >99% 45.
Knowing how to diagnose macrophage activation syndrome (MAS) could be life-saving because of its short therapeutic window of opportunity. Even if the full diagnostic criteria are not met, treatment should be started as soon as possible to silence the cytokine storm and prevent hyperinflammatory complications, critical illness and death. Cross-specialty collaboration is the key to success.
Once a diagnosis of macrophage activation syndrome (MAS) has been made, serum ferritin concentrations are useful for monitoring disease activity and response to treatment. Very high peak levels as well as a limited decrease (less than 50% from first measurement near diagnosis) after initiation of treatment are associated with high mortality in pediatric patients 46.
Table 2. HScore † for diagnosis of macrophage activation syndrome (MAS)
| Variable | Number of Points |
|---|---|
| Temperature | |
| <38.4 °C | 0 |
| 38.4–39.4 °C | 33 |
| >39.4 °C | 49 |
| Organomegaly | |
| None | 0 |
| Hepatomegaly or splenomegaly | 23 |
| Hepatomegaly and splenomegaly | 38 |
| Cytopenia | |
| One lineage | 0 |
| Two lineages | 24 |
| Three lineages | 34 |
| Triglycerides (mmol/L) | |
| <1.5 | 0 |
| 1.5–4.0 | 44 |
| >4.0 | 64 |
| Fibrinogen (g/L) | |
| >2.5 | 0 |
| ≤2.5 | 30 |
| Ferritin (ng/mL) | |
| <2000 | 0 |
| 2000–6000 | 35 |
| >6000 | 50 |
| Serum aspartate aminotransferase (IU/L) | |
| <30 | 0 |
| ≥30 | 19 |
| Hemophagocytosis on bone marrow aspirate | |
| No | 0 |
| Yes | 35 |
| Known immunosuppression | |
| No | 0 |
| Yes | 18 |
Footnotes: † The probability of having macrophage activation syndrome (MAS) ranges from <1% with an HScore of ≤90 to >99% with an HScore of ≥250.
[Source 47 ]Adult Still’s disease causes
The exact underlying cause of adult onset Still’s disease is not fully understood 2. The hypothesis remains that adult onset Still’s disease is a reactive syndrome in which various infectious agents may act as triggers. Some researchers suspect the adult onset Still’s disease might be triggered by a viral or bacterial infection in a genetically predisposed host 48. Both genetic factors and a variety of viruses, bacteria like Yersinia enterocolitica and Mycoplasma pneumonia and other infectious factors have been suggested as important 49. A French study of 62 patients showed the association of adult onset Still’s disease with HLA antigen subtypes 50.
The immune dysregulation plays a central role in adult-onset Still’s disease and is characterized by pathogenic involvement of both arms of the immune system. No risk factors for adult onset Still’s disease have been identified.
In the mid-2000s, McGonagle and McDermott 51, 52 formulated the hypothesis of two main pathogenic mechanisms underlying immune-mediated inflammation against the self and proposed a new classification for immunological diseases that distinguished autoimmunity from autoinflammation. The term autoimmunity was used to refer to adaptive immunity and was defined as aberrant dendritic cell, B cell and T cell responses in primary and secondary lymphoid organs leading to a break in tolerance and development of immune reactivity towards native antigens (with autoantibodies in most cases). The term autoinflammation was used to refer to innate immunity and was defined as dysregulated activation of macrophages and neutrophils in response to a danger signal leading to tissue damage. These categories represent a continuum, which enabled a new classification of immune-mediated inflammatory disorders to be refined in subsequent years 53.
The diagnosis of autoimmune diseases is often supported by the presence of autoantibodies or autoantigen-specific T cells and B cells. By contrast, no specific biomarker exists for systemic autoinflammatory disorders (SAID). The definition mainly relies on similarities with monogenic, hereditary periodic fever syndromes, which were at the origin of the concept. Autoinflammatory diseases have several key features, including intense inflammation with periodic fever, tissue inflammation depending on the disease, increased leukocyte and neutrophil counts, elevated erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level, a pathogenic function of the inflammasome and a therapeutic response to IL-1 blockade 54. Aside from monogenic familial syndromes, Crohn’s disease was the first classified non-familial polygenic autoinflammatory disorder, mainly tissue specific, with predominant gut involvement 55. A few years later, Still’s disease (systemic-onset juvenile idiopathic arthritis (SJIA) and adult onset Still’s disease) was described as another non-familial autoinflammatory disorder, becoming one of the most characteristic polygenic systemic autoinflammatory disorders (SAID) 56, 57.
There is a high degree of similarity between infections and the onset of adult onset Still’s disease for fever, leucocytosis and elevated C-reactive protein (CRP). Logically, many investigators focused on identifying infectious triggers and described the occurrence of adult onset Still’s disease after infection with cytomegalovirus, Epstein-Barr, influenza, Mycoplasma, hepatitis, etc. 58. Scientists now know that cytomegalovirus (CMV) may also trigger a relapse of adult onset Still’s disease 59. Blood cultures and polymerase chain reaction (PCR) tests may, therefore, be useful for a differential diagnosis, although no specific diagnostic algorithms exist to date. It is currently still not clear which pathogenic viruses and bacteria should be included in the diagnostic workup. Remarkably, procalcitonin is not a reliable marker, since patients suffering from adult onset Still’s disease can show elevated procalcitonin levels without confirmed infection 60.
Other studies have examined the relationship between cancer and adult onset Still’s disease 61 and reported malignancy-mediated autoinflammation in breast cancer 62, thyroid cancer 63, melanoma, lung cancer and haematological malignancies, mostly lymphomas 64.
A role for infections as a trigger
Bacteria or viruses are the usual suspects for the danger signals. Numerous case reports describe the occurrence of adult onset Still’s disease after viral infection (rubella; measles; mumps; Epstein-Barr virus; hepatitis A, B, or C virus; HIV; cytomegalovirus; parvovirus B19; adenovirus; echovirus; human herpes virus 6; influenza and parainfluenza viruses; Coxsackie virus) or bacterial infection (Yersinia enterocolitica, Campylobacter jejuni, Chlamydia trachomatis or pneumoniae, Mycoplasma pneumoniae, Borrelia burgdorferi) 65. In addition, patients often experience odynophagia or pharyngitis just before the start of the disease or the relapse, which could correspond to the infectious danger signal that will trigger Toll-like receptors and start the intense inflammatory process.
A role for a genetic background
In contrast to monogenic, hereditary, periodic fever syndromes, the underlying genetic background of adult onset Still’s disease is largely unknown. The disease is present in different geographic regions and different ethnic groups 65. Association studies have suggested a potential predisposing genetic background. HLA-Bw35 was the first identified as a susceptibility antigen and associated with a mild self-limiting pattern of the disease 66. HLA-DR4 was found more prevalent in adult onset Still’s disease cases versus healthy controls, and HLA-DRw6 was associated with the occurrence of proximal arthralgia 67. However, no functional analysis has confirmed these hypotheses 65.
Recently, two major findings have been reported in systemic-onset juvenile idiopathic arthritis (SJIA) patients. Firstly, a homoallelic missense mutation in the enzyme laccase (multicopper oxidoreductase) domain-containing 1—LACC1—has been identified in 13 systemic-onset juvenile idiopathic arthritis (SJIA) patients from five Saudi consanguineous families 68. Although familial systemic-onset juvenile idiopathic arthritis (SJIA) is not the rule, this study raises the potential role of this laccase in the pathogenesis of systemic-onset juvenile idiopathic arthritis (SJIA). Secondly, a large association study performed in 982 systemic-onset juvenile idiopathic arthritis (SJIA) children and 8010 healthy controls identified a strong association between systemic-onset juvenile idiopathic arthritis (SJIA) and different HLA-DRB1∗11 haplotypes which all contain glutamate 58 69. This finding is more challenging since it would involve adaptive immunity in the adult onset Still’s disease pathogenesis which was not expected 70.
Finally, substantial advances have been made in exploring the human genome, which will facilitate the identification of potential mutations in genes involved in autoinflammatory pathways, including de novo germinal mutations or somatic mutations occurring during fetus development or after birth 71.
Adult onset Still’s disease pathogenesis
Current scientific understanding of the mechanisms underlying adult onset Still’s disease pathogenesis is mostly hypothetical, but studies of patients with systemic-onset juvenile idiopathic arthritis (SJIA) and a generally growing understanding of systemic autoinflammatory disorders (SAID) suggest the involvement of a pro-inflammatory cascade (see Figure 3) 3. Scientists still do not know what exactly triggers pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). The causal inferences between genetics and adult onset Still’s disease are controversial. Human genetic factors apparently contribute to systemic-onset juvenile idiopathic arthritis (SJIA) in children, whereas the underlying genomic susceptibility in the adult onset Still’s disease (AOSD) is unclear 72.
Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) stimulate macrophages and neutrophils, leading to activation of specific inflammasomes via Toll-like receptors. Inflammasomes are multiprotein units that act as catalysts by activating the caspase pathway immediately after they come into contact with damage or illness. Caspase enzymes lead to overproduction of IL-1β, the hallmark of adult onset Still’s disease, and IL-18. IL-1β and IL-18 then promote further abnormal inflammation by several cytokine bursts, including IL-6, IL-8, IL-17, IL-18 and TNF-α. At this point, the patient is experiencing heavy systemic symptoms 73.
Furthermore, activated macrophages stimulate the release of excessive levels of ferritin. In addition to functioning as an iron storage molecule, ferritin also plays a central role in many conditions with an amplified inflammatory response, currently called “hyperferritinemic syndromes”, such as adult onset Still’s disease, macrophage activation syndrome (MAS), catastrophic antiphospholipid syndrome and septic shock 74. Ferritin has a key role in inflammation by promoting cytokine production, and at the same time, cytokines can regulate ferritin synthesis.
Moreover, analysis of accumulating data over the past years showed an enhancement of neutrophil extracellular traps (NET) in adult onset Still’s disease, which promotes the acute phase response by activating the NLRP3 inflammasome 75.
Additionally, dysfunctional natural killer (NK) cells, elevated T-helper Th1 and Th17 cells, enhanced IFN-γ and IL-17 levels, different alarmins, such as the S100 proteins, significantly higher IFN-γ-producing Th1 cells and Th1/Th2 cells ratios and advanced glycation end products complete the proinflammatory environment in many ways, which favors the abnormal response of the human immune system 76.
The massive release of cytokines in patients with adult onset Still’s disease over a prolonged period of time can be fatal. Deficient resolution of inflammation may be mostly due to failures in immune system self-regulation. Deficient regulatory T cells, decreased or defective natural killer (NK) cells, insufficient production of anti-inflammatory cytokines or problematic circulation of advanced glycation end products (AGEs) have been hypothesized to cause these complex problems 77. Surprisingly, the anti-inflammatory cytokine IL-10 levels are elevated during the higher state of inflammation and correlate with disease activity in adult onset Still’s disease 78.
Figure 3. Adult onset Still’s disease pathogenesis
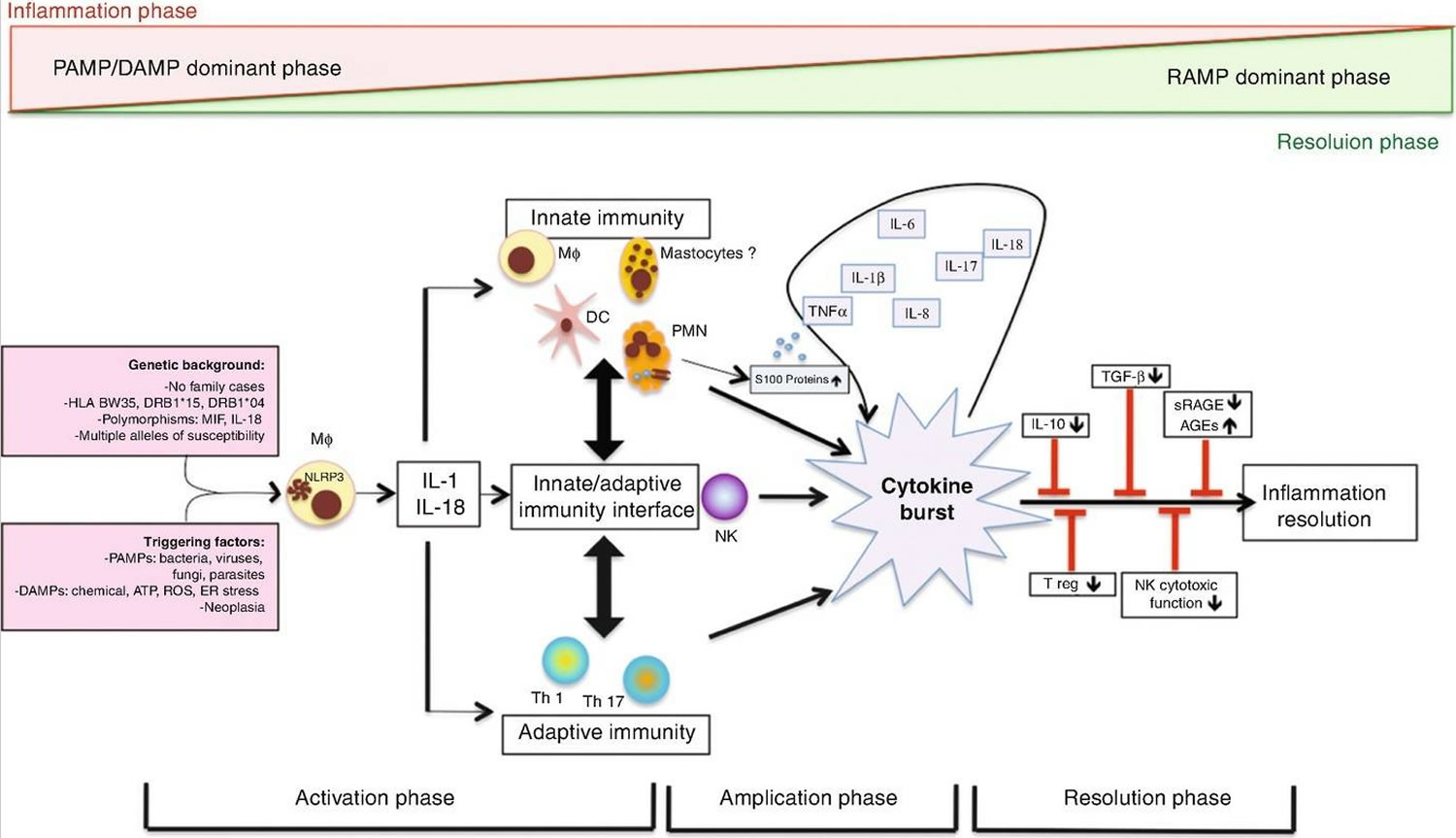
Footnotes: Danger signals (pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), such as infections or environmental chemical factors) are transmitted to macrophages and neutrophils through Toll-like receptors (TLRs), which excessively activate the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome in patients with a predisposing genetic background. This excessive activation of NLRP3 seems to be central and leads to intense production of IL-1β and IL-18. These cytokines intensely stimulate innate immune cell activation, as well as adaptive immune cells, leading to overproduction of several pro-inflammatory cytokines including IL-6, IL-8, IL-17 and TNF, as well as further production of IL-1β and IL-18. Several factors actively contribute to this amplified inflammatory response, which is often referred to as the cytokine ‘burst’ or ‘storm’. In addition to IL-1β itself conferring retrograde activation of macrophages and neutrophils, alarmins such as the S100 proteins and advanced glycation end products (AGEs) are involved. Aside from the amplification mechanisms, deficiency or failure in regulatory or anti-inflammatory mechanisms might be involved in the pathogenesis of autoinflammatory diseases, including deficiency in regulatory T (Treg) cells or natural killer (NK) cells, insufficient IL-10 production and deficiency in production of resolving lipid mediators, soluble receptors of AGEs (sRAGEs) or other resolution-associated molecular patterns (RAMPs). Hence, adult-onset Still’s disease (AoSD) pathogenesis is related to an imbalance between inflammation and resolution.
Abbreviations: AGE = advanced glycosylated end products; ATP = adenosine triphosphate; ER = endoplasmic reticulum; DAMP = damage-associated molecular pattern; DC = dendritic cells; HLA = human leukocyte antigen; IL = interleukin, MΦ = macrophages; MIF = macrophage inhibitory factor; NK = natural killer; PAMP = pathogen-associated molecular pattern; PMN = polymorphonuclear neutrophil; RAMP = resolution-associated molecular pattern; ROS = reactive oxygen species; sRAGE = soluble receptors of AGE products; TGF = transforming growth factor; Th1 = T-helper 1 cells; Treg = T-regulatory cells
[Source 3 ]Adult Still’s disease diagnosis
Adult onset Still’s disease is a diagnosis of exclusion. Adult Still’s disease can only be diagnosed after many other diseases such as infections and cancer are ruled out. The process of eliminating similar medical conditions is most likely to take a considerable amount of time. Adult Still’s disease diagnosis is based on review of your symptoms, medical history, physical examination and possibly laboratory tests. There is no single test that can diagnose adult Still’s disease. Instead, blood tests are used to rule out other diseases with similar symptoms. Other tests, such as X-rays, may be done to check for joint inflammation or damage.
A physical exam may show a fever, rash, and arthritis. Your health care provider will use a stethoscope to listen for changes in the sound of your heart or lungs.
Adult Still’s disease testing
The following blood tests can be helpful in diagnosing adult onset Still’s disease:
- Complete blood count (CBC), may show a high number of white blood cells (granulocytes) and reduced number of red blood cells.
- C-reactive protein (CRP), a measure of inflammation, will be higher than normal.
- ESR (sedimentation rate), a measure of inflammation, will be higher than normal.
- Ferritin level will be very high. Ferritin is a protein that stores iron, a nutrient that is necessary for the production of healthy red blood cells and the distribution of oxygen throughout the body. When your body uses iron, a small amount of ferritin is released from cells and circulates in the blood. Your ferritin level reflects the total amount of iron stored in your body. A serum ferritin level of more than 1000 ng/ml is common in adult onset Still’s disease. Ferritin levels are generally higher than five times the upper limits of normal in patients with adult onset Still’s disease. Elevated ferritin suggests the presence of the disease with an 80% sensitivity and 46% specificity. If combined with a decrease in the proportion of glycosylated ferritin (GF) <20%, the specificity will rise to 93% 48.
- Fibrinogen level will be high.
- Liver function tests will show high levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT).
- Rheumatoid factor (RF) and antinuclear antibodies (ANA) test will be negative. Less than 10% of the patients have antinuclear antibodies (ANA) and rheumatoid factor (RF) but generally only in low titer.
- Blood cultures and viral studies will be negative.
Other tests may be needed to check for inflammation of the joints, chest, liver, and spleen:
- Abdominal ultrasound
- CT scan of the abdomen
- X-rays of the joints, chest, or stomach area (abdomen). Radiographs early in the disease typically are either normal or show slight joint space narrowing or periarticular osteopenia. Narrowing of the wrist carpometacarpal and intercarpal joint spaces which may progress to bone ankylosis is a classic radiographic finding of adult-onset Still’s disease (AOSD) 79.
- Synovial fluid is usually inflammatory with synovial fluid analysis displays high cellularity, >2000 cells/mm³ (mean leukocyte range of 100 to 48,000 cells/microL), which confirms joint inflammation. When performed, synovium biopsy reveals only nonspecific synovitis.
The lab findings discussed below are characteristic of adult onset Still’s disease, but not pathognomonic (specifically characteristic or indicative for a particular disease) and hence their presence along with clinical manifestations will help the clinician in establishing the diagnosis after ruling out alternate causes.
Ferritins are the major biomarkers for adult onset Still’s disease 3. Serum ferritin level is a key biomarker to assess disease activity, but probably less relevant for diagnosis since its specificity is limited and no clear threshold has been identified so far 80. The diagnostic value of glycosylated ferritin (GF) level, with the threshold of glycosylated ferritin≤20%, is much more interesting 81.
Inflammatory markers, ESR and CRP are elevated in almost all the patients 50. Leukocytosis, generally more than 15,000 cells/microliter with a predominance of neutrophils greater than 80%, normocytic normochromic anemia and thrombocytosis are the hematological findings. These hematological abnormalities can be severe enough to mimic primary hematologic disease. Bone marrow biopsy has been reported to show hyperplasia of granulocytic precursors and hypercellularity and hemophagocytosis in some cases. Hepatic transaminases can be elevated in 75 percent of patients and aldolase can also elevate in some due to liver inflammation.
Procalcitonin, a marker of severe systemic infection, was also found elevated in patients with active adult onset Still’s disease and does not appear relevant to distinguish acute infection from adult onset Still’s disease flare 82.
Serum level of calprotectin (i.e., S100A8/S100A9 protein) could be an additional disease activity biomarker because alarmins seem to play a key role in adult onset Still’s disease pathogenesis. However, it is not specific to adult onset Still’s disease and may be elevated in many other inflammatory conditions 83. Serum amyloid A protein (SAA) is an inflammatory biomarker found predictive of amyloidosis 82.
Despite increasing sophistication in the diagnostic workup for possible cancers, there are no universally accepted guidelines for patients with adult onset Still’s disease, which makes daily clinical work more difficult. Positron emission tomography and computed tomography (PET/CT) scanning could be useful in difficult case scenarios to rule out solid tumours or large vessel vasculitis mimicking adult onset Still’s disease, but it is not routine practice because of the relatively high costs 84. Bone marrow examination can rule out a haematologic malignancy or support the diagnosis of macrophage activation syndrome (MAS).
Table 3. Most relevant serum biomarkers identified to date in adult-onset Still’s disease cohorts and their intended potential use
| Biomarker | Diagnosis a | Disease activity monitoring | Prognosis : severity (i.e., predictive of life-threatening complications) | Prognosis: evolution (i.e., predictive of evolution pattern b (systemic versus arthritis; monophasic, recurrent, or progressive, either systemic or joint) | References |
|---|---|---|---|---|---|
| Routine biomarkers | |||||
| CRP | High sensitivity No specificity | + | − | − | 82 |
| Ferritins | |||||
| Ferritin >ULN ≥5ULN (≥1000 μg/L) | High sensitivity No specificity | + | + | ± | 85 |
| Se 40.8% Sp 80% | + | + | High levels associated with systemic pattern c | ||
| Glycosylated ferritin (GF) ≤20% | Se 79.5% Sp 66.4% | − | + | NA | 81 |
| Ferritin >ULN and GF ≤20% | Se 70.5% Sp 83.2% | − | − | − | 86 |
| Ferritin >5ULN and GF ≤20% | Se 70.5% Sp 92.9% | − | − | − | 81 |
| Calprotectin (S100A8/S100A9 proteins) | ±d | + | NA | NA | 83 |
| Procalcitonin | Weak (rule out sepsis) | NA | NA | NA | 87 |
| SAA | NA | NA | + Predictive of AA amyloidosis | NA | 82 |
| Nonroutine biomarkers | |||||
| IL-18 > 150 ng/L >366 ng/L | Se 88% Sp 78% Se 91.7% Sp 99.1% | + | + High levels are associated with RHL, hepatitis, and steroid dependence | + High levels potentially associated with systemic pattern | 88 |
| IL-1β∗ | − e | + | ± | ± High levels potentially associated with systemic pattern | 89 |
| IL-6∗ | − e | + | ± High levels potentially associated with RHL | ± High levels potentially associated with arthritis pattern | 90 |
| TNF-α∗ | − e | − | − | − | 90 |
| S100A12 protein∗ | ± f | + | NA | NA | 91 |
| AGEs and sRAGE∗ | ± | + | NA | + Higher AGE levels in serum with polycyclic or chronic articular patterns (compared to monocyclic pattern) | 92 |
| CXCL10∗ | + | + | NA | NA | 93 |
| CXCL13∗ | + | + | NA | NA | 93 |
| sCD163∗ | ± | NA | NA | NA | 94 |
| MIF∗ | + | + | NA | NA | 95 |
| ICAM1∗ | ± | + | NA | NA | 96 |
Footnotes: + yes, − no, ± tested but contradictory results, NA not assessed, ULN upper level of normal, se sensitivity, sp. specificity, ∗reference range according to manufacturer
a A good diagnostic biomarker helps rule out the differential diagnoses of infection, malignancy, and other inflammatory disorders
b Identifying the disease subset might orientate the therapeutic strategy
c Serum ferritin levels are significantly higher in the systemic subtype 97, but high ferritin levels after adequate treatment may predict chronic articular course 85
d Calprotectin levels help rule out rheumatoid arthritis, but further studies are needed to validate it as a diagnostic biomarker because of no statistical difference between adult onset Still’s disease and septic patients, although the populations were small 82
e Elevated plasma levels of IL-1β, IL-6, and TNFα have been found during adult onset Still’s disease, but the cytokine profile is not specific and cannot differentiate adult onset Still’s disease patients from those with sepsis
f S100A12 was found an efficient diagnostic and monitoring biomarker in systemic juvenile arthritis, but further studies are needed for validation in adult onset Still’s disease
Abbreviations: AA = amyloid A; AGEs = advanced glycation end products; CRP = C-reactive protein; GF = glycosylated ferritin; ICAM1 = intracellular adhesion molecule-1; IL = interleukin; MIF = macrophage inhibitory factor; RHL = reactive hemophagocytic lymphohistiocytosis; SAA = serum amyloid A protein; sRAGE = soluble receptors for AGEs; TNF = tumor necrosis factor
Potential biomarkers for future clinical research
Several research works assessed serum cytokine levels. High serum levels of IL-1 , IL-6, and IL-18 have been found in systemic forms of adult onset Still’s disease and can be considered activity biomarkers. However, their additional value on top of CRP level and other inflammatory biomarkers is unclear, and these cytokines are clearly not specific to adult onset Still’s disease. Thus, they are not recommended in routine investigation. Of importance, IL-18 seems to play a key role in reactive hemophagocytic lymphoproliferation (RHL).
Advanced glycation end (AGE) products and soluble receptors for advanced glycation end products (RAGEs), involved in adult onset Still’s disease pathogenesis, may be elevated in other inflammatory disorders. High serum levels have been associated with polycyclic or chronic/progressive evolution patterns 82. Serum level of soluble CD163, a macrophage activation biomarker, is elevated in adult onset Still’s disease but is not specific to the disease. Finally, several chemokines (C-X-C motif chemokine ligands 10 and 13, macrophage inhibitory factor) have been found to be diagnostic biomarkers for adult onset Still’s disease, which needs to be confirmed in larger patient samples 82.
Adult onset Still’s disease differential diagnosis
Differential diagnosis of adult onset Still’s disease 98:
- Infections
- Bacteria
- Pyogenic bacterial septicemia
- Infectious endocarditis
- Biliary, colic, or urinary occult infections,
- Tuberculosis
- Brucellosis, yersiniosis
- Viruses
- HIV
- Viral hepatitis
- Epstein-Barr virus
- Cytomegalovirus
- Influenza
- Parvovirus B19
- Herpesviridae
- Measles, rubella
- Parasites
- Toxoplasmosis, abscessed parasitosis
- Bacteria
- Cancers
- Lymphoma, Castleman disease, myeloproliferative disorders, melanoma and colon, breast, lung, kidney and thyroid cancer
- In pediatrics: leukemia
- Systemic diseases
- Systemic lupus erythematosus (SLE), idiopathic inflammatory myopathies, vasculitis, hereditary autoinflammatory syndromes, neutrophilic dermatosis, Sweet syndrome, reactive arthritis, sarcoidosis, Schnitzler syndrome, Kikuchi-Fujimoto disease
- In pediatrics: other types of inflammatory arthritis
Adult Still’s disease criteria
Adult Still’s disease criteria may be helpful, although they have been developed more for clinical research than diagnosis 3. At least seven different variety of classification criteria for adult onset Still’s disease are there for use in research. Two sets of adult Still’s disease criteria have been validated (Table 4). The Yamaguchi criteria for classification of adult Still’s disease 99, published in 1992, is the most widely used and has the highest sensitivity; however, it contains exclusion criteria that are problematic in clinical practice. The Fautrel criteria set has the advantage of including ferritin and glycosylated ferritin (GF) levels as diagnostic biomarkers and does not require exclusion criteria 10. In a recent validation study, both sets showed high sensitivity and specificity 100.
Table 4. Classification criteria for adult-onset Still’s disease
| Yamaguchi et al. 99 | Fautrel et al. 10 |
|---|---|
| Major criteria | |
| 1. Fever ≥39 °C lasting 1 week or more 2. Arthralgia lasting 2 weeks or more 3. Typical skin rash: maculopapular, nonpruritic, salmon-pink rash with concomitant fever spikes 4. Leukocytosis ≥10,000/mm³ with neutrophil polymorphonuclear count ≥80% | 1. Spiking fever ≥39 °C 2. Arthralgia 3. Transient erythema 4. Pharyngitis 5. Neutrophil polymorphonuclear count ≥80% 6. Glycosylated ferritin fraction ≤20% |
| Minor criteria | |
| 1. Pharyngitis or sore throat 2. Lymphadenopathy and/or splenomegaly 3. Liver enzyme abnormalities (aminotransferases) 4. Negative for rheumatoid factor or antinuclear antibodies | 1. Typical rash 2. Leukocytosis ≥10,000/mm³ |
| Exclusion criteria | |
| 1. Absence of infection, especially sepsis and Epstein-Barr viral infection 2. Absence of malignant diseases, especially lymphomas 3. Absence of inflammatory disease, especially polyarteritis nodosa | None |
| At least five criteria, including two major criteria And No exclusion criteria | Four major criteria Or Three major criteria and two minor criteria |
| Classification performance a Sensitivity 96.3%; Specificity 98.2%; PPV, 94.6%; NPV, 99.3% Modified Yamaguchi criteria Yamaguchi criteria and – Ferritin > N:Sensitivity, 100%; Specificity 97.1%; PPV, 87.1%; NPV, 100% – Glycosylated ferritin (GF) ≤ 20%:Sensitivity, 98.2%; Specificity, 98.6%; PPV, 93%; NPV, 99.6% | Classification performance a Sensitivity 87.0%; Specificity 97.8%; PPV, 88.7%; NPV, 97.5% |
Footnote: aLebrun D et al. Semin Arthritis Rheum 2018 9
Yamaguchi criteria for Still’s disease
The Yamaguchi criteria for classification of adult Still’s disease 99, published in 1992, is the most widely used; however, it contains exclusion criteria that are problematic in clinical practice.
Yamaguchi criteria for Still’s disease 99:
At least five criteria, including two major criteria and No exclusion criteria
Major criteria
- Fever ≥39 °C lasting 1 week or more
- Arthralgia lasting 2 weeks or more
- Typical skin rash: maculopapular, nonpruritic, salmon-pink rash with concomitant fever spikes
- Leukocytosis ≥10,000/mm³ with neutrophil polymorphonuclear count ≥80%
Minor criteria
- Pharyngitis or sore throat
- Lymphadenopathy and/or splenomegaly
- Liver enzyme abnormalities (aminotransferases)
- Negative for rheumatoid factor or antinuclear antibodies
Exclusion criteria
1. Absence of infection, especially sepsis and Epstein-Barr viral infection
2. Absence of malignant diseases, especially lymphomas
3. Absence of inflammatory disease, especially polyarteritis nodosa
Classification performance a 9
- Se, 96.3%; Sp, 98.2%; PPV, 94.6%; NPV, 99.3%
Modified Yamaguchi criteria
Yamaguchi criteria and
- Ferritin > N:Se, 100%; Sp 97.1%; PPV, 87.1%; NPV, 100%
- Glycosylated ferritin (GF) ≤ 20%:Se, 98.2%; Sp, 98.6%; PPV, 93%; NPV, 99.6%
Adult Still’s disease treatment
Adult onset Still’s disease is rare and no randomized controlled trial has been performed 3. Thus, the only information on therapy is from observational studies and retrospective case series.
The goal of treatment for adult onset Still’s disease is to control the symptoms of arthritis (see Figure 4). Aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, are most often used first. Corticosteroids, such as prednisone may be used for more severe cases. The use of corticosteroids is limited by the known long-term safety issues, especially with higher doses. For at least 30–40% of adult onset Still’s disease patients, the disease cannot be controlled by corticosteroids even when combined with conventional disease-modifying antirheumatic drugs (DMARDs), such as methotrexate. Recently, the management of adult onset Still’s disease benefited from the proofs of the efficacy of targeted biotherapies.
Although nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, have been effective in systemic-onset juvenile idiopathic arthritis (SJIA), they are rarely effective in adult onset Still’s disease; only 20% of patients have achieved control with this therapy 23.
Of the NSAIDs, indometacin 150–250 mg/day seems to be the most effective 23. Liver enzymes must be monitored at the initial stages of the disease because severe hepatitis has been suggested to be related to treatment with NSAIDs 4.
Acetaminophen may help to reduce fever, although it is often not enough to suspend it. Other analgesics might be necessary to relieve intense myalgia and joint paint.
If the adult onset Still’s disease is severe or persists for a long time (becomes chronic), medicines that suppress your immune system might be needed. Such medicines include:
- Methotrexate
- Methotrexate is used as an immunomodulatory agent in rheumatoid arthritis (RA). Methotrexate is efficient in controlling adult onset Still’s disease disease activity and allowing for steroid dose sparing 101. However, whether methotrexate can prevent or limit joint structural damage in the erosive form of adult onset Still’s disease, as has been shown for rheumatoid arthritis (RA), is unclear. Of importance, methotrexate can be associated with anti-IL-1- or anti-IL-6-targeted therapies. The presence of liver enzyme abnormalities does not contraindicate methotrexate prescription, but close biological monitoring is necessary 4.
- Anakinra (interleukin-1 receptor agonist)
- Tocilizumab (interleukin 6 inhibitor)
- Janus kinase (JAK) Inhibitors
- Janus kinase (JAK) inhibitors block a wide variety of proinflammatory cells and can therefore become a very promising treatment approach in heterogeneous disorders, such as adult onset Still’s disease. In a study with 14 patients with refractory adult onset Still’s disease, seven of them achieved complete remission under tofacitinib, while six responded partially. This trial also showed the steroid sparing effect of tofacitinib, especially in the articular phenotype 102. Furthermore, a reported case of adult onset Still’s disease complicated by macrophage activation syndrome (MAS) describes remission with tofacitinib after failure of response to tocilizumab 103. Another case report describes successful treatment of adult onset Still’s disease with tofacitinib in a HIV-positive female patient 104. Baricitinib could also be an option, although current data are debatable 105.
- Cyclosporine A
- Cyclosporine A has been proposed before the era of biotherapies, in patients with systemic features of adult onset Still’s disease or with macrophage activation syndrome (MAS), with an interesting efficacy. However, the tolerance of this drug limits its use, but it certainly can be of interest in complex or refractory situations.
- Tumor necrosis factor (TNF) inhibitors such as etanercept (Enbrel)
- Stimulated by encouraging results from other chronic inflammatory joint diseases, especially rheumatoid arthritis (RA), TNF inhibitors have been the first biologics used for adult onset Still’s disease 106. However, driven by the deceptive impression that adult onset Still’s disease, with a predominant arthritic phenotype, is a subgroup of seronegative rheumatoid arthritis (RA), the results from uncontrolled trials involving usually small cohorts of patients were inconsistent. Since favorable outcomes have been seen in only a few patients without any specific characteristics, TNF inhibitors can only be considered third-line drugs, preferentially for patients with chronic arthritis. TNF inhibitors should probably only be prescribed for patients in the end stage of the articular type to inhibit erosion progression 107.
Figure 4. Suggested strategy for management of adult onset Still’s disease (diameter of the circles represents the challenge in clinical practice)
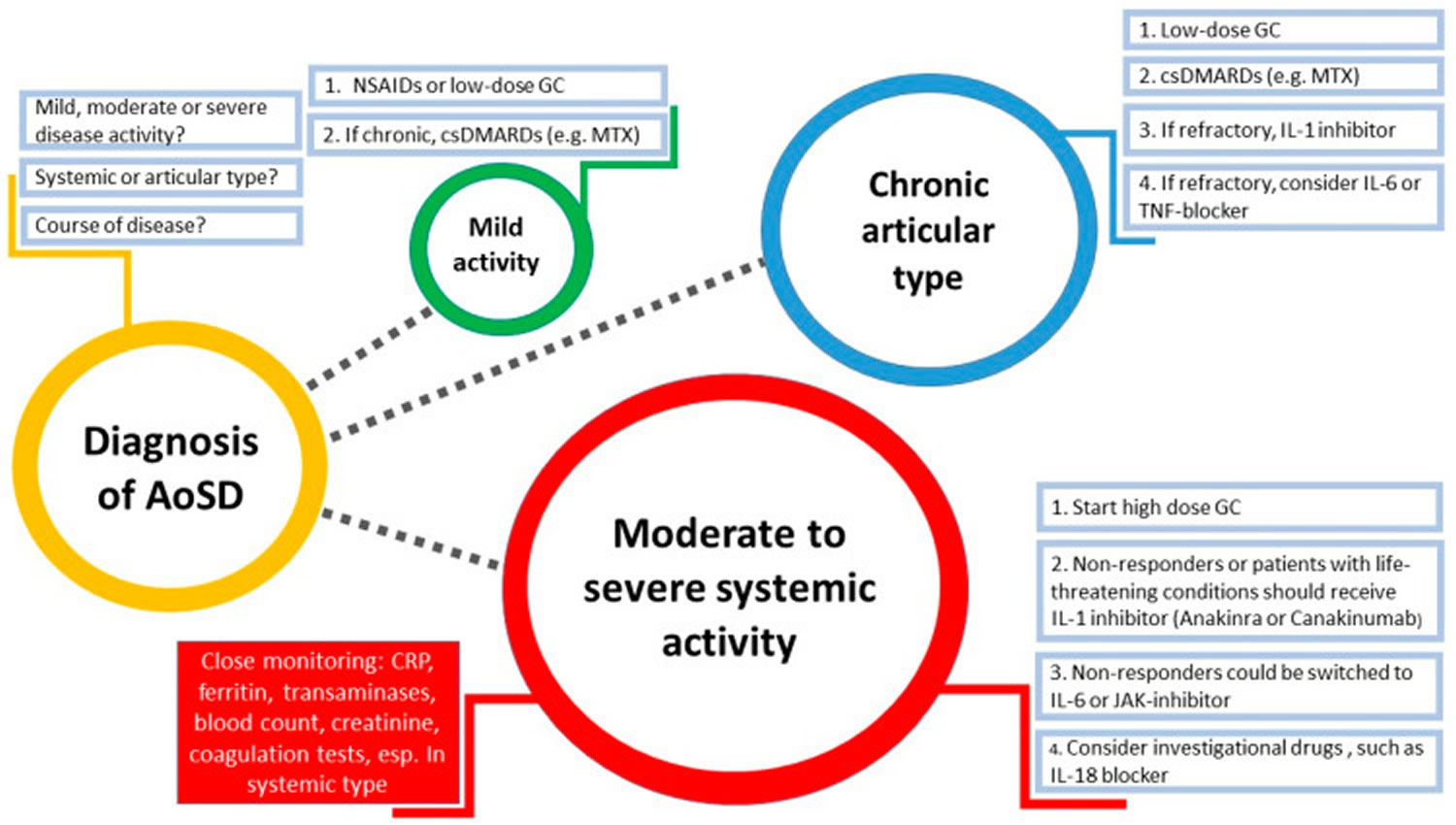
Abbreviations: AoSD = Adult-onset Still’s disease; NSAIDs = Non-steroidal anti-inflammatory drugs; MTX = Methotrexate; csDMARDs = Conventional synthetic disease-modifying antirheumatic drugs; IL = Interleukin; TNF = Tumor necrosis factor; JAK = Janus kinase; GC = Glucocorticoids; CRP = C-reactive protein.
[Source 2 ]Adult onset Still’s disease medication
Corticosteroids
Once the diagnosis is established, corticosteroids are usually required to induce symptom remission. Optimal dosing relies on medium to high doses (i.e., 0.5–1 mg/kg/day of prednisone equivalent) 4. Patients with serious visceral involvement might achieve a quick response with intravenous infusion of high-dose methylprednisolone 4. Response to corticosteroids is often dramatic—within a couple of hours or a few days 4. If you’re taking high doses of prednisone, talk to your doctor about taking more calcium and vitamin D supplements to help prevent osteoporosis.
The consensus is lacking on a therapeutic tapering scheme once clinical remission is achieved. However, owing to the potentially serious side effects of cumulative corticosteroid therapy, many others currently recommend to achieve the dose of 0.1 mg/kg/day after 6 weeks of therapy and to stop completely after three months. If this is not possible, the response should be considered inadequate, and a disease-modifying treatment, especially a targeted therapy, should be started 108.
Targeted therapies
Different targeted therapies have been used to treat refractory adult onset Still’s disease (Table 5).
Table 5. Adult onset Still’s disease targeted therapies
| Targeted therapy | International nonproprietary name and usual dose | Reported no. of patients | Preferential AoSD pattern | Follow-up (months) | Complete remission (%) | Steroid dose reduction |
|---|---|---|---|---|---|---|
| IL-1 receptor antagonists | Anakinra 100 (sometimes 200) mg/day SC | >250 | Systemic and articular | >12 | 80 | Yes |
| Anti-IL-1ß | Canakinumab 150 (sometimes 300) mg/month SC | <5 | Systemic and articular | >12 | 100 | Yes |
| IL-1 trap | Rilonacept Loading dose of 100–320 mg and then 100–320 mg/week | <5 | Systemic and articular | >12 | 100 | Yes |
| TNF blockers | Infliximab 3–7.5 mg/kg at W0–W2–W6 and then every 6–8 weeks IV Or Etanercept 50 mg/7 days SC | <100 | Articular | >12 | 0–100 | n.k. |
| IL-6 receptor antagonists | Tocilizumab 8 mg/kg/month IV Or 162 mg/week SC | <150 | Systemic and articular | >12 | 60–85 | Yes |
| Anti-IL-18 | Tadekinig alfa 80 mg/week SC Or 160 mg/week SC | <25 | NA | 4 | n.k. | n.k. |
| B cell directed | Rituximab 1 g D1–D15 every 6 months | Single case | NA | 12 | n.k. | n.k. |
| T cell directed | Abatacept 500–1000 mg/month IVa Or 125/week SC | Single case | NA | >12 | n.k. | n.k. |
Footnotes: a Abatacept dose depends on patient’s weight: <60 kg =500 mg, 60–100 kg = 750 mg, ≥100 kg = 1000 mg
Abbreviations: D = day; IV = intravenously; SC = subcutaneously; W = week; NA = not available; IL = interleukin; TNF = tumor necrosis factor; n.k. = not known
[Source 3 ]IL-1 Inhibition
The use of IL-1 inhibitors for adult onset Still’s disease contributed to the revival of considering this mode of action in rheumatology and now represents the primary choice for treating autoinflammatory diseases in general. However, it took surprisingly long until clinical development moved forward from cohort studies to randomized, placebo-controlled trials. Currently, two IL-1 blocking compounds are the focus of phase 2/phase 3 studies with different designs.
Anakinra, a recombinant IL-1 receptor antagonist, was the first biologic showing beneficial effects in treating the systemic and articular features of adult onset Still’s disease in many case series, uncontrolled trials, and different national surveys. Although the evidence for its efficacy has not been proven by controlled trials, the overall number of more than 250 published cases provides convincing results. In fact, a clear and sustained improvement was described for systemic and also arthritic symptoms in most treated cases 109. Systemic features seem to show a more rapid response, and longer exposure seems usually required for improvement of arthritis. Of major importance for long-term safety were consistent reports of a reduction or even discontinuation of glucocorticoids use as well as NSAIDs. In this context, meta-analyses from eight case series and three national surveys meeting predefined quality standards and including more than 100 anakinra-treated adult onset Still’s disease patients revealed a remission rate of approximately 80% and a reduced use of steroids in approximately 35% of patients 110. Recently, a large observational retrospective multicenter study from Italy added evidence for a strong impact on disease activity with the Pouchot score as well as clinical and serological features regardless of sex, age, disease pattern, or co-medication 111. Since September 2017, a randomized, double-blind, placebo-controlled, multicenter, phase 3 study in the United States has recruited patients for investigating two different doses of anakinra (2 mg/kg/day [max 100 mg/day] or 4 mg/kg/day [max 200 mg/day]) to evaluate its efficacy and safety in patients newly diagnosed with systemic-onset juvenile idiopathic arthritis (SJIA) and adult onset Still’s disease. The primary end point, American College of Rheumatology 30 (ACR30) response, has already been evaluated at week 2; however, the overall treatment period is short, with only 12 weeks of exposure followed by a 4-week safety follow-up 112. Of note, anakinra is the only IL-1 blocker for which we have substantial long-term results in terms of efficacy and safety in adult onset Still’s disease 113, and it has now been approved in this indication. In contrast to monogenic autoinflammatory diseases, in adult onset Still’s disease, remission can continue in some cases even after treatment is stopped. However, a relatively high withdrawal rate of 40% has been reported owing to loss of response over time and also to frequent injection site reactions to the required daily administrations.
The other approved biologic for adult onset Still’s disease is canakinumab, a fully human antibody against IL-1β 114. Furthermore, canakinumab is also approved for treating other autoinflammatory diseases including cryopyrin-associated periodic syndromes, familial Mediterranean fever, TNF receptor-associated periodic syndrome, and hyper-IgD syndrome as well as systemic-onset juvenile idiopathic arthritis (SJIA). Although the overall experience with canakinumab in adult onset Still’s disease is still limited, the reported response of systemic features and arthritis was rapid and sustained over many months to years in most patients, frequently allowing for tapering of steroids 115. Of note, canakinumab was found highly effective for patients with adult onset Still’s disease who were difficult to treat, including those showing failure of NSAIDs, glucocorticoids, and other IL-1 inhibitors. With these promising results, a placebo-controlled, randomized, multicenter, phase 2 study of canakinumab in adult onset Still’s disease is ongoing 116. The dosage of canakinumab with monthly injections of up to 4 mg/kg body weight (maximum dos of 150 mg) is based on the pharmacokinetic profile in adolescent patients with systemic-onset juvenile idiopathic arthritis (SJIA). The primary aim is to investigate the efficacy of canakinumab in patients with adult onset Still’s disease and active joint involvement in terms of the proportion of patients with a clinically significant reduction in disease activity (Disease Activity Score in 28 joints [DAS28] > 1.2) following a treatment period of 12 weeks. The overall placebo-controlled period is 24 weeks, but nonresponders can be unblinded and show treatment rescue at week 12. The core study is followed by an optional long-term extension over 2 years to provide additional safety results in the adult population.
In summary, the published data for anti-IL-1 agents in refractory adult onset Still’s disease clearly show a consistently high rate of full or partial remission with the additional achievement of lowering or stopping glucocorticoid medication 117.
IL-6 Inhibition
Inhibition of IL-6 signaling is possible by two different approaches, the direct neutralization of the cytokine or the blockade of the respective receptor. None of these mechanisms has been investigated in adult onset Still’s disease in a controlled setting of clinical trials in detail. For the two different IL-6 receptor antagonists currently available in daily practice for treating rheumatic diseases, only the tocilizumab adult onset Still’s disease case series has been published and reported at conferences 118. To summarize, the observed anti-inflammatory effects were strong, rapid, and sustained for most of the cases. Usually, the systemic features disappeared entirely, but also the arthritic manifestations improved, and the serologic activity decreased 119. However, because of limited data, the likelihood of response cannot be estimated as clearly as for IL-1 inhibitors and seems to be between 60% to 80% in terms of full remission. A recently published meta-analysis of ten original studies (147 individuals) on the efficacy of tocilizumab and adult onset Still’s disease revealed overall high partial and complete remission rates of 85–77%, respectively. Tocilizumab prevented new flares, was well tolerated, and could significantly reduce the need for corticosteroids 120.
Whether other IL-6 inhibitors have similar effects remains unknown. As shown for rheumatoid arthritis (RA) treatment, with adult onset Still’s disease, differences could exist with respect to safety, especially in comparison to the direct cytokine inhibitors. Also, the advantages or disadvantages that distinguish IL-6 and IL-1 inhibition are unclear. Although clearly IL-6 as well as IL-1 inhibitors have high response rates and represent alternative approaches in adult onset Still’s disease, we have no way to predict the individual effects and define the ideal treatment algorithm.
IL-18 Inhibition
A novel compound is Tadekinig alfa, a recombinant human IL-18 binding protein 121. This drug was tested in healthy volunteers and patients with psoriasis and rheumatoid arthritis (RA) in phase 1 studies and demonstrated a good safety and tolerability profile with only mild adverse events at the injection site. Because of the postulated role of IL-18 in the pathogenesis of adult onset Still’s disease, it was a logical step to investigate the effects of Tadekinig alfa in this condition. A first open-label, dose-finding phase 2 study involving multiple centers in Europe was designed to capture safety information as the primary outcome. Two doses (80 and 160 mg) were administered during 12 weeks, and patients were followed up for 4 more weeks. As a secondary outcome measurement, the efficacious dose at 3 weeks was defined as normalization of body temperature and decrease of CRP level by 50% or more of baseline values.
Although limited by the low patient number and short observation period in the study, the safety profile of Tadekinig alfa was overall acceptable, with injection site reactions and infections being the most frequent adverse events. In terms of efficacy, reduced level of CRP as well as other inflammatory markers (including IL-18, ferritin) was detected in some patients. Furthermore, an improvement in skin rash and a slight but significant reduction in the prednisolone doses were reported. In summary, inhibition of IL-18 by Tadekinig alfa could be a promising new approach in adult Still’s disease that needs to be investigated in a controlled setting.
Life-threatening complications treatment and management
Reactive Hemophagocytic Lymphohistiocytosis (RHL)
Management should include, firstly, symptomatic care in a medicine department or intensive care unit (ICU); secondly, active and rapid search for an infection; and, thirdly, introduction of high-dose steroids. Reactive hemophagocytic lymphohistiocytosis (RHL) has described under IL-1 therapy, it appears prudent to introduce this treatment only after reactive hemophagocytic lymphohistiocytosis (RHL) control by a few days of high-dose steroids.
Coagulation Disorders
Disseminated intravascular coagulation (DIC) is a medical emergency whose care combines:
- Supportive measures, which could include platelet transfusion, coagulation factors (or fresh frozen plasma), and fibrinogen (in cryoprecipitate) infusion to control severe bleeding or sometimes heparin if no active bleeding is present 122.
- Specific anti-inflammatory agents often decided in multidisciplinary rounds, with mainly high-dose corticosteroids, either intravenous (methylprednisolone 15 mg/kg/day) or oral (prednisone 1 mg/kg/day) 28. To limit steroid exposure, the addition of other immunomodulatory agents, such as IL-1 or IL-6 blockers or cyclosporine, is recommended 110.
Thrombotic microangiopathy (TMA) also called thrombocytic thrombocytopenic purpura (TTP) or Moschcowitz syndrome treatment is complex and mandatorily includes 34:
- Supportive care in an ICU, with plasma exchange being central 123
- High-dose steroids intravenously and then orally
Other immunomodulatory agents proposed for refractory cases include anakinra 35, tocilizumab 124, intravenous immunoglobulins, cyclophosphamide, azathioprine, cyclosporine A, and rituximab 35. Despite this treatment, mortality associated with TMA remains high, up to 20% 34.
Cardiac and Pulmonary Involvements
Their treatment combines supportive care, high-dose steroids, and frequently IL-1 or IL-6 inhibitors.
Regarding pulmonary arterial hypertension (PAH), early diagnosis is central for the prognosis of this complication, with mortality remaining as high as 40% 34. The diagnosis includes 125:
- Hospitalization in an ICU and rapid referral to a specific reference center for multidisciplinary round discussions.
- Vasodilating therapies frequently combining endothelin receptor antagonists (bosentan, ambrisentan), phosphodiesterase-5 inhibitors and other guanylate cyclase stimulators (sildenafil or others), and prostacyclin analogues.
- High-dose steroids, initially intravenously and then orally.
- Rapid introduction of biologic immunomodulatory agents targeting IL-1 or IL-6 or other immunosuppressive agents, such as methotrexate, azathioprine, cyclophosphamide, cyclosporine, or rituximab. However, these treatments must be carefully monitored because a few cases of pulmonary arterial hypertension (PAH) worsening have been described after their introduction.
Pulmonary arterial hypertension (PAH) deserves to be well known by physicians caring for adult onset Still’s disease patients because it was not described in most of the initial adult onset Still’s disease case series for several reasons: now better diagnostic tools for pulmonary arterial hypertension (PAH); differential efficacy of large-spectrum immunomodulatory agents, such as steroids, as compared with more targeted therapies, such as IL-1 or IL-6 blockers; and exposure to microorganisms as an additional target whose ecology may have evolved in recent years.
Amyloidosis
Even administered late, adult onset Still’s disease therapy, such as steroids and above all IL-1 and IL-6 blockers, may at least in part improve the clinical and biological symptoms.
Adult Still’s disease natural treatment
There is a case report of adult-onset Still’s disease being successfully treated with Chinese herbal medicine 126. In this case, we report a 28-year-old male badminton coach presenting with a 15-day history of fever and skin rash, accompanied by sore throat, fatigue, myalgia and chills. Additionally, hepatosplenomegaly, multiple lymphadenopathies, aminotransferase abnormality, and elevated inflammatory factor levels were observed during hospitalization. Not benefitting from antibiotic therapy, the patient was finally diagnosed with adult-onset Still’s disease, after a careful examination, then showed rapid remission after a six-week treatment with Chinese herbal medicine granules based on Xiaochaihu Decoction and Yinqiao Powder 126. After stopping the treatment, there was no relapse within a 15-month follow-up period. This is the first well-documented case of adult Still’s disease being successfully treated natural treatment. The case report suggests that Chinese herbal medicine granules based on Xiaochaihu Decoction and Yinqiao Powder may be a reliable choice for complementary and alternative therapy for adult-onset Still’s disease, but confirming the utility of Chinese herbal medicine for adult onset Still’s disease requires further support from prospective studies.
Arthritis diet
While there’s no miracle diet for arthritis, many foods can help fight inflammation and improve joint pain and other symptoms. For starters, a diet rich in whole foods, including fruits, vegetables, fish, nuts and beans, but low processed foods and saturated fat, is not only great for overall health, but can also help manage disease activity.
A traditional Mediterranean dietary pattern, which typically has a high ratio of monounsaturated (MUFA) and Omega-3 to Omega-6 polyunsaturated fatty acid (PUFAs) and supplies an abundance of fruits, vegetables, legumes, and grains, has been shown to have anti-inflammatory effects when compared with typical North American and Northern European dietary patterns in most observational and interventional studies 127.
The Mediterranean Diet is characterized by 128:
- An abundance of plant food (fruit, vegetables, breads, cereals, potatoes, beans, nuts, and seeds);
- Minimally processed, seasonally fresh, locally grown foods;
- Desserts comprised typically of fresh fruit daily and occasional sweets containing refined sugars or honey;
- Olive oil (high in polyunsaturated fat) as the principal source of fat;
- Daily dairy products (mainly cheese and yogurt) in low to moderate amounts;
- Fish and poultry in low to moderate amounts;
- Up to four eggs weekly;
- Red meat rarely; and
- Wine in low to moderate amounts with meals.
Mediterranean Diet benefits
Studies confirm that eating foods commonly part of the Mediterranean diet can do the following:
- Lower blood pressure
- Protect against chronic conditions, ranging from cancer to stroke
- Help arthritis by curbing inflammation
- Benefit your joints as well as your heart
- Lead to weight loss, which can lessen joint pain
Below are key foods from the Mediterranean diet and why they’re so good for joint health.
Fish
- How much: Health authorities like the American Heart Association and the Academy of Nutrition and Dietetics recommend three to four ounces of fish, twice a week. Arthritis experts claim more is better.
- Why: Some types of fish are good sources of inflammation-fighting omega-3 fatty acids. One study found those who had the highest consumption of omega-3s had lower levels of two inflammatory proteins: C-reactive protein (CRP) and interleukin-6. More recently, researchers have shown that taking fish oil supplements helps reduce joint swelling and pain, duration of morning stiffness and disease activity among people who have rheumatoid arthritis (RA).
- Best sources: Salmon, tuna, sardines, herring, anchovies, scallops and other cold-water fish. Hate fish? Take a supplement. Studies show that taking 600 to 1,000 mg of fish oil daily eases joint stiffness, tenderness, pain and swelling.
Nuts and seeds
- How much: Eat 1.5 ounces of nuts daily (one ounce is about a handful).
- Why: Multiple studies confirm the role of nuts in an anti-inflammatory diet. One study found that over a 15-year period, men and women who consumed the most nuts had a 51% lower risk of dying from an inflammatory disease (like rheumatoid arthritis) compared with those who ate the fewest nuts. Another study found that subjects with lower levels of vitamin B6 – found in most nuts – had higher levels of inflammatory markers. Nuts are also jam-packed with inflammation-fighting monounsaturated fat (MUFA). And though they’re relatively high in fat and calories, studies show snacking on nuts promotes weight loss because their protein, fiber and monounsaturated fats are satiating. Just keep in mind that more is not always better.
- Best sources: Walnuts, pine nuts, pistachios and almonds.
Fruits and vegetables
- How much: Aim for nine or more servings daily (one serving equals one cup of most veggies or fruit or two cups of raw leafy greens).
- Why: Fruits and vegetables are loaded with antioxidants. These potent chemicals act as the body’s natural defense system, helping to neutralize unstable molecules called free radicals that can damage cells. Research has shown that anthocyanins found in cherries and other red and purple fruits like strawberries, raspberries, blueberries and blackberries have an anti-inflammatory effect. Citrus fruits – like oranges, grapefruits and limes – are rich in vitamin C. Research shows getting the right amount of that vitamin aids in preventing inflammatory arthritis and maintaining healthy joints. Other research suggests eating vitamin K-rich veggies like broccoli, spinach, lettuce, kale and cabbage dramatically reduces inflammatory markers in the blood.
- Best sources: Colorful fruits and veggies – the darker or more brilliant the color, the more antioxidants it has. Good ones include blueberries, cherries, spinach, kale and broccoli.
Olive oil
- How much: Two to three tablespoons daily.
- Why: Olive oil is loaded with heart-healthy fats, as well as oleocanthal, which has properties similar to nonsteroidal anti-inflammatory drugs (NSAIDs). Oleocanthal inhibits activity of cyclooxygenase (COX) enzymes, with a pharmacological action similar to ibuprofen. Inhibiting these enzymes dampens the body’s inflammatory processes and reduces pain sensitivity.
- Best sources: Extra virgin olive oil goes through less refining and processing, so it retains more nutrients than standard varieties. And it’s not the only oil with health benefits. Avocado and safflower oils have shown cholesterol-lowering properties, while walnut oil has 10 times the omega-3s that olive oil has.
Beans
- How much: About one cup, twice a week (or more).
- Why: Beans are loaded with fiber and phytonutrients, which help lower CRP, an indi¬cator of inflammation found in the blood. At high levels, CRP could indicate anything from an infection to rheumatoid arthritis (RA). In a study scientists analyzed the nutrient content of 10 common bean varieties and identified a host of antioxidant and anti-inflammatory compounds. Beans are also an excellent and inexpensive source of protein and have about 15 grams per cup, which is important for muscle health.
- Best sources: Small red beans, red kidney beans and pinto beans rank among the U.S. Department of Agriculture’s top four antioxidant-containing foods (wild blueberries take the number 2 spot).
Whole grains
- How much: Eat a total of six ounces of grains per day; at least three of which should come from whole grains. One ounce of whole grain would be equal to ½ cup cooked brown rice or one slice of whole-wheat bread.
- Why: Whole grains contain plenty of filling fiber – which can help you maintain a healthy weight. Some studies have also shown that fiber and fiber-rich foods can lower blood levels of CRP, an inflammatory marker.
- Best sources: Eat foods made with the entire grain kernel, like whole-wheat flour, oatmeal, bulgur, brown rice and quinoa. Some people may need to be careful about which whole grains they eat. Gluten – a protein found in wheat and other grains – has been linked to inflammation for people with celiac disease or gluten sensitivity.
Nightshade vegetables
- Why: Nightshade vegetables, including eggplant, tomatoes, red bell peppers and potatoes, are disease-fighting powerhouses that boast maximum nutrition for minimal calories.
- Why not: They also contain solanine, a chemical that has been branded the culprit in arthritis pain. There’s no scientific evidence to suggest that nightshades trigger arthritis flares.
- Test it: Some experts believe these vegetables contain a potent nutrient mix that helps inhibit arthritis pain. However, many people do report symptom relief when they avoid nightshade vegetables. So, if you notice that your arthritis pain flares after eating them, consider eliminating all nightshade vegetables from your diet for a few weeks to see if it makes a difference. Then slowly add them back into your diet to see if symptoms worsen or stay the same.
Anti inflammatory diet research
Scientific evidence shows that eating mostly plant-based foods — variety of vegetables, fruits, whole grains, beans and other plant foods — plays a big role in preventing cancer and contributing to a healthier life. That’s because plant-based foods are high in the types of fiber, nutrients, minerals, vitamins and phytochemicals (natural substances) that may help to prevent cancer. Plus, plant-foods can help you manage your weight, and give you the energy you need to enjoy physical activity. A healthy diet emphasizes foods such as a variety of vegetables, fruits, whole grains, and beans that can reduce your risk for cancer and other chronic diseases. These foods are rich in fiber, vitamins, and other natural substances called phytochemicals that help keep you in good health, and protect against cancer. They are also naturally low in calories. So make whole grains, vegetables, fruits and pulses (legumes) such as beans and lentils a major part of your normal diet. Get started by covering at least two-thirds (2/3) of your plate with plant foods such as whole grains, vegetables, fruit and beans. The remaining third (1/3) of your plate may be filled with animal-based protein rich foods such as seafood, poultry and dairy foods and occasionally with lean red meat. The other most important consideration for any anti-inflammatory diet is calorie restriction 129. Any reduction of excess calorie intake will lead to a decrease in systemic oxidative stress. Calorie restriction has been the most successful therapeutic intervention to improve healthspan (defined as longevity minus years of disability) in virtually every species studied 130. Significant metabolic benefits have been achieved by calorie restriction in healthy overweight and normal-weight individuals who participated in the various CALERIE (Comprehensive Assessment of the Long-Term Effects of Reducing Intake of Energy) studies 131, 132.
Whole grains are healthier than refined grains because the process of refining carbohydrates results in the elimination of much of the fiber, vitamins, minerals, phytonutrients, and essential fatty acids 133. Furthermore, refined starches and sugars can rapidly alter blood glucose and insulin levels 133 and postprandial hyperglycemia can increase production of free radicals as well as proinflammatory cytokines 134.
There is consistent evidence that consumption of whole grains is protective against incident type 2 diabetes and cardiovascular disease 135, 136. However, it is unclear how these protective effects are mediated. Dietary fiber intake may reduce the risk of these diseases by mediating the pro-inflammatory process 137, 138. Two mechanistic hypotheses have emerged. First, dietary fiber may decrease oxidation of glucose and lipids, while maintaining a healthy intestinal environment. Second, dietary fiber may prevent inflammation by altering adipocytokines in adipose tissue and increasing enterohepatic circulation of lipids and lipophilic compounds 139. The link between dietary fiber intake and reduced high sensitivity serum C-reactive protein (hs-CRP) has been observed in several recent studies, including two analyses using cross-sectional data from NHANES 1999–2000 140, 141, an analysis using a longitudinal cohort of 524 healthy adults 142 and a small clinical trial 143.
Various dietary components including long chain omega-3 fatty acids, antioxidant vitamins, plant flavonoids, prebiotics and probiotics have the potential to modulate predisposition to chronic inflammatory conditions. These components act through a variety of mechanisms including decreasing inflammatory mediator production through effects on cell signaling and gene expression (omega-3 fatty acids, vitamin E, plant flavonoids), reducing the production of damaging oxidants (vitamin E and other antioxidants), and promoting gut barrier function and anti-inflammatory responses (prebiotics and probiotics) 144. In a large Danish study 145 involving over 7000 men and women who were followed over 15 years (1982-1998). What that study found was that in both men and women who frequently consume wholemeal bread, vegetables, fruits, and fish, was associated with a better overall survival rate as well as better cardiovascular survival 145.
A low-fat diet (≤30% of total calories) is still considered by many physicians to be a healthy choice for both primary and secondary prevention of cardiovascular disease (coronary heart disease) 146. An unintended consequence of emphasizing low-fat diets may have been to promote unrestricted carbohydrate intake, which reduces high-density lipoprotein cholesterol (HDL “good” cholesterol) and raises triglyceride levels, exacerbating the metabolic manifestations of the insulin resistance syndrome, also known as the metabolic syndrome 147.
Arachidonic acid (AA) derived (omega-6) eicosanoids (primarily from refined vegetable oils such as corn, sunflower, and safflower) increase the production of proinflammatory cytokines IL-1, TNF-α, and IL-6, operating as precursors of the proinflammatory eicosanoids of the prostaglandin (PG)2-series 148. In contrast, the omega-3 (n-3) polyunsaturated fatty acids (PUFAs), found in fish, fish oil, walnuts, wheat germ, and some dietary supplements such as flax seed products can curb the production of Arachidonic acid (AA)-derived eicosanoids 149. The Omega-6 and Omega-3 polyunsaturated fatty acids (PUFAs) compete for the same metabolic pathways, and thus their balance is important 150. Accordingly, it is not surprising that both higher levels of Omega-3 polyunsaturated fatty acids (PUFAs) as well as lower Omega-6:Omega-3 ratios are associated with lower proinflammatory cytokine production 151.
Furthermore, in another study a healthy dietary pattern rich in fruit, vegetables and olive oil, such as the Mediterranean diet, is associated with lower levels of inflammatory markers, perhaps because of the anti-inflammatory properties of antioxidants 133.
The antioxidant properties of vegetables and fruits are thought to be one of the fundamental mechanisms underlying their anti-inflammatory dietary contributions 133. Oxidants such as superoxide radicals or hydrogen peroxide that are produced during the metabolism of food can activate the NF-κB pathway, promoting inflammation 152. Higher fruit and vegetable intakes are associated with lower oxidative stress and inflammation 152. In fact, some evidence suggests that the addition of antioxidants or vegetables may limit or even reverse proinflammatory responses to meals high in saturated fat 153.
To overcome silent inflammation requires an anti-inflammatory diet (with omega-3s and polyphenols). The most important aspect of such an anti-inflammatory diet is the stabilization of insulin and reduced intake of omega-6 fatty acids. The ultimate treatment lies in reestablishing hormonal and genetic balance to generate satiety instead of constant hunger. Anti-inflammatory nutrition with caloric restriction, should be considered as a form of gene silencing technology, in particular the silencing of the genes involved in the generation of silent inflammation. To this anti-inflammatory diet foundation supplemental omega-3 fatty acids at the level of 2–3 g of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) per day should be added. Finally, a diet rich in colorful, non-starchy vegetables would contribute adequate amounts of polyphenols to help not only to inhibit nuclear factor (NF)-κB (primary molecular target of inflammation) but also activate AMP kinase 154.
Exercises for arthritis
Although you might not want to work out if your joints ache, exercise is recommended for all types of arthritis. Exercise can help you maintain your range of motion and relieve pain and stiffness.
Ask your doctor for specific suggestions, including how long and hard you should exercise. Your doctor may be able to refer you to exercise programs in your area. If she’s unable to do so, seek the help of a physical therapist or certified professional trainer who has extensive experience working with people with arthritis.
Certain forms of exercise are particularly effective for individuals with arthritis. Here are five you may want to consider:
- Walking. Walking is safe for almost everyone, even those with severe arthritis. It only requires is a pair of comfortable, supportive shoes; and it involves zero training. If walking a hilly route is too strenuous stick with level ground like an indoor mall or gym with a flat track so you can walk safely and comfortably in any weather. To get the most from your walking workout, consider the Arthritis Foundation’s Walk With Ease program (https://www.arthritis.org/health-wellness/healthy-living/physical-activity/walking/walk-with-ease). It helps you develop a walking plan suited for your needs, helps you stay motivated and teaches you to exercise safely. You can do it in a group with a trained instructor, or on your own with guidance from a book that provides instructions, checklists, progress charts and other helpful information. Because it’s an evidence-based program, it’s safe and effective for people with arthritis.
- Water workouts. Swimming or water aerobics are especially great for people who are heavier or who have advanced arthritis. If you can find a heated pool, all the better – warm water almost instantly relieves painful joints.
- Stationary or recumbent cycling. Both recumbent and stationary bikes allow you to get your heart rate up, but they put very little pressure on the hip and knee joints. A recumbent bike is a safer choice if your balance is doubtful or if you’re overweight, new to exercise or exercising post-knee surgery. An upright bike allows you to spin faster and is best reserved for an injury-free, experienced exerciser.
- Yoga and tai chi. Tai chi and yoga improve flexibility and balance – two areas which individuals with arthritis often struggle with – and both are gentle on joints, too. Check your local community and fitness centers for yoga and tai chi classes, or you can exercise on your own time with guidance from a Tai Chi available from Youtube.
- Resistance training. Resistance training is an absolute must for people with arthritis. Contrary to popular belief, weights are an excellent choice: the key to using them safely is to have proper form and to lift the correct amount of weight for your strength level. If you’re not sure, ask a physical therapist or personal trainer to teach you. Rubber resistance bands and strengthening equipment at the gym are good ways to build lean muscle mass. Strong muscles absorb the shock that would otherwise affect your joints. It’s like the difference between walking barefoot on a cold floor and wearing warm, padded slippers.
Adult onset Still’s disease prognosis
Adult onset Still’s disease prognosis can be quite diverse and currently is impossible to predict 3. The prognosis of adult onset Still’s disease is dominated by potential life-threatening events as well as progressive functional impairment. In many people with adult onset Still’s disease, symptoms may come back several times over the next few years. Symptoms can continue for a long time (chronic) in about one third of people with adult Still disease.
Several disease patterns have been observed in adult Still’s disease patient cohort studies. For approximately 19–44% of affected patients, adult onset Still’s disease has a monocyclic course without relapses 2. A polycyclic course is identified in 10–41% of affected patients and is characterized by unpredictable periods of exacerbation after a few months or years. Approximately 35–57% of affected patients show a chronic progressive course, which is the most frequent one, which is characterized by steady progression, continuous inflammation and often erosive joint involvement 155.
However, recent studies have introduced a new approach by grouping patients with adult onset Still’s disease into only two phenotypes: one with predominantly systemic features and one with a chronic articular disease course. Treatment of the systemic form is different from the treatment used for adults with progressive joint involvement, due to a higher inflammatory status and possible multi-organ damage with haematological complications. The non-systemic subgroup, on the other hand, may begin with systemic symptoms and evolve to a disease resembling rheumatoid arthritis at the end stage. This phenotypic dichotomy may also simplify the design of future clinical trials 156.
Predictive factors for the systemic subset of adult onset Still’s disease include high fever (>39 °C) and high levels of liver enzymes or CRP, while female sex, polyarthritis at disease onset and steroid dependence are associated with the chronic articular subgroup 157. This simplified theory of dichotomous adult Still’s disease courses is supported, at least partially, by studies on cytokine profiles and responses to biologic treatments 158.
Self-Limited, Monocyclic Evolution
Self-Limited Monocyclic Evolution combines systemic manifestations and joint involvement: the initial flare evolves over a few weeks; remission is achieved with steroids or other immunomodulatory agents after a few days or weeks; and treatments can be progressively tapered and then stopped without relapse after a few months. The pattern may represent 19–44% of cases 3.
Recurrent or Polycyclic Evolution
Recurrent or Polycyclic Evolution is characterized by adult onset Still’s disease relapses after a few months or years, under immunomodulatory treatment or after its discontinuation. In this pattern, one classical presentation is a first flare during childhood-diagnosed systemic-onset juvenile idiopathic arthritis (SJIA), followed by sustained drug-free remission for several years, and then a relapse in adulthood. In most of these cases, recurrences combine systemic and joint involvement. The pattern represents 10–41% of cases 3.
Chronic and Progressive Evolution
In this form, a continuous inflammation is responsible for chronic and frequently erosive joint involvement with regular systemic flares. This pattern was the most frequent, estimated at 35–67% of cases, in old series 23. Although no new estimates are available, this pattern should be less likely with the most recent targeted therapies.
Life prognosis
The life prognosis is dominated by the severity of the following visceral involvements, in isolation or combination: hematological complications, such as DIC, TMA, or severe RHL; fulminant hepatitis with rapid liver failure; and acute respiratory distress syndrome, extensive myocarditis, or multiorgan failure 28. Importantly, exacerbation of these serious symptoms has been reported within the first days of treatment initiation, especially of biological therapies. This necessitates a close and very careful monitoring of such patients at the start of treatment. In addition to organ involvements, the development of secondary amyloidosis of the amyloidosis A type remains possible in long-standing refractory chronic articular adult onset Still’s disease 28.
Functional prognosis
Functional prognosis depends mainly on erosion and joint destruction in chronic articular adult onset Still’s disease, which affects approximately one-third of patients 21. No clear predictor of erosive joint disease has been identified, but, as joint involvement is often limited, functional prognosis in adult onset Still’s disease seems to be less severe than in RA or other inflammatory arthritides 159.
Corticosteroid side effects
Corticosteroid side effects are also common because steroids are usually prescribed in high doses and the long term, which might contribute to the overall prognosis in adult onset Still’s disease.
References- Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still’s disease. J Autoimmun. 2018 Sep;93:24-36. doi: 10.1016/j.jaut.2018.07.018
- Tomaras, S., Goetzke, C. C., Kallinich, T., & Feist, E. (2021). Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. Journal of clinical medicine, 10(4), 733. https://doi.org/10.3390/jcm10040733
- Mitrovic, S., Feist, E., & Fautrel, B. (2019). Adult-Onset Still’s Disease. Periodic and Non-Periodic Fevers, 93–132. https://doi.org/10.1007/978-3-030-19055-2_6
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22(5):773–792. doi: 10.1016/j.berh.2008.08.006
- Adult Still’s Disease. https://www.arthritis.org/diseases/adult-stills-disease
- Magadur-Joly G, Billaud E, Barrier JH, Pennec YL, Masson C, Renou P, et al. Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54(7):587–590. doi: 10.1136/ard.54.7.587
- Wakai K, Ohta A, Tamakoshi A, Ohno Y, Kawamura T, Aoki R, et al. Estimated prevalence and incidence of adult Still’s disease: findings by a nationwide epidemiological survey in Japan. J Epidemiol. 1997;7(4):221–225. doi: 10.2188/jea.7.221
- Bywaters E. G. (1971). Still’s disease in the adult. Annals of the rheumatic diseases, 30(2), 121–133. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1005739/pdf/annrheumd00002-0002.pdf
- Lebrun D, Mestrallet S, Dehoux M, Golmard JL, Granger B, Georgin-Lavialle S, et al. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin Arthritis Rheum. 2018;47(4):578–585. doi: 10.1016/j.semarthrit.2017.07.005
- Fautrel B, Zing E, Golmard J-L, Le Moel G, Bissery A, Rioux C, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore) 2002;81(3):194–200. doi: 10.1097/00005792-200205000-00003
- Takahashi H, Ohara M, Imai K. Gastrointestinal manifestation. Jpn J Clin Immunol. 2004;27:1145-1155.
- Hori, H, Yabe, H, Fukuchi, T, Sugawara, H. A woman with adult-onset Still’s disease and acute intestinal pseudo-obstruction. Clin Case Rep. 2021; 9: 153– 157. https://doi.org/10.1002/ccr3.3488
- El Younsi S, Perlemuter G, Clerc D, Buffet C, Pelletier G. Still’s disease and intestinal pseudo-obstruction. Gastroenterol Clin Biol. 2014;28:309-310.
- Shinohara T, Hidaka T, Matsuki Y, Suzuki K, Ohsuzu F. Calcinosis cutis and intestinal pseudoobstruction in a patient with adult onset Still’s disease associated with recurrent relapses of disordered coagulopathy. Intern Med. 1999;38:516-520.
- Ravelli A, Martini A. Macrophage activation syndrome. In: Lehman TH, Cimaz R, editors. Pediatric rheumatology. Amsterdam: Elsevier; 2008. p. 55–63.
- Parodi A, Davi S, Pringe AB, Pistorio A, Ruperto N, Magni-Manzoni S, et al, for the Lupus Working Group of the Paediatric Rheumatology European Society. Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum 2009; 60:3388–99.
- Kumar S, Vaidyanathan B, Gayathri S, Rajam L. Systemic onset juvenile idiopathic arthritis with macrophage activation syndrome misdiagnosed as Kawasaki disease: case report and literature review. Rheumatol Int 2013;33:1065–9.
- Rossi-Semerano L, Hermeziu B, Fabre M, Kone-Paut I. Macrophage activation syndrome revealing familial Mediterranean fever. Arthritis Care Res (Hoboken) 2011;63:780–3.
- Ravelli A, Minoia F, Davì S on behalf of the Paediatric Rheumatology International Trials Organisation, the Childhood Arthritis and Rheumatology Research Alliance, the Pediatric Rheumatology Collaborative Study Group, and the Histiocyte Society, et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis. Annals of the Rheumatic Diseases 2016;75:481-489. https://www.rheumatology.org/Portals/0/Files/A-and-R-Classification-Criteria-Macrophage-Activation-Syndrome-2016.pdf
- Crispín JC, Martínez-Baños D, Alcocer-Varela J. Adult-onset Still disease as the cause of fever of unknown origin. Medicine (Baltimore) 2005;84(6):331–337. doi: 10.1097/01.md.0000188009.47085.76
- Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014;13(7):708–722. doi: 10.1016/j.autrev.2014.01.058
- Zuelgaray E, Battistella M, Sallé de Chou C, Vignon-Pennamen M-D, Rybojad M, Petit A, et al. Increased severity and epidermal alterations in persistent versus evanescent skin lesions in adult onset Still’s disease. J Am Acad Dermatol. 2018;79(5):969–971. doi: 10.1016/j.jaad.2018.05.020
- Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore) 1991;70(2):118–136. doi: 10.1097/00005792-199103000-00004
- Park J-H, Bae JH, Choi Y-S, Lee H-S, Jun J-B, Jung S, et al. Adult-onset Still’s disease with disseminated intravascular coagulation and multiple organ dysfunctions dramatically treated with cyclosporine A. J Korean Med Sci. 2004;19(1):137–141. doi: 10.3346/jkms.2004.19.1.137
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–2620. doi: 10.1002/art.38690
- Drepper M, Rubbia-Brandt L, Spahr L. Tocilizumab-induced acute liver injury in adult onset Still’s disease. Case Rep Hepatol. 2013;2013:964828
- Bae, C. B., Jung, J. Y., Kim, H. A., & Suh, C. H. (2015). Reactive hemophagocytic syndrome in adult-onset Still disease: clinical features, predictive factors, and prognosis in 21 patients. Medicine, 94(4), e451. https://doi.org/10.1097/MD.0000000000000451
- Mitrovic S, Fautrel B. Complications of adult-onset Still’s disease and their management. Expert Rev Clin Immunol. 2018;14(5):351–365. doi: 10.1080/1744666X.2018.1465821
- Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol 2012; 31:1223–1230.
- Hot A, Toh ML, Cope B, et al. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical feature and long-term outcome: a case-conrol study of 8 patients. Medicine (Baltimore) 2010; 89:37–46.
- Burgi U, Mendez A, Hasler P, et al. Hemophagocytic syndrome in adult-onset Still’s disease (AOSD): a must for biologics? Case report and brief review of the literature. Rheumatol Int 2012; 32:3269–3272.
- Tristano AG. Macrophage activation syndrome: a frequent but under-diagnosed complication associated with rheumatic diseases. Med Sci Monit 2008; 14:RA27–36.
- Gerfaud-Valentin M, Maucort-Boulch D, Hot A, Iwaz J, Ninet J, Durieu I, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore) 2014;93(2):91–99. doi: 10.1097/MD.000000000000002
- Efthimiou P, Kadavath S, Mehta B. Life-threatening complications of adult-onset Still’s disease. Clin Rheumatol. 2014;33(3):305–314. doi: 10.1007/s10067-014-2487-4
- El Karoui K, Karras A, Lebrun G, Charles P, Arlet JB, Jacquot C, et al. Thrombotic microangiopathy and purtscher-like retinopathy associated with adult-onset Still’s disease: a role for glomerular vascular endothelial growth factor? Arthritis Rheum. 2009;61(11):1609–1613. doi: 10.1002/art.24826
- Hirata S, Okamoto H, Ohta S, Kobashigawa T, Uesato M, Kawaguchi Y, et al. Deficient activity of von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura in the setting of adult-onset Still’s disease. Rheumatology (Oxford) 2006;45(8):1046–1047. doi: 10.1093/rheumatology/kel176
- Reginato AJ, Schumacher HR, Baker DG, O’Connor CR, Ferreiros J. Adult onset Still’s disease: experience in 23 patients and literature review with emphasis on organ failure. Semin Arthritis Rheum. 1987;17(1):39–57. doi: 10.1016/0049-0172(87)90015-1
- Uppal SS, Al-Mutairi M, Hayat S, Abraham M, Malaviya A. Ten years of clinical experience with adult onset Still’s disease: is the outcome improving? Clin Rheumatol. 2007;26(7):1055–1060. doi: 10.1007/s10067-006-0440-x
- Ben Ghorbel I, Khanfir M, Houman MH. Amyloidosis in adult onset Still’s disease. Rev Med Interne. 2004;25(9):675–677. doi: 10.1016/j.revmed.2004.04.025
- Hot A., Toh M.L., Coppéré B., Perard L., Madoux M.H., Mausservey C., Desmurs-Clavel H., Ffrench M., Ninet J. Reactive hemophagocytic syndrome in adult-onset Still disease: Clinical features and long-term outcome: A case-control study of 8 patients. Medicine. 2010;89:37–46. doi: 10.1097/MD.0b013e3181caf100
- Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004 Aug 1. 104(3):735-43.
- Mehta P., Cron R.Q., Hartwell J., Manson J.J., Tattersall R.S. Silencing the cytokine storm: The use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020;2:e358–e367. doi: 10.1016/S2665-9913(20)30096-5
- Macrophage Activation Syndrome Clinical Presentation. https://emedicine.medscape.com/article/1380671-clinical
- Minoia F, Davì S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis A multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014 Jul 30.
- Fardet L., Galicier L., Lambotte O., Marzac C., Aumont C., Chahwan D., Coppo P., Hejblum G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613–2620. doi: 10.1002/art.38690
- Lin T.F., Ferlic-Stark L.L., Allen C.E., Kozinetz C.A., McClain K.L. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatric Blood Cancer. 2011;56:154–155. doi: 10.1002/pbc.22774
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, Coppo P, Hejblum G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014 Sep;66(9):2613-20. doi: 10.1002/art.38690 http://saintantoine.aphp.fr/score/
- Bhargava J, Panginikkod S. Still Disease. [Updated 2021 Jul 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538345
- Ohta A, Yamaguchi M, Tsunematsu T, Kasukawa R, Mizushima H, Kashiwagi H, Kashiwazaki S, Tanimoto K, Matsumoto Y, Akizuki M, et al. Adult Still’s disease: a multicenter survey of Japanese patients. J Rheumatol. 1990 Aug;17(8):1058-63.
- Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, Hill RO, Gutkowski A, Harth M, Myhal D, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991 Mar;70(2):118-36.
- McGonagle, D., & McDermott, M. F. (2006). A proposed classification of the immunological diseases. PLoS medicine, 3(8), e297. https://doi.org/10.1371/journal.pmed.0030297
- McGonagle D, Savic S, McDermott MF. The NLR network and the immunological disease continuum of adaptive and innate immune-mediated inflammation against self. Semin Immunopathol. 2007 Sep;29(3):303-13. doi: 10.1007/s00281-007-0084-1
- Peckham D, Scambler T, Savic S, McDermott MF. The burgeoning field of innate immune-mediated disease and autoinflammation. J Pathol. 2017 Jan;241(2):123-139. doi: 10.1002/path.4812
- Maria ATJ, et al. Adult onset Still’s disease (AOSD) in the era of biologic therapies: dichotomous view for cytokine and clinical expressions. Autoimmun. Rev. 2014;13:1149–1159.
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, Cho JH. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001 May 31;411(6837):603-6. doi: 10.1038/35079114
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008 Oct;22(5):773-92. doi: 10.1016/j.berh.2008.08.006
- Vastert SJ, Kuis W, Grom AA. Systemic JIA: new developments in the understanding of the pathophysiology and therapy. Best Pract. Res. Clin. Rheumatol. 2009;23:655–664.
- Gerfaud-Valentin M., Jamilloux Y., Iwaz J., Sève P. Adult-onset Still’s disease. Autoimmun. Rev. 2014;13:708–722. doi: 10.1016/j.autrev.2014.01.058
- Jia J., Shi H., Liu M., Liu T., Gu J., Wan L., Teng J., Liu H., Cheng X., Ye J., et al. Cytomegalovirus Infection May Trigger Adult-Onset Still’s Disease Onset or Relapses. Front. Immunol. 2019;10:898. doi: 10.3389/fimmu.2019.00898
- Scirè C.A., Cavagna L., Perotti C., Bruschi E., Caporali R., Montecucco C. Diagnostic value of procalcitonin measurement in febrile patients with systemic autoimmune diseases. Clin. Exp. Rheumatol. 2006;24:123–128.
- Fukuoka K., Miyamoto A., Ozawa Y., Ikegaya N., Maesono T., Komagata Y., Kaname S., Arimura Y. Adult-onset Still’s disease-like manifestation accompanied by the cancer recurrence after long-term resting state. Mod. Rheumatol. 2019;29:704–707. doi: 10.1080/14397595.2016.1259547
- Neishi J., Tsukada Y., Maehara T., Ueki K., Maezawa A., Nojima Y. Adult Still’s disease as a paraneoplastic manifestation of breast cancer. Scand. J. Rheumatol. 2000;29:328–330. doi: 10.1080/030097400447741
- Ahn J.K., Oh J.M., Lee J., Kim S.W., Cha H.S., Koh E.M. Adult onset Still’s disease diagnosed concomitantly with occult papillary thyroid cancer: Paraneoplastic manifestation or coincidence? Clin. Rheumatol. 2010;29:221–224. doi: 10.1007/s10067-009-1305-x
- Liozon E., Ly K.H., Vidal-Cathala E., Fauchais A.L. Adult-onset Still’s disease as a manifestation of malignancy: Report of a patient with melanoma and literature review. La Rev. De Med. Interne. 2014;35:60–64. doi: 10.1016/j.revmed.2013.02.014
- Jamilloux Y, Gerfaud-Valentin M, Martinon F, Belot A, Henry T, Sève P. Pathogenesis of adult-onset Still’s disease: new insights from the juvenile counterpart. Immunol Res. 2015;61(1–2):53–62. doi: 10.1007/s12026-014-8561-9
- Terkeltaub R, Esdaile JM, Décary F, Harth M, Lister J, Lapointe N. HLA-Bw35 and prognosis in adult Still’s disease. Arthritis Rheum. 1981;24(12):1469–1472. doi: 10.1002/art.1780241203
- Wouters JM, van de Putte LB. Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med. 1986;61(235):1055–1065.
- Wakil SM, Monies DM, Abouelhoda M, Al-Tassan N, Al-Dusery H, Naim EA, et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2015;67(1):288–295. doi: 10.1002/art.38877
- Ombrello MJ, Remmers EF, Tachmazidou I, Grom A, Foell D, Haas J-P, et al. HLA-DRB1∗11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U S A. 2015;112(52):15970–15975. doi: 10.1073/pnas.1520779112
- Aksentijevich I, McDermott MF. Lessons from characterization and treatment of the autoinflammatory syndromes. Curr Opin Rheumatol. 2017;29(2):187–194. doi: 10.1097/BOR.0000000000000362
- Sarrabay G, Touitou I. Autoinflammation. Management of hereditary recurrent fevers—SHARE experience. Nat Rev Rheumatol. 2015;11(10):567–569. doi: 10.1038/nrrheum.2015.114
- Li, Z., Liu, H. L., Chen, J., Zeng, T., He, L., Li, M., Luo, C., Liu, S., Ding, T. T., Yimaiti, K., Teng, J., Li, X., Ding, Y., Cheng, X., Zhou, J., Ye, J., Ji, J., Su, Y. T., Shi, H., Sun, Y., … Yang, C. (2020). Both HLA class I and II regions identified as genome-wide significant susceptibility loci for adult-onset Still’s disease in Chinese individuals. Annals of the rheumatic diseases, 79(1), 161–163. https://doi.org/10.1136/annrheumdis-2019-215239
- Wang M.Y., Jia J.C., Yang C.D., Hu Q.Y. Pathogenesis, disease course, and prognosis of adult-onset Still’s disease: An update and review. Chin. Med. J. 2019;132:2856–2864. doi: 10.1097/CM9.0000000000000538
- Ruddell R.G., Hoang-Le D., Barwood J.M., Rutherford P.S., Piva T.J., Watters D.J., Santambrogio P., Arosio P., Ramm G.A. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology. 2009;49:887–900. doi: 10.1002/hep.22716
- Hu Q., Shi H., Zeng T., Liu H., Su Y., Cheng X., Ye J., Yin Y., Liu M., Zheng H., et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019;21:9. doi: 10.1186/s13075-018-1800-z
- Chen D.Y., Chen Y.M., Lan J.L., Lin C.C., Chen H.H., Hsieh C.W. Potential role of Th17 cells in the pathogenesis of adult-onset Still’s disease. Rheumatology. 2010;49:2305–2312. doi: 10.1093/rheumatology/keq284
- Shimojima Y., Kishida D., Ueno K.I., Ushiyama S., Ichikawa T., Sekijima Y. Characteristics of Circulating Natural Killer Cells and Their Interferon-γ Production in Active Adult-onset Still Disease. J. Rheumatol. 2019;46:1268–1276. doi: 10.3899/jrheum.181192
- Sun Y., Wang Z., Chi H., Hu Q., Ye J., Liu H., Cheng X., Shi H., Zhou Z., Teng J., et al. Elevated serum levels of interleukin-10 in adult-onset Still’s disease are associated with disease activity. Clin. Rheumatol. 2019;38:3205–3210. doi: 10.1007/s10067-019-04642-x
- Björkengren AG, Pathria MN, Sartoris DJ, Terkeltaub R, Esdaile JM, Weisman M, Resnick D. Carpal alterations in adult-onset Still disease, juvenile chronic arthritis, and adult-onset rheumatoid arthritis: comparative study. Radiology. 1987 Nov;165(2):545-8. doi: 10.1148/radiology.165.2.3659381
- Fautrel B, Le Moël G, Saint-Marcoux B, Taupin P, Vignes S, Rozenberg S, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. 2001;28(2):322–329.
- Vignes S, Le Moël G, Fautrel B, Wechsler B, Godeau P, Piette JC. Percentage of glycosylated serum ferritin remains low throughout the course of adult onset Still’s disease. Ann Rheum Dis. 2000;59(5):347–350. doi: 10.1136/ard.59.5.347
- Mitrovic S, Fautrel B. New markers for adult-onset Still’s disease. Joint Bone Spine. 2018;85(3):285–293. doi: 10.1016/j.jbspin.2017.05.011
- Kim Hyoun-Ah, Han Jae, Kim Woo-Jung, Noh Hyun, An Jeong-Mi, Yim Hyunee, Jung Ju-Yang, Kim You-Sun, Suh Chang-Hee. TLR4 Endogenous Ligand S100A8/A9 Levels in Adult-Onset Still’s Disease and Their Association with Disease Activity and Clinical Manifestations. International Journal of Molecular Sciences. 2016;17(8):1342. doi: 10.3390/ijms17081342
- Kim Y.J., Kim S.I., Hong K.W., Kang M.W. Diagnostic value of 18F-FDG PET/CT in patients with fever of unknown origin. Intern. Med. J. 2012;42:834–837. doi: 10.1111/j.1445-5994.2012.02828.x
- Colina M, Zucchini W, Ciancio G, Orzincolo C, Trotta F, Govoni M. The evolution of adult-onset Still disease: an observational and comparative study in a cohort of 76 Italian patients. Semin Arthritis Rheum. 2011;41(2):279–285. doi: 10.1016/j.semarthrit.2010.12.006
- Van Reeth C, Le Moel G, Lasne Y, Revenant MC, Agneray J, Kahn MF, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still’s disease. J Rheumatol. 1994;21(5):890–895.
- Scirè CA, Cavagna L, Perotti C, Bruschi E, Caporali R, Montecucco C. Diagnostic value of procalcitonin measurement in febrile patients with systemic autoimmune diseases. Clin Exp Rheumatol. 2006;24(2):123–128.
- Priori R, Colafrancesco S, Alessandri C, Minniti A, Perricone C, Iaiani G, et al. Interleukin 18: a biomarker for differential diagnosis between adult-onset Still’s disease and sepsis. J Rheumatol. 2014;41(6):1118–1123. doi: 10.3899/jrheum.130575
- Rau M, Schiller M, Krienke S, Heyder P, Lorenz H, Blank N. Clinical manifestations but not cytokine profiles differentiate adult-onset Still’s disease and sepsis. J Rheumatol. 2010;37(11):2369–2376. doi: 10.3899/jrheum.100247
- Chen D-Y, Lan J-L, Lin F-J, Hsieh T-Y. Proinflammatory cytokine profiles in sera and pathological tissues of patients with active untreated adult onset Still’s disease. J Rheumatol. 2004;31(11):2189–2198.
- Bae C-B, Suh C-H, An J-M, Jung J-Y, Jeon J-Y, Nam J-Y, et al. Serum S100A12 may be a useful biomarker of disease activity in adult-onset Still’s disease. J Rheumatol. 2014;41(12):2403–2408. doi: 10.3899/jrheum.140651
- Chen D-Y, Chen Y-M, Lin C-C, Hsieh C-W, Wu Y-C, Hung W-T, et al. The potential role of advanced glycation end products (AGEs) and soluble receptors for AGEs (sRAGE) in the pathogenesis of adult-onset still’s disease. BMC Musculoskelet Disord. 2015;16:111. doi: 10.1186/s12891-015-0569-3
- Han JH, Suh C-H, Jung J-Y, Nam J-Y, Kwon JE, Yim H, et al. Association of CXCL10 and CXCL13 levels with disease activity and cutaneous manifestation in active adult-onset Still’s disease. Arthritis Res Ther. 2015;17:260. doi: 10.1186/s13075-015-0773-4
- Colafrancesco S, Priori R, Alessandri C, Astorri E, Perricone C, Blank M, et al. sCD163 in AOSD: a biomarker for macrophage activation related to hyperferritinemia. Immunol Res. 2014;60(2–3):177–183. doi: 10.1007/s12026-014-8563-7
- Zou Y-Q, Lu L-J, Li S-J, Zeng T, Wang X-D, Bao C-D, et al. The levels of macrophage migration inhibitory factor as an indicator of disease activity and severity in adult-onset Still’s disease. Clin Biochem. 2008;41(7–8):519–524. doi: 10.1016/j.clinbiochem.2008.01.008
- Chen D-Y, Lan J-L, Lin F-J, Hsieh T-Y. Association of intercellular adhesion molecule-1 with clinical manifestations and interleukin-18 in patients with active, untreated adult-onset Still’s disease. Arthritis Rheum. 2005;53(3):320–327. doi: 10.1002/art.21164
- Ichida H, Kawaguchi Y, Sugiura T, Takagi K, Katsumata Y, Gono T, et al. Clinical manifestations of adult-onset Still’s disease presenting with erosive arthritis: association with low levels of ferritin and interleukin-18. Arthritis Care Res. 2014;66(4):642–646. doi: 10.1002/acr.22194
- Feist, E., Mitrovic, S., & Fautrel, B. (2018). Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nature reviews. Rheumatology, 14(10), 603–618. https://doi.org/10.1038/s41584-018-0081-x
- Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, Kashiwazaki S, Tanimoto K, Matsumoto Y, Ota T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992 Mar;19(3):424-30.
- Lebrun D, Mestrallet S, Dehoux M, Golmard JL, Granger B, Georgin-Lavialle S, et al. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin Arthritis Rheum. 2018;47(4):578–585. https://doi.org/10.1016/j.semarthrit.2017.07.005
- Fautrel B, Borget C, Rozenberg S, Meyer O, Le Loët X, Masson C, et al. Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1999;26(2):373–378.
- Hu Q., Wang M., Jia J., Teng J., Chi H., Liu T., Liu H.L., Cheng X., Ye J., Su Y., et al. Tofacitinib in refractory adult-onset Still’s disease: 14 cases from a single centre in China. Ann. Rheum. Dis. 2020;79:842–844. doi: 10.1136/annrheumdis-2019-216699
- Honda M., Moriyama M., Kondo M., Kumakura S., Murakawa Y. Tofacitinib-induced remission in refractory adult-onset Still’s disease complicated by macrophage activation syndrome. Scand. J. Rheumatol. 2020;49:336–338. doi: 10.1080/03009742.2020.1729405
- Hoff P., Walther M., Wesselmann H., Weinerth J., Feist E., Ohrndorf S. Successful treatment of adult Still’s disease with tofacitinib in a HIV-2 positive female patient. Z. Fur Rheumatol. 2020 doi: 10.1007/s00393-020-00853-9
- Kacar M., Fitton J., Gough A.K., Buch M.H., McGonagle D.G., Savic S. Mixed results with baricitinib in biological-resistant adult-onset Still’s disease and undifferentiated systemic autoinflammatory disease. RMD Open. 2020:6. doi: 10.1136/rmdopen-2020-001246
- Kiyonaga Y, Maeshima K, Imada C, Haranaka M, Ishii K, Shibata H. Steroid-sparing effects of etanercept in a patient with steroid-dependent adult-onset Still’s disease. Intern Med. 2014;53(11):1209–1213. doi: 10.2169/internalmedicine.53.1488
- Fautrel B., Sibilia J., Mariette X., Combe B. Tumour necrosis factor alpha blocking agents in refractory adult Still’s disease: An observational study of 20 cases. Ann. Rheum. Dis. 2005;64:262–266. doi: 10.1136/ard.2004.024026
- Feist Eugen, Mitrovic Stéphane, Fautrel Bruno. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nature Reviews Rheumatology. 2018;14(10):603–618. doi: 10.1038/s41584-018-0081-x
- Giampietro C, Ridene M, Lequerre T, Costedoat Chalumeau N, Amoura Z, Sellam J, et al. Anakinra in adult-onset Still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res. 2013;65(5):822–826. doi: 10.1002/acr.21901
- Hong D, Yang Z, Han S, Liang X, Ma K, Zhang X. Interleukin 1 inhibition with anakinra in adult-onset Still disease: a meta-analysis of its efficacy and safety. Drug Des Devel Ther. 2014;8:2345–2357.
- Colafrancesco S, Priori R, Valesini G, Argolini L, Baldissera E, Bartoloni E, et al. Response to interleukin-1 inhibitors in 140 Italian patients with adult-onset Still’s disease: a multicentre retrospective observational study. Front. Pharmacol., 13 June 2017. https://www.frontiersin.org/articles/10.3389/fphar.2017.00369/full
- A Study to Evaluate Efficacy and Safety of Anakinra in the Treatment of Still’s Disease (SJIA and AOSD) (anaSTILLs). https://clinicaltrials.gov/ct2/show/NCT03265132
- Ortiz-Sanjuán F, Blanco R, Riancho-Zarrabeitia L, Castañeda S, Olivé A, Riveros A, et al. Efficacy of anakinra in refractory adult-onset Stillʼs disease: multicenter study of 41 patients and literature review. Medicine (Baltimore) 2015;94(39):e1554. doi: 10.1097/MD.0000000000001554
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor, in refractory adult-onset Still’s disease. Semin Arthritis Rheum. 2012;42(2):201–205. doi: 10.1016/j.semarthrit.2012.03.004
- Barsotti S, Neri R, Iacopetti V, d’Ascanio A, Talarico R, Tripoli A, et al. Successful treatment of refractory adult-onset still disease with canakinumab: a case report. J Clin Rheumatol. 2014;20(2):121. doi: 10.1097/RHU.0000000000000082
- Canakinumab for Treatment of Adult-onset Still’s Disease (CONSIDER). https://clinicaltrials.gov/ct2/show/NCT02204293
- Junge G, Mason J, Feist E. Adult onset Still’s disease—the evidence that anti-interleukin-1 treatment is effective and well-tolerated (a comprehensive literature review) Semin Arthritis Rheum. 2017;47(2):295–302. doi: 10.1016/j.semarthrit.2017.06.006
- Cipriani P, Ruscitti P, Carubbi F, Pantano I, Liakouli V, Berardicurti O, et al. Tocilizumab for the treatment of adult-onset Still’s disease: results from a case series. Clin Rheumatol. 2014;33(1):49–55. doi: 10.1007/s10067-013-2381-5
- Song ST, Kim JJ, Lee S, Kim H-A, Lee EY, Shin KC, et al. Efficacy of tocilizumab therapy in Korean patients with adult-onset Still’s disease: a multicentre retrospective study of 22 cases. Clin Exp Rheumatol. 2016;34(6):S64–S71.
- Ma Y, Wu M, Zhang X, Xia Q, Yang J, Xu S, et al. Efficacy and safety of tocilizumab with inhibition of interleukin-6 in adult-onset Still’s disease: a meta-analysis. Mod Rheumatol. 2018;28(5):849–857. doi: 10.1080/14397595.2017.1416924
- Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kötter I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018;77(6):840–847.
- Sakata N, Shimizu S, Hirano F, Fushimi K. Epidemiological study of adult-onset Still’s disease using a Japanese administrative database. Rheumatol Int. 2016;36(10):1399–1405. doi: 10.1007/s00296-016-3546-8
- Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043–4049. doi: 10.1182/blood-2003-11-4035
- Masuyama A, Kobayashi H, Kobayashi Y, Yokoe I, Sugimura Y, Maniwa K, et al. A case of adult-onset Still’s disease complicated by thrombotic thrombocytopenic purpura with retinal microangiopathy and rapidly fatal cerebral edema. Mod Rheumatol. 2013;23(2):379–385. doi: 10.3109/s10165-012-0650-9
- Lowther GH, Chertoff J, Cope J, Alnuaimat H, Ataya A. Pulmonary arterial hypertension and acute respiratory distress syndrome in a patient with adult-onset Still’s disease. Pulm Circ. 2017;7(4):797–802. doi: 10.1177/2045893217712710
- Lyu MS, Li DY, Zhou SZ, Ban CJ, Yan J. Adult-onset Still’s disease successfully treated with Chinese herbal medicine: A case report with 15-month follow-up. J Integr Med. 2020 Nov;18(6):530-534. https://doi.org/10.1016/j.joim.2020.08.004
- Diet and Inflammation. Nutrition in Clinical Practice Vol 25, Issue 6, pp. 634 – 640. First published date: December-07-2010. 10.1177/0884533610385703
- F.B. Hu. The Mediterranean Diet and mortality—olive oil and beyond. N Engl J Med, 348 (2003), pp. 2595-2596
- Sears, B., & Saha, A. K. (2021). Dietary Control of Inflammation and Resolution. Frontiers in nutrition, 8, 709435. https://doi.org/10.3389/fnut.2021.709435
- Most, J., Tosti, V., Redman, L. M., & Fontana, L. (2017). Calorie restriction in humans: An update. Ageing research reviews, 39, 36–45. https://doi.org/10.1016/j.arr.2016.08.005
- Martin, C. K., Das, S. K., Lindblad, L., Racette, S. B., McCrory, M. A., Weiss, E. P., Delany, J. P., Kraus, W. E., & CALERIE Study Team (2011). Effect of calorie restriction on the free-living physical activity levels of nonobese humans: results of three randomized trials. Journal of applied physiology (Bethesda, Md. : 1985), 110(4), 956–963. https://doi.org/10.1152/japplphysiol.00846.2009
- Das, S. K., Roberts, S. B., Bhapkar, M. V., Villareal, D. T., Fontana, L., Martin, C. K., Racette, S. B., Fuss, P. J., Kraus, W. E., Wong, W. W., Saltzman, E., Pieper, C. F., Fielding, R. A., Schwartz, A. V., Ravussin, E., Redman, L. M., & CALERIE-2 Study Group (2017). Body-composition changes in the Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy (CALERIE)-2 study: a 2-y randomized controlled trial of calorie restriction in nonobese humans. The American journal of clinical nutrition, 105(4), 913–927. https://doi.org/10.3945/ajcn.116.137232
- The effects of diet on inflammation: emphasis on the metabolic syndrome. Giugliano D, Ceriello A, Esposito K. J Am Coll Cardiol. 2006 Aug 15; 48(4):677-85. https://www.ncbi.nlm.nih.gov/pubmed/16904534/
- Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Circulation. 2002 Oct 15; 106(16):2067-72. http://circ.ahajournals.org/content/106/16/2067.long
- Mellen PB, Walsh TF, Herrington DM. Whole grain intake and cardiovascular disease: a meta-analysis. Nutr Metab Cardiovasc Dis. 2008;18:283–90. https://www.ncbi.nlm.nih.gov/pubmed/17449231
- Priebe M, van Binsbergen J, de Vos R, Vonk RJ. Whole grain foods for the prevention of type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2008, Issue 1. Art. No.: CD006061. DOI: 10.1002/14651858.CD006061.pub2.
- Pereira MA, O’Reilly E, Augustsson K, Fraser GE, Goldbourt U, Heitmann BL, Hallmans G, Knekt P, Liu S, Pietinen P, Spiegelman D, Stevens J, Virtamo J, Willett WC, Ascherio A. Dietary fiber and risk of coronary heart disease: a pooled analysis of cohort studies. Arch Intern Med. 2004;164:370–376. https://www.ncbi.nlm.nih.gov/pubmed/14980987
- Liu S. Whole-grain foods, dietary fiber, and type 2 diabetes: searching for a kernel of truth. The American journal of clinical nutrition. 2003;77:527–529. http://ajcn.nutrition.org/content/77/3/527.long
- King DE. Dietary fiber, inflammation, and cardiovascular disease. Mol Nutr Food Res. 2005;49:594–600. https://www.ncbi.nlm.nih.gov/pubmed/15884088
- Ajani UA, Ford ES, Mokdad AH. Dietary fiber and C-reactive protein: findings from national health and nutrition examination survey data. The Journal of nutrition. 2004;134:1181–1185. http://jn.nutrition.org/content/134/5/1181.long
- King DE, Egan BM, Geesey ME. Relation of dietary fat and fiber to elevation of C-reactive protein. Am J Cardiol. 2003;92:1335–1339. https://www.ncbi.nlm.nih.gov/pubmed/14636916
- Ma Y, Griffith JA, Chasan-Taber L, et al. Association between dietary fiber and serum C-reactive protein-. The American journal of clinical nutrition. 2006;83(4):760-766. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1456807/
- King DE, Egan BM, Woolson RF, Mainous AG, 3rd, Al-Solaiman Y, Jesri A. Effect of a high-fiber diet vs a fiber-supplemented diet on C-reactive protein level. Arch Intern Med. 2007;167:502–506. https://www.ncbi.nlm.nih.gov/pubmed/17353499
- Inflammatory disease processes and interactions with nutrition. Br J Nutr. 2009 May;101 Suppl 1:S1-45. doi: 10.1017/S0007114509377867. https://www.ncbi.nlm.nih.gov/pubmed/19586558/
- Dietary patterns and mortality in Danish men and women: a prospective observational study. British Journal of Nutrition (2001), 85, 219-225. DOI: 10.1079/BJN2000240. https://www.cambridge.org/core/services/aop-cambridge-core/content/view/2C582B42A8FEC3F37A7EEE62875078B1/S0007114501000307a.pdf
- Diets and cardiovascular disease. An evidence-based assessment. J Am Coll Cardiol, 45 (2005), pp. 1379-1387.
- The diet-heart hypothesis: a critique. J Am Coll Cardiol, 43 (2004), pp. 731-733.
- In humans, serum polyunsaturated fatty acid levels predict the response of proinflammatory cytokines to psychologic stress. Maes M, Christophe A, Bosmans E, Lin A, Neels H. Biol Psychiatry. 2000 May 15; 47(10):910-20. https://www.ncbi.nlm.nih.gov/pubmed/10807964/
- Habitual dietary intake of n-3 and n-6 fatty acids in relation to inflammatory markers among US men and women. Pischon T, Hankinson SE, Hotamisligil GS, Rifai N, Willett WC, Rimm EB. Circulation. 2003 Jul 15; 108(2):155-60. http://circ.ahajournals.org/content/108/2/155.long
- Depressive symptoms, omega-6:omega-3 fatty acids, and inflammation in older adults. Kiecolt-Glaser JK, Belury MA, Porter K, Beversdorf DQ, Lemeshow S, Glaser R. Psychosom Med. 2007 Apr; 69(3):217-24. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2856352/
- Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. Ferrucci L, Cherubini A, Bandinelli S, Bartali B, Corsi A, Lauretani F, Martin A, Andres-Lacueva C, Senin U, Guralnik JM. J Clin Endocrinol Metab. 2006 Feb; 91(2):439-46. https://www.ncbi.nlm.nih.gov/pubmed/16234304/
- Inflammatory disease processes and interactions with nutrition. Calder PC, Albers R, Antoine JM, Blum S, Bourdet-Sicard R, Ferns GA, Folkerts G, Friedmann PS, Frost GS, Guarner F, Løvik M, Macfarlane S, Meyer PD, M’Rabet L, Serafini M, van Eden W, van Loo J, Vas Dias W, Vidry S, Winklhofer-Roob BM, Zhao J. Br J Nutr. 2009 May; 101 Suppl 1():S1-45. https://www.ncbi.nlm.nih.gov/pubmed/19586558/
- Effect of dietary antioxidants on postprandial endothelial dysfunction induced by a high-fat meal in healthy subjects. Esposito K, Nappo F, Giugliano F, Giugliano G, Marfella R, Giugliano D. Am J Clin Nutr. 2003 Jan; 77(1):139-43. http://ajcn.nutrition.org/content/77/1/139.long
- Anti-inflammatory Diets. Journal of the American College of Nutrition Vol. 34 , Iss. sup1,2015. http://dx.doi.org/10.1080/07315724.2015.1080105
- Pouchot J., Sampalis J.S., Beaudet F., Carette S., Décary F., Salusinsky-Sternbach M., Hill R.O., Gutkowski A., Harth M., Myhal D., et al. Adult Still’s disease: Manifestations, disease course, and outcome in 62 patients. Medicine. 1991;70:118–136. doi: 10.1097/00005792-199103000-00004
- Maria A.T., Le Quellec A., Jorgensen C., Touitou I., Rivière S., Guilpain P. Adult onset Still’s disease (AOSD) in the era of biologic therapies: Dichotomous view for cytokine and clinical expressions. Autoimmun. Rev. 2014;13:1149–1159. doi: 10.1016/j.autrev.2014.08.032
- Chen D.Y., Lan J.L., Lin F.J., Hsieh T.Y. Proinflammatory cytokine profiles in sera and pathological tissues of patients with active untreated adult onset Still’s disease. J. Rheumatol. 2004;31:2189–2198.
- Franchini S., Dagna L., Salvo F., Aiello P., Baldissera E., Sabbadini M.G. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010;62:2530–2535. doi: 10.1002/art.27532
- Sampalis JS, Esdaile JM, Medsger TA, Partridge AJ, Yeadon C, Senécal JL, et al. A controlled study of the long-term prognosis of adult Still’s disease. Am J Med. 1995;98(4):384–388. doi: 10.1016/S0002-9343(99)80318-0





{kind=link}