
What is albinism
Albinism is a group of inherited genetic disorder that results in little or no production of the pigment melanin in the skin, hair and eyes. The term “albinism” originated from the Latin word “albus” meaning white 1. People with albinism are sometimes called “albinos”. However, “a person with albinism” is the preferred term. Melanin is a term used to describe a large group of related molecules responsible for many biological functions, including pigmentation of skin and hair and photoprotection of skin and eye 2. Melanin is a brown/black or red/yellow polymer produced by within melanocytes (pigment cells) which are located at the base of the epidermis, it is packaged in small, round membrane-bound organelles called melanosomes. Melanosomes are transported from melanocytes to neighboring keratinocytes via tentacle-like dendritic processes. Melanosomes arriving in keratinocytes are positioned superficially to cell nuclei, which serves to protect from incoming ultraviolet (UV) radiation 3. Melanin is carried by keratinocytes to the skin surface. Melanin content of skin is the main determining factor of skin and hair color; hair is considered a form of skin with regards to pigmentation. Whether you have dark skin or light skin depends on the amount and type of melanin produced in your skin. The melanocytes of dark-skinned people produce more melanin than those of people with light skin.
People with albinism may experience the following symptoms:
- Very pale skin, hair and eyes.
- Patches of missing skin pigment.
- Crossed eyes (strabismus).
- Rapid eye movements (nystagmus).
- Vision problems.
- Light sensitivity (photophobia).
Albinism, in any of its forms, is the result of heritable mutations in one of several genes that lead to defective melanocytes, unable to properly synthesize melanin or distribute it through dermal tissues. The exact number of melanocytes in the skin are preserved, unlike conditions such as piebaldism and vitiligo, where melanocytes are absent 4. Symptoms of albinism can include light skin or changes in skin color; very white to brown hair; very light blue to brown eye color that may appear red in some light and may change with age; sensitivity to sun exposure; and increased risk of developing skin cancer 5. Because most albino people have fair complexions, they are particularly susceptible to solar damage and these people must utilize lifelong sun protection precautions such as wearing sunscreen or sunblock, hats, sunglasses and sun-protective clothing. People with albinism should be educated on avoidance of prolonged ultraviolet (UV) light exposure and avoidance of medications that increase photosensitivity.
Melanin also plays a role in the development of certain optical nerves, so all forms of albinism cause problems with the development and function of the eyes and vision problems are associated with albinism 6. A common myth is that people with albinism have red eyes. Although lighting conditions can allow the blood vessels at the back of the eye to be seen, which can cause the eyes to look reddish or violet, most people with albinism have blue eyes, and some have hazel or brown eyes 7. There are different types of albinism and the amount of pigment in the eyes varies.
Albinism occurs in all racial and ethnic groups throughout the world. In the U.S., approximately one in 18,000 to 20,000 people has some type of albinism 7. In other parts of the world, the occurrence can be as high as one in 3,000. Most children with albinism are born to parents whose hair and eye color are typical for their ethnic backgrounds.
Albinism is caused by mutations in one of several genes, and most types are inherited in an autosomal recessive manner. Although there’s no cure, albino people can take steps to improve vision and avoid too much sun exposure.
A person with albinism can protect your skin, hair and eyes by:
- Staying out of the sun.
- Wearing sunglasses.
- Covering up with sun-protective clothing.
- Wearing hats.
- Applying sunscreen regularly.
If you have crossed eyes (strabismus), a surgeon may be able to correct the issue with surgery.
Is albinism a disease?
Albinism isn’t a disease. Albinism is a genetic condition that people are born with. It’s not contagious, and it can’t be spread.
Is albinism genetic?
Yes, albinism is passed down (inherited) through families. People are born with albinism when they inherit an albinism gene from their parents.
In oculocutaneous albinism, both parents must carry an albinism gene for their child to be born with albinism. The child has a 1 in 4 chance of being born with albinism. If just one parent has an albinism gene, the child won’t have oculocutaneous albinism. But they’ll have a 50% chance of being a carrier of the gene themselves.
What assistance do children with albinism need?
All children with albinism benefit from sunglasses and hats to reduce glare and prevent sunburn. Children with albinism may need some form of visual aid, depending on the type and extent of the visual condition. Glasses or contact lenses can correct for short or long sightedness or astigmatism. Older children may need a monocular for distance viewing and some may need large print or a magnifier for reading.
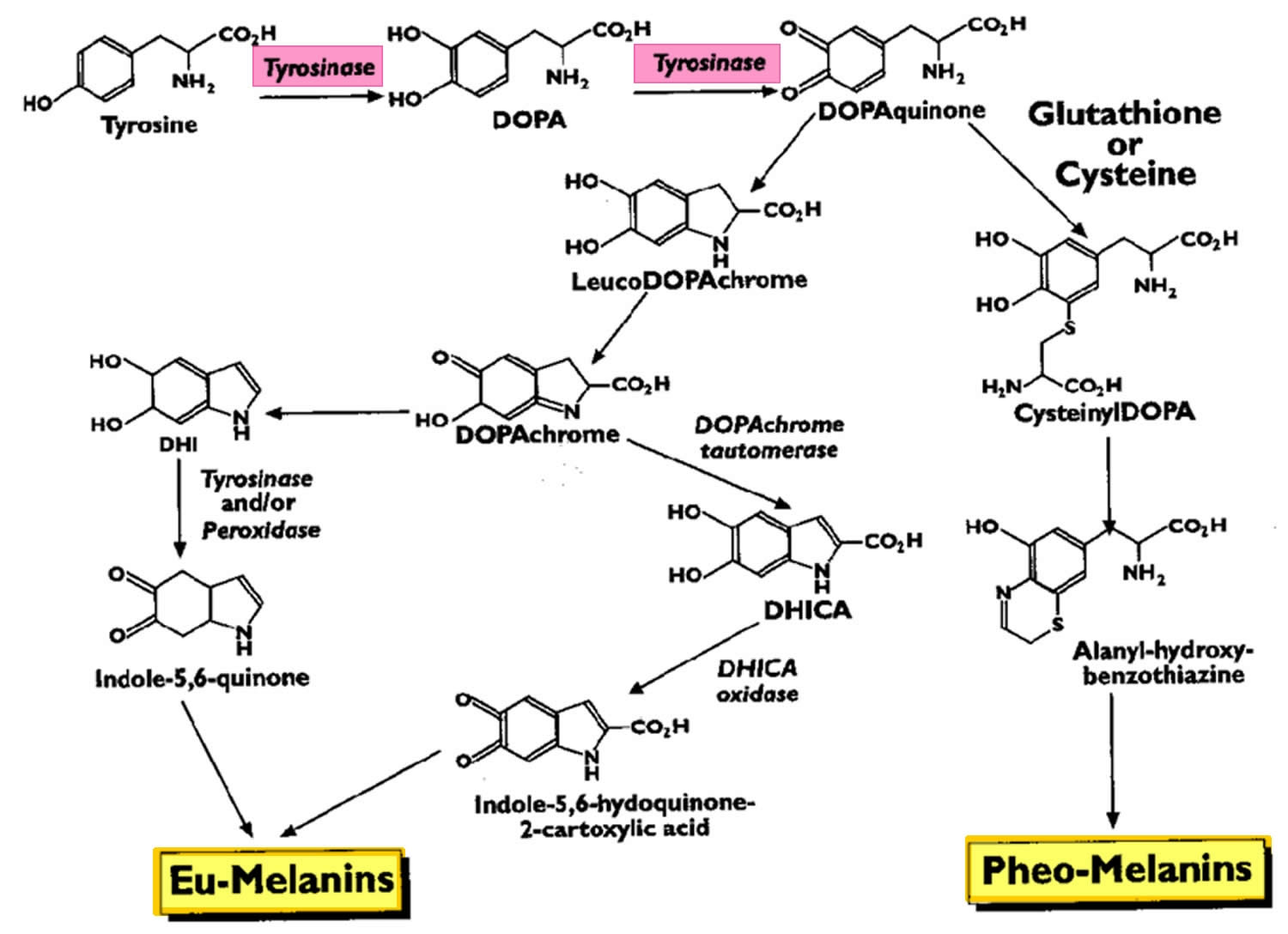
Melanin biosynthesis
In humans, melanin exists as three forms: eumelanin (which is subdivided further into black and brown forms), pheomelanin, and neuromelanin 2. Eumelanin and pheomelanin are produced in various amounts in the basal layer of the epidermis within cells called melanocytes. The first step of biosynthesis of both eumelanin and pheomelanin begins the same way. Tyrosine is converted into dihydroxyphenylalanine (DOPA), which requires tyrosine hydroxylase and tetrahydrobiopterin as a cofactor. The enzyme tyrosinase then converts dihydroxyphenylalanine into dopaquinone, which can follow a variety of pathways to form the eumelanin or pheomelanin (Figure 1).
The primary stimulus for melanogenesis and subsequent melanosome production is ultraviolet (UV) radiation, which upregulates melanocyte production of pro-opiomelanocortin (POMC) and its downstream products, alpha-melanocyte-stimulating hormone (alpha-MSH) and adrenocorticotropic hormone (ACTH) 2. The overall effect is to increase eumelanin production. Interestingly, people with pro-opiomelanocortin mutations have red hair and Fitzpatrick skin type 1 due to the relative increase in pheomelanin to eumelanin expression.
Neuromelanin is a dark pigment produced by dopaminergic and noradrenergic cells of the substantia nigra and locus coeruleus as a breakdown product of dopamine 8.
Figure 1. Melanin biosynthesis
Melanin function
In its various forms, melanin fulfills a variety of biological functions, including skin and hair pigmentation and photoprotection of the skin and eye.
Pigmentation of the skin results from the accumulation of melanin-containing melanosomes in the basal layer of the epidermis. Differences in skin pigmentation result both from the relative ratio of eumelanin (brown–black) to pheomelanin (yellow–red), as well as the number of melanosomes within melanocytes. Pheomelanin accounts for the pinkish skin constituting the lips, nipples, vagina, and glans of the penis. In general, lightly pigmented skin tends to contain melanocytes with clusters of two to three melanosomes, whereas darkly pigmented skin tends to contain individual melanosomes which can melanize neighboring keratinocytes more readily. The overall melanin density correlates with the darkness of skin as well as Fitzpatrick skin type.
The interplay between melanin and UV radiation is complex. Researchers widely believe that melanin production in melanocytes increased as an evolutionary adaptation to the widespread loss of human body hair more than a million years ago. Populations living closer to the equator tended to develop a greater proportion of eumelanin, which is a UV–absorbent, antioxidant, and free radical scavenger. Conversely, populations living further from the equator are relatively richer in pheomelanin, which produces free radicals in response to UV radiation, accelerating carcinogenesis. As the main stimulus for cutaneous vitamin D production is UV light exposure, it follows that dark-skinned individuals also tend to have lower levels of vitamin D and should be screened accordingly.
Less clear is the link between melanin, the sun, and cutaneous immunology. Both acute and chronic UV light exposure induces immunosuppression; UVA light is used therapeutically for a large number of skin conditions, including psoriasis. Intriguingly, melanin is believed to have immunomodulatory and even anti-bacterial properties, although the underlying mechanisms have not yet been fully elucidated. Malignant melanocytes rich in melanin are less sensitive to chemo-, radio-, or photodynamic therapy, and amelanotic melanomas have longer disease-free and overall survival than melanotic ones. Therefore, some have suggested inhibition of melanogenesis as a therapy for malignant melanoma.
Just as melanin protects the skin from photodamage, it also protects the eye. Melanin is concentrated in the iris and choroid, and those with grey, blue, and green eye colors, as well as albinos, have more sun-related ocular issues.
Hair color is determined by the relative proportion of various forms of melanin:
- Black and brown hair results from varying degrees of black and brown eumelanin
- Blonde hair results from a small amount of brown eumelanin in the absence of black eumelanin
- Red hair results from roughly equal amounts of pheomelanin as eumelanin. Strawberry blonde hair results from brown eumelanin in the presence of pheomelanin.
Types of albinism
There are several different types of albinism. Levels of pigmentation vary depending on which type of albinism you have. The different types of albinism include:
- Oculocutaneous albinism (OCA): Oculocutaneous albinism or OCA, is the most common type of albinism. People with oculocutaneous albinism (OCA) have extremely pale hair, skin and eyes. There are seven different subtypes of oculocutaneous albinism (OCA), caused by mutations in one of seven genes (OCA1 to OCA7).
- Ocular albinism: Ocular albinism or OA, is much less common than oculocutaneous albinism (OCA). Ocular albinism affects only your eyes. People with ocular albinism usually have blue eyes. Sometimes your irises (colored part of your eyes) are very pale, so your eyes may appear red or pink. This is because the blood vessels inside your eyes show through the irises. Your skin and hair color are usually normal.
- Hermansky-Pudlak syndrome: Hermansky-Pudlak syndrome or HPS, is a type of albinism that includes a form of oculocutaneous albinism (OCA) along with blood disorders, bruising issues and lung, kidney or bowel diseases.
- Chediak-Higashi syndrome: Chediak-Higashi syndrome (CHS) is a type of albinism that includes a form of oculocutaneous albinism (OCA) along with immune and neurological issues.
Oculocutaneous albinism (OCA)
Oculocutaneous albinism or OCA, involves dilution of the color of the hair, skin and eyes and is the most common type of albinism. People with oculocutaneous albinism (OCA) have white or light-colored hair, very fair skin and eyes. Long-term sun exposure greatly increases the risk of skin damage and skin cancers, including an aggressive form of skin cancer called melanoma, in people with this condition. Oculocutaneous albinism (OCA) also reduces pigmentation of the colored part of the eye (the iris) and the light-sensitive tissue at the back of the eye (the retina). People with this condition usually have vision problems such as reduced sharpness; rapid, involuntary eye movements (nystagmus); and increased sensitivity to light (photophobia).
Over the years, researchers have identified multiple types of oculocutaneous albinism (OCA), which are distinguished by their specific skin, hair, and eye color changes and by their genetic cause. Oculocutaneous albinism type 1 is characterized by white hair, very pale skin, and light-colored irises. Type 2 is typically less severe than type 1; the skin is usually a creamy white color and hair may be light yellow, blond, or light brown. Type 3 includes a form of albinism called rufous oculocutaneous albinism, which usually affects dark-skinned people. Affected individuals have reddish-brown skin, ginger or red hair, and hazel or brown irises. Type 3 is often associated with milder vision abnormalities than the other forms of oculocutaneous albinism. Type 4 has signs and symptoms similar to those seen with type 2. In general, these systems contrasted types of albinism having almost no pigmentation with types having slight pigmentation. In less pigmented types of albinism, hair and skin are cream-colored and vision is often in the range of 20/200. In types with slight pigmentation, hair appears more yellow or has a reddish tinge and vision may be better.
DNA tests can determine the precise type of albinism. Research on albinism genes is ongoing. To date there are seven different subtypes of oculocutaneous albinism (OCA) caused by mutations in one of seven genes – OCA1, OCA2, OCA3, OCA4, OCA5, OCA6 and OCA7. Some are further divided into subtypes. Oculocutaneous albinism type 1 (OCA-1) and type 2 (OCA-2) are the most common types of oculocutaneous albinism. All seven types of oculocutaneous albinism (OCA) are inherited in an autosomal recessive manner and are thus found in both males and females, which is in contrast to that of X-linked recessive inheritance of ocular albinism (OA). Ocular nystagmus is seen in virtually all forms of oculocutaneous albinism (OCA) 10.
Researchers have also identified several other genes that result in albinism with other features. One group includes at least 10 genes leading to Hermansky-Pudlak Syndrome (HPS). In addition to albinism, Hermansky-Pudlak Syndrome (HPS) is associated with bleeding problems and bruising. Some forms are also associated with lung and bowel disease. Hermansky-Pudlak Syndrome (HPS) is a less common form of albinism but should be suspected if a person with albinism shows unusual bruising or bleeding or if a genetic test for a type of OCA produces inconclusive results.
Other albinism-related syndromes include Chediak-Higashi syndrome and Griscelli syndrome.
Overall, an estimated 1 in 20,000 people worldwide are born with oculocutaneous albinism (OCA). The condition affects people in many ethnic groups and geographical regions. Types 1 and 2 are the most common forms of this condition; types 3 and 4 are less common. Type 2 occurs more frequently in African Americans, some Native American groups, and people from sub-Saharan Africa. Type 3, specifically rufous oculocutaneous albinism, has been described primarily in people from southern Africa. Studies suggest that type 4 occurs more frequently in the Japanese and Korean populations than in people from other parts of the world.
OCA Type 1
OCA1 is also known as tyrosinase-related albinism or tyrosinase-related OCA, constitutes several subtypes that occur due to mutation in TYR gene for tyrosinase (chromosome 11q14-q21) 11. Tyrosinase enzyme catalyzes the first two steps in the melanin biosynthesis pathway, responsible for converting tyrosine to DOPA and subsequently to DOPAquinone (see Figure 1 above). Depending on differential production of the end product melanin, the severity of phenotype varies. A point mutation of TYR gene drives the conformation of the tyrosinase enzyme, which results in either absent or decreased enzyme activity. Almost 200 mutations in TYR gene are known to date.
OCA 1A
OCA 1A was formerly known as “tyrosinase-negative” albinism is due to the tyrosinase enzyme being inactive and no melanin is produced, leading to white hair and very light skin 12. Patients affected by OCA 1A thus have the most profound phenotype of all oculocutaneous albinisms with the greatest risk for skin cancer and highest frequency of visual symptoms and acuity loss. Hair bulbs of the affected individuals remain DOPA-negative throughout their lives, indicating that the tyrosinase activity is fully inactive 13. Due to the relative severity of ocular and dermatologic problems, close ophthalmic and dermatologic care is essential.
OCA 1B
OCA-1B was known as the “yellow variant” albinism due to reduced, but not absent, tyrosinase activity that produce some melanin pigment. Patients with 1B may be indistinguishable from OCA-1A at birth due to lack of any detectable pigment early on 14, but during the first several years of life OCA-1B patients accumulate yellow pigment in their skin, eye, and hair. In OCA1B, the hair may darken to blond, yellow/orange or even light brown, as well as slightly more pigment in the skin. Their vision is moderately to severely reduced.
OCA 1MP
OCA 1MP stands for “OCA 1 Mimal Pigment.” King et al in 1986 15 first proposed oculocutaneous albinism (OCA) minimal pigment as a subtype of OCA-1 by showing disordered TYR activity and clinically detectable accumulation of pigment only in the irises that increases with age. The most recent update of the definition proposed by Kono et al 16 claimed that the sites of pigment accumulation include hair and lentigo. With life-long negative DOPA staining of hair bulbs, however, OCA-1MP phenotype contrasts with OCA-1B that has positive DOPA staining.
OCA 1 TS
OCA 1 TS is the Temperature Sensitive albinism that earned its name from variable tyrosinase activity depending on skin temperature. In this subtype, tyrosinase activity is inversely proportional to temperature, leading to dark hair on the cooler extremities but white hairs near the body core such as the armpits, pubic area, and scalp. As in OCA-1B, the patients can be indistinguishable at birth from OCA-1A 14.
OCA Type 2
OCA type 2 (OCA-2) is the most common type of albinism throughout the world with the highest frequency in equatorial Africa 17. The characteristic gene involvement for OCA-2 is the OCA2 gene, previously known as P gene. OCA2 gene is responsible for melanosome function as it helps regulate the influx of tyrosine and other internal environment of the melanosome thus affecting the functionality of the organelle and melanin production. Of note, non-pathologic polymorphisms of the OCA2 contribute to iris colors in normal individuals 14. This disorder is generally a milder form of OCA type 1 (OCA-1), with skin pigmentation ranging from white to fair; hair color ranging from yellow to even black; and generally milder visual dysfunction compared to OCA type 1 (OCA-1). At times the affected individual may be indistinguishable from having OCA-1B and OCA-1MP 18. Interestingly, about 1% of patients with either Prader-Willi syndrome or Angelman syndrome are hypopigmented though often without ocular features of albinism. This phenomenon is due to the position of the OCA2 gene between the genes whose deletions are responsible for the two syndromes 19.
OCA 2 Brown Albinism (BOCA or brown oculocutaneous albinism) is a part of the clinical spectrum of OCA type 2 (OCA-2). This variant of OCA type 2 (OCA-2) was initially identified in Africans of Nigerian and Ghanan ancestry and in and African Americans with light brown hair and skin 20. OCA 2 Brown Albinism (BOCA) patients can present anywhere between the classic phenotype (yellow/blond hair, creamy skin, blue/hazel irides) to brown-pigmentation. Affected individuals from other ethnic groups can have moderate to near-normal skin pigmentation 21.
OCA Type 3
OCA 3 occurs due to a mutation in TYRP1 (tyrosinase-related protein) gene. This subtype of OCA is seen in African-born blacks but rarely in other ethnicities 22. Phenotypes overlap with OCA-2 Brown Albinism (BOCA or brown oculocutaneous albinism) and with red/rufous OCA (ROCA) 23, although as the phenotype names imply, the BOCA can be differentiated from ROCA based on skin and hair color. ROCA phenotype is characterized by red-bronze skin color, ginger hair, and blue or brown irises 24. Studies have since shown that TYRP1 contributes to melanin synthesis as well as melanocyte proliferation and apoptosis 25.
OCA Type 4
Recessive mutation in membrane-associated transport protein gene (MATP), also known as SLC45A2 , is responsible for OCA type 4. Although the gene was first identified in a Turkish patient 26, it has since been found in albino subjects in Japan and elsewhere in Europe as well. This is the third most common type of OCA after OCA2 and OCA1 27.
OCA Type 5
A new OCA gene called OCA5 was found to be responsible for the typical OCA phenotype in a consanguinous Pakistani family 28. This gene is mapped onto chromosome 4q24 and the genotype was designated OCA 5. Further studies on this gene are underway.
OCA Type 6
In 2013, a Chinese group of researchers, Wei et al 29 found mutations in SLC24A5 on 15q21.1 among patients of Chinese origin who presented with characteristic phenotype of non-syndromic OCA. This gene, previously studied but not linked directly to OCA phenotype, is believed to impair or disrupt melanosomal maturation 30. OCA due to mutation in SLC24A5 gene has been since termed OCA type 6. Morice-Picard et al 31 have since shown that this mutation is seen in different ethnic groups, indicating that the mutation is not limited to the Chinese population.
OCA Type 7
As the latest addition to the subtype of oculocutaneous albinism (OCA), OCA type 7 was identified in 2013 by Karen Grønskov and Thomas Rosenberg based on homozygosity mapping and gene sequencing of a consanguineous family with oculocutaneous albinism (OCA) symptoms on the Faroe Islands of Denmark 30. The probands as well as unrelated but affected individuals on the island had homozygous non-sense mutation in C10orf11, a previously unknown gene that has been since shown to contribute to melanocyte differentiation. Production of OCA phenotype due to this gene mutation is thus termed OCA type 7 32.
Ocular albinism (OA)
Ocular albinism (OA) is much less common than oculocutaneous albinism (OCA), accounting for 10-15% of all albinism cases. Ocular albinism affects only your eyes with melanin pigment mainly missing from the eyes while the skin and hair appear normal or only slightly lighter. Ocular albinism (OA) reduces the coloring (pigmentation) of the iris, which is the colored part of the eye, and the retina, which is the light-sensitive tissue at the back of the eye. People with ocular albinism (OA) usually have blue eyes. Sometimes your irises (colored part of your eyes) are very pale, so your eyes may appear red or pink. This is because the blood vessels inside your eyes show through the irises. Your skin and hair color are usually normal.
Ocular albinism is characterized by severely impaired sharpness of vision (visual acuity) and problems with combining vision from both eyes to perceive depth (stereoscopic vision). Although the vision loss is permanent, it does not worsen over time. Other eye abnormalities associated with this condition include rapid, involuntary eye movements (nystagmus); eyes that do not look in the same direction (strabismus); and increased sensitivity to light (photophobia). Many affected individuals also have abnormalities involving the optic nerves, which carry visual information from the eye to the brain.
Unlike some other forms of albinism, ocular albinism (OA) does not significantly affect the color of the skin and hair. People with ocular albinism may have a somewhat lighter complexion than other members of their family, but these differences are usually minor.
There are two known types of ocular albinism (OA) without cutaneous involvement. The most common form of ocular albinism is known as the Nettleship-Falls ocular albinism or ocular albinism type 1 (OA-1). Other forms of ocular albinism are much rarer and may be associated with additional signs and symptoms, such as hearing loss.
The most common form of this disorder, ocular albinism type 1 (OA-1), affects at least 1 in 60,000 males. The classic signs and symptoms of this condition are much less common in females.
OA Type 1
Ocular albinism type 1 (OA-1) also known as Nettleship-Falls ocular albinism, is inherited in an X-linked recessive manner and thus occurs only in boys. Ocular albinism type 1 (OA-1) is the most common form of ocular albinism representing 10% of all albinism and estimated prevalence of 1 in 50,000 to 150,000 live births 33. Mutations in GPR143 gene at Xp22.3-22.2 are known to be causative 34. The affected gene codes for intracellular GPCR (G-protein coupled receptor) that controls the melanosome transport in pigment cells, disruption of which affects the number and size of the melanosomes. OA1 patients produce macromelanosomes seen on skin biopsy 14.
The clinical manifestation of OA1 depend on the ethnic background of the individual, with those who are of darkly pigmented background being less severely affected than those from light pigmentation groups 35. Albinism of the eye with OA tends to be severe with poor vision. Diagnostic methods include molecular analysis of OA1 gene (GPR143), family pedigree analysis, and examination of the carrier mother. Due to lyonization of one X chromosome in each somatic cell, carrier females may exhibit evidence of speckled albinism. In up to 90% of carrier females, mud-splattered fundus is seen 36 due to the patchy dissemination of amelanotic patches of retinal pigment epithelium (RPE) among melanotic patches. Other potential findings are iris transillumination in 75%, hypopigmented macules of the skin, and biopsy evidence of macromelanosomes in those skin segments 37. Of note, OA1 has been associated with late-onset sensorineural deafness (OASD) 38. The syndrome is likely the consequence of a contiguous gene defect that includes OA1 gene. Late-onset sensorineural deafness (OASD) shares similarities but is but etiologically distinct from Waardenburg syndrome, which is characterized by patchy depigmentation of hair and skin, heterochromia irides, and congenital deafness 39.
OA Type 2
Ocular albinism type 2 (OA-2) also known as Aland Island eye disease or Forsius-Eriksson type ocular albinism, is a rare X-linked disorder with similar clinical manifestations as OA1 with the additional protan color vision defect and defective dark adaptation. In addition to such differences, biopsy examination of skin samples from OA2 individuals revealed that OA2 is morphologically distinct from OA1 40. Additionally, female carriers of OA2 do not exhibit characteristic fundus pattern seen in those of OA1 carriers. Based on molecular analysis, the defective gene that gives rise to this disorder is likely in Xp21.3-p21.2 for CACNA1F gene 41. The clinical findings, however, overlap with congenital stationary night blindness (CSNB2A). Due to similar features of OA2 and CSNB2A, Hawksworth et al 42 raised the question of whether these two entities are the same.
Hermansky-Pudlak Syndrome
Ocular albinism is a component of Hermansky-Pudlak syndrome (HPS), a rare autosomal recessive disorder that is further characterized by bleeding tendency due to platelet storage deficiency, interstitial lung disease, and granulomatous colitis (depending on the subtype) 43. Hermansky-Pudlak syndrome (HPS) is believed to be due to an abnormality in the formation of intracellular vesicles such as melanosomes and the dense bodies of platelets most commonly due to a mutation in HPS1 gene on chromosome 10 14. Because Hermansky-Pudlak syndrome (HPS) is seen most commonly among those of Swiss or Puerto Rican descent at the prevalence of 1:1800, it should be suspected in patients with albinism with the relevant ethnic background plus the associated systemic symptoms 44.
Chediak-Higashi Syndrome
Chediak-Higashi syndrome (CHS) is a rare autosomal recessive disorder boasts less than 500 reported cases in the literature in the past 20 years 45 and is among the triad of rare diseases grouped together as “silvery hair syndromes” 46. Chediak-Higashi syndrome (CHS) presents with variable but often intermediate form of hypopigmentation recurrent severe pyogenic infections that is ultimately lethal accompanied by progressive neurologic abnormalities, coagulation defects, and mucosal defects such as gingivitis, oral ulcerations, and periodontal diseases . The underlying defect of Chediak-Higashi syndrome (CHS) is a mutation in the lysosomal trafficking regulator in the CHS1/LYST gene 45. The resultant abnormal protein trafficking leads to dysfunctional fusion of vesicles and failure of transport of lysosomes to the appropriate intracellular site. The diagnosis should be considered in a young patient—many patients do not live past early childhood—presenting with oculocutaneous albinism (OCA) with the aforementioned systemic symptoms. Blood smear showing giant azurophilic granules in neutrophils, eosinophils, and other granulocytes is pathognomonic for Chediak-Higashi syndrome (CHS) 47. Bone marrow transplant is curative in some patients 48.
Griscelli Syndrome
Another member of the silvery hair syndrome triad, Griscelli syndrome (GS) is a rare autosomal-recessive disorder that results in hypopigmentation of skin and hair (silver hair), neurologic deficit, with or without immunologic impairment or hematophagocytic syndrome. The severity of the diseases is determined by the involved gene– MYO5A (GS type 1), RAB27A (GS type 2), and melanophilin (Mlph) (GS type 3). Griscelli syndrome (GS) can be distinguished from Chediak-Higashi syndrome (CHS) due to lack of giant intracellular granules that are seen in Chediak-Higashi syndrome.
Albinism causes
Albinism is caused by a genetic mutation that is usually passed from parents to child. Mutations in several different genes, on different chromosomes, can cause different types of albinism. A total of 15 genes are currently associated with various types of albinism 27 with as many as 729 mutations reported in the Human Gene Mutation Database associated with any of those loci 49. The mutation disrupts the production of melanin, the pigment that protects the skin from UV rays. Melanin is also important for the proper development of the eye. Without melanin, the retina and the optic nerve may not develop properly. The retina is the light-sensitive tissue lining the back of the eye, and the optic nerve fibers, help relay images to the brain.
Oculocutaneous albinism (OCA) can result from mutations in several genes, including TYR, OCA2, TYRP1, and SLC45A2. Changes in the TYR gene cause OCA type 1; mutations in the OCA2 gene are responsible for OCA type 2; TYRP1 mutations cause OCA type 3; and changes in the SLC45A2 gene result in OCA type 4. Mutations in additional genes likely underlie the other forms of this disorder (see Table 1 below). The genes associated with oculocutaneous albinism (OCA) are involved in producing a pigment called melanin, which is the substance that gives skin, hair, and eyes their color. In the retina, melanin also plays a role in normal vision. Mutations in any of these genes disrupt the ability of cells to make melanin, which reduces pigmentation in the skin, hair, and eyes. A lack of melanin in the retina leads to the vision problems characteristic of oculocutaneous albinism.
Alterations in the MC1R gene can change the appearance of people with oculocutaneous albinism type 2 (OCA-2). This gene helps regulate melanin production and is responsible for some normal variation in pigmentation. People with genetic changes in both the OCA2 and MC1R genes have many of the usual features of oculocutaneous albinism type 2, including light-colored eyes and vision problems; however, they typically have red hair instead of the usual yellow, blond, or light brown hair seen with this condition.
Some individuals with oculocutaneous albinism do not have mutations in any of the known genes. In these people, the genetic cause of the condition is unknown.
Oculocutaneous albinism (OCA) is inherited in an autosomal recessive manner (Figure 2). This means that two mutations are necessary for an individual to have OCA. Individuals normally have two copies of each numbered chromosome and the genes on them – one inherited from the father, the other inherited from the mother. Neither of these gene copies is functional in people with albinism. Each unaffected parent of an individual with an autosomal recessive condition carries one functional copy of the causative gene and one nonfunctional copy. They are referred to as carriers, and do not typically show signs or symptoms of the condition. Both parents must carry a defective OCA gene to have a child with albinism. When two individuals who are carriers for the same autosomal recessive condition have children, with each pregnancy there is a 25% (1 in 4) risk for the child to have the condition, a 50% (1 in 2) risk for the child to be an unaffected carrier like each of the parents, and a 25% chance for the child to not have the condition and not be a carrier.
Ocular albinism type 1 (OA-1) results from mutations in the GPR143 gene. The GPR143 gene provides instructions for making a protein that plays a role in pigmentation of the eyes and skin. It helps control the growth of melanosomes, which are cellular structures that produce and store a pigment called melanin. Melanin is the substance that gives skin, hair, and eyes their color. In the retina, this pigment also plays a role in normal vision. Most mutations in the GPR143 gene alter the size or shape of the GPR143 protein. Many of these genetic changes prevent the protein from reaching melanosomes to control their growth. In other cases, the protein reaches melanosomes normally but mutations disrupt the protein’s function. As a result of these changes, melanosomes in skin cells and the retina can grow abnormally large. Researchers are uncertain how these giant melanosomes are related to vision loss and other eye abnormalities in people with ocular albinism.
Rare cases of ocular albinism are not caused by mutations in the GPR143 gene. In these cases, the genetic cause of the condition is often unknown.
Ocular albinism type 1 (OA-1) is inherited in an X-linked pattern (Figure 3). A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome and one Y), one altered copy of the causative gene (GPR143 gene) in each cell is sufficient to cause the characteristic features of ocular albinism, because males do not have another X chromosome with a working copy of the gene. Because females have two copies of the X chromosome, women with only one copy of a GPR143 mutation in each cell usually do not experience vision loss or other significant eye abnormalities. They may have mild changes in retinal pigmentation that can be detected during an eye examination 50.
A less common type of ocular albinism (OA) shows a different pattern of inheritance, autosomal recessive. With this type of inheritance, both parents of a child with autosomal recessive ocular albinism carry the gene for it (Figure 2). Boys and girls are equally affected. If both mother and father carry the gene, then at each birth there is a one in four chance that the child will have ocular albinism. Newer research suggests that autosomal recessive ocular albinism (OA) is a variant of oculocutaneous albinism (OCA). The skin and hair color may be somewhat lighter than that of other family members. Autosomal recessive ocular albinism may be a variant of either tyrosinase-related (type 1) or P gene (type 2) oculocutaneous albinism.
Researchers have also identified several other genes in which mutations can result in albinism with other features. One group of these includes at least nine genes (on different chromosomes) leading to Hermansky-Pudlak syndrome (HPS). In addition to albinism, Hermansky-Pudlak syndrome (HPS) is associated with bleeding problems and bruising. Some forms are also associated with lung and bowel disease. Like oculocutaneous albinism (OCA), Hermansky-Pudlak syndrome (HPS) is inherited in an autosomal recessive manner.
Figure 2. Oculocutaneous albinism autosomal recessive inheritance pattern
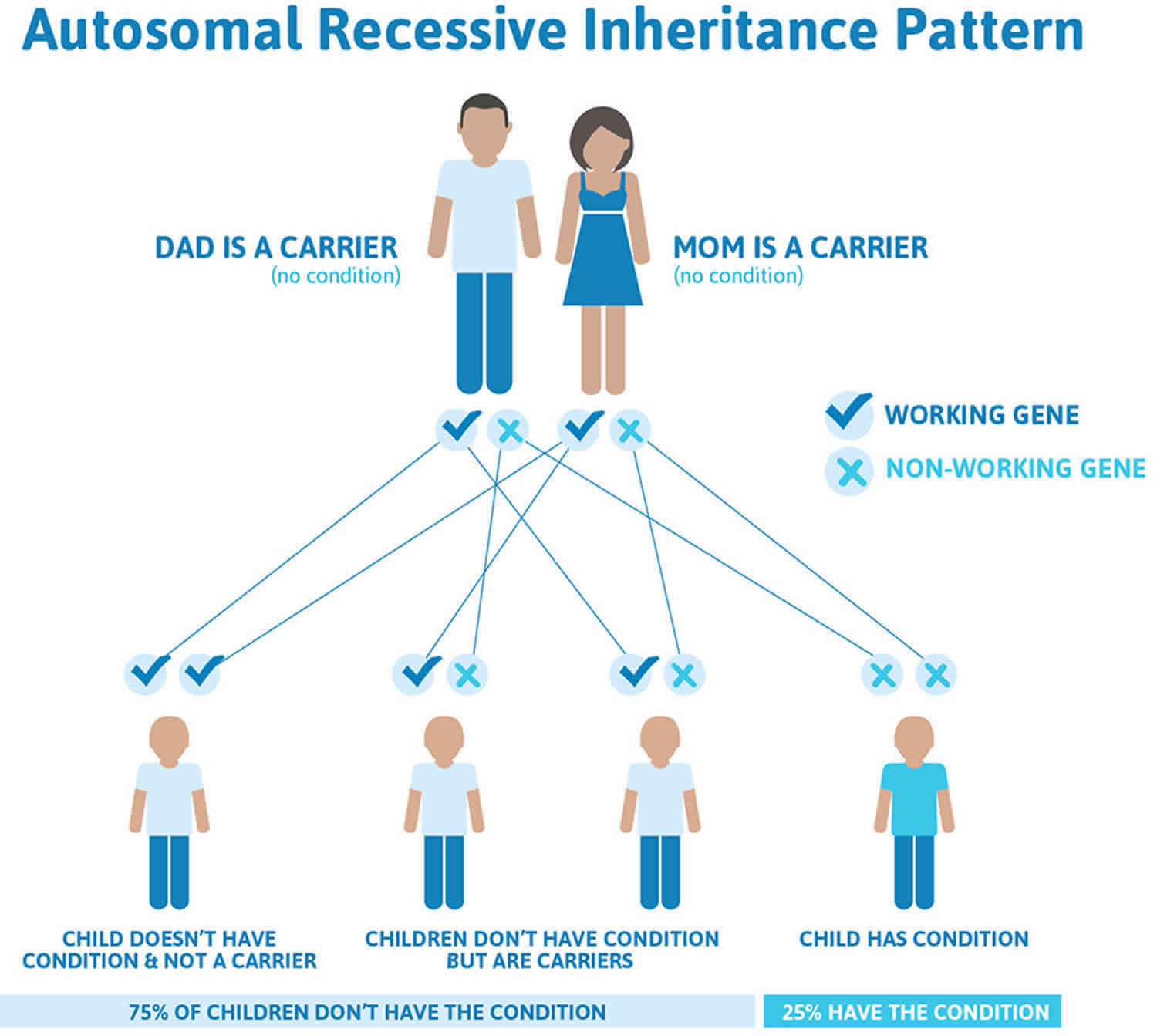
Footnotes: To have an autosomal recessive disorder, you inherit two mutated genes, one from each parent (who are carriers of the mutated gene). These disorders are usually passed on by two carriers. Carriers of the mutated gene is rarely affected, but they have one mutated gene (recessive gene) and one normal gene (dominant gene) for the condition. Two carriers have a 25% chance of having an unaffected child with two normal genes (left), a 50% chance of having an unaffected child who also is a carrier (middle), and a 25% chance of having an affected child with two recessive genes (right).
Figure 3. Ocular albinism X-linked inheritance pattern
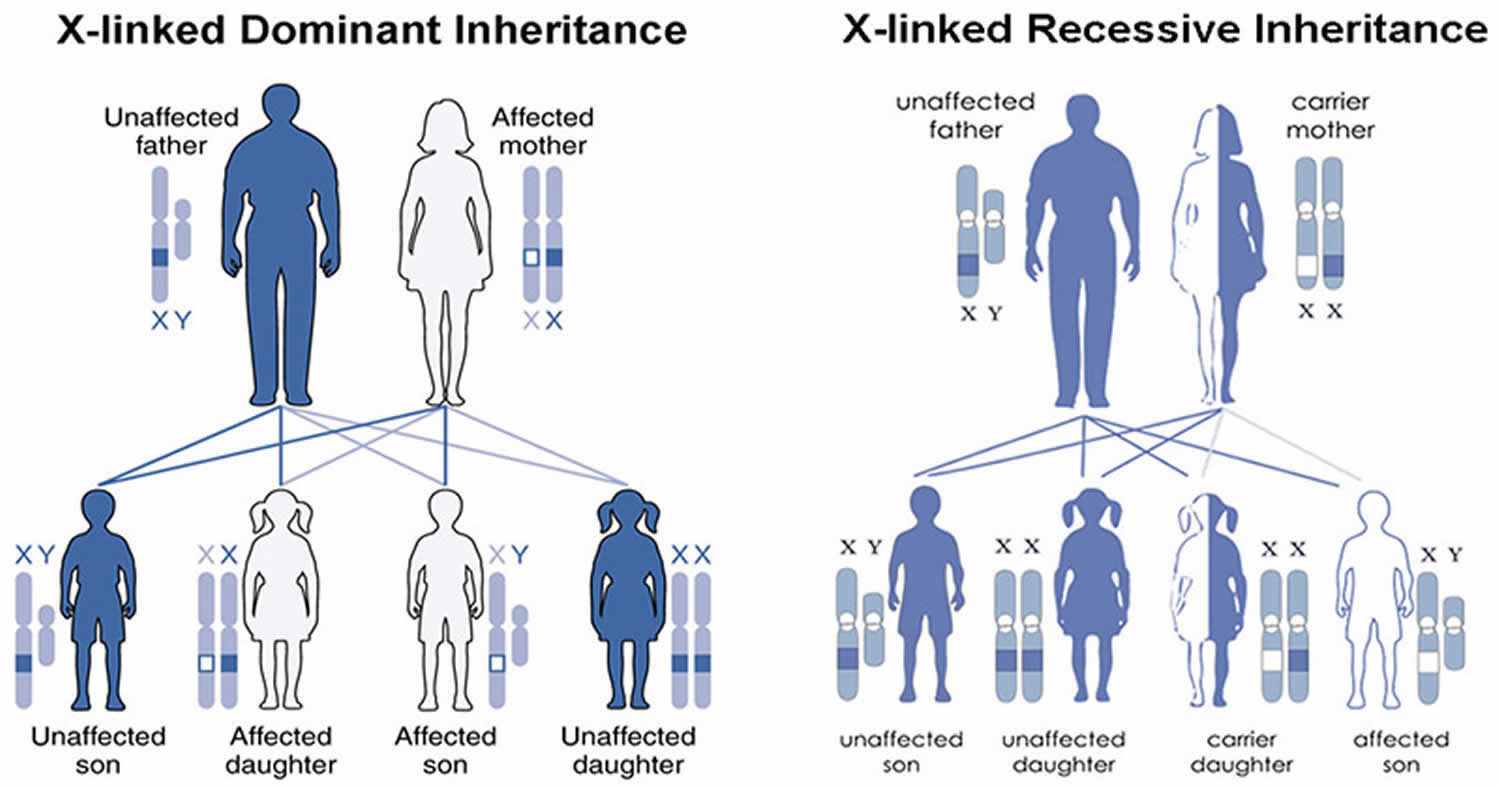
Footnotes: A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the GPR143 gene in each cell is sufficient to cause the characteristic features of ocular albinism. Because females have two copies of the X chromosome, women with only one copy of a GPR143 mutation in each cell usually do not experience vision loss or other significant eye abnormalities. They may have mild changes in retinal pigmentation that can be detected during an eye examination.
Albinism genetics
The genes for oculocutaneous albinism (OCA) are located on “autosomal” chromosomes. Autosomes are the 22 pairs of chromosomes that contain genes for our general body characteristics, compared to the one pair of sex chromosomes. You normally have two copies of these chromosomes and the many genes on them – one inherited from your father, the other inherited from your mother. For a recessive trait (like most types of albinism) to occur, both of the person’s chromosomes must carry that trait. That means that most types of albinism result from inheriting an albinism trait from both the mother and the father who often have typical pigmentation. In this case, the mother and father are considered to be carriers of the albinism trait because they each carry a recessive gene for the condition but do not manifest the condition themselves. When both parents carry the albinism gene (and neither parent has albinism) there is a one in four chance at each pregnancy that the baby will be born with albinism. This type of inheritance is called “autosomal recessive” inheritance.
Ocular albinism (OA1) is caused by a change in the GPR143 gene that plays a signaling role that is especially important to pigmentation in the eye. OA1 follows a simpler pattern of inheritance because the gene for OA1 is on the X chromosome. Females have two copies of the X chromosome while males have only one copy (and a Y chromosome that makes them male). To have ocular albinism, a male only needs to inherit one changed copy of the gene for ocular albinism from his carrier mother. Therefore almost all of the people with OA1 are males. Parents should be suspicious if a female child is said to have ocular albinism. While possible if the mother is a carrier of ocular albinism and the father has ocular albinism, it is extremely rare.
For couples who have not had a child with albinism, there is no simple test to determine whether a person carries a gene for albinism. Researchers have analyzed the DNA of many people with albinism and found the changes that cause albinism, but these changes are not always in exactly the same place, even for a given type of albinism. Moreover, many of the tests do not find all possible changes. Therefore, the tests for the albinism gene may be inconclusive. If parents have had a child with albinism previously, and if that affected child has had a confirmed diagnosis by DNA analysis, there is a way to test in subsequent pregnancies to see if the fetus has albinism. The test uses either amniocentesis (placing a needle into the uterus to draw off fluid) or chorionic villous sampling (CVS). Cells in the fluid are examined to see if they have an albinism gene from each parent.
For specific information and genetic testing, seek the advice of a qualified geneticist or genetic counselor. Those considering prenatal testing should be made aware that people with albinism usually adapt quite well to their disabilities and lead very fulfilling lives.
Table 1. Genes associated with albinism
| Full name | Abbreviation | Type | Gene/protein | Locus | Inheritance |
|---|---|---|---|---|---|
| Tyrosine negative | OCA 1A | OCA | Tyrosinase | 11q14-q21 | AR |
| Yellow variant | OCA 1B | OCA | Tyrosinase | 11q14-q21 | AR |
| Minimal pigment | OCA 1MP | OCA | Tyrosinase | 11q14-q21 | AR |
| Temperature sensitive | OCA 1TS | OCA | Tyrosinase | 11q14-q21 | AR |
| OCA Type 2 | OCA 2 | OCA | P gene / OCA2 | 15q11.2-q12 | AR |
| Brown albinism | BOCA | OCA | P gene / OCA2 | 15q11.2-q12 | 15q11.2-q12 |
| OCA Type 3 | OCA 3 / ROCA | OCA | Tyrosinase related protein (TYRP1) | 9p23 | AR |
| OCA Type 4 | OCA 4 | OCA | Membrane-associated transporter protein (MATP) = SLC45A2 | 5p13.3 | AR |
| OCA Type 5 | OCA 5 | OCA | Unknown | 4q24 | AR |
| OCA Type 6 | OCA 6 | OCA | SLC24A5 | 15q21.1 | AR |
| OCA Type 7 | OCA 7 | OCA | C10orf11 | 10q22 | AR |
| Oculoalbinism 1 aka Nettleship-Falls ocular albinism | OA 1 | OA | GPR143 | Xp22.3 | XR |
| Oculoalbinism 2 aka Forsius-Eriksson type ocular albinism | OA 2 | OA | CACNA1F | Xp21.3-p21.2 | XR |
Footnotes: *OMIM indicates Online Mendelian Inheritance in Man
Abbreviations: AD = autosomal dominant; AR = autosomal recessive; OA = ocular albinism; OCA = oculocutaneous albinism; XR = X-linked recessive.
[Source 14 ]People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Albinism prevention
Albinism is an inherited condition. If a family member has albinism, a genetic counselor can help you understand the type of albinism and the chances of having a future child with albinism. He or she can also explain the available tests.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Albinism symptoms
Signs and symptoms of albinism involve skin, hair, and eye color and vision. Some children with albinism are born with pinkish-white skin and white hair. Their eyes are usually light grey, blue or hazel, although they can look pink in the light.
People with albinism usually have poor vision. Glasses can help, but some have poor vision even with glasses. Some people with albinism also have nystagmus, which is involuntary flicking movements of the eye.
Skin
The most recognizable form of albinism results in white hair and very light-colored skin compared with siblings. Skin coloring (pigmentation) and hair color can range from white to brown, and may be nearly the same as that of parents or siblings without albinism.
With exposure to the sun, some people may develop:
- Freckles
- Moles, with or without pigment — moles without pigment are generally pink-colored
- Large freckle-like spots (lentigines)
- Sunburn and the inability to tan
For some people with albinism, skin pigmentation never changes. For others, melanin production may begin or increase during childhood and the teen years, resulting in slight changes in pigmentation.
Hair
Hair color can range from very white to brown. People of African or Asian descent who have albinism may have hair color that’s yellow, reddish or brown. Hair color may also darken by early adulthood or stain from exposure to normal minerals in water and the environment, and appear darker with age.
Symptoms of ocular albinism
Vision impairment is a key feature of all types of albinism. Eye problems and issues may include:
- Poor vision: Vision can range from normal for those minimally affected to legal blindness or worse (vision less than 20/200) for those with more severe forms of albinism. Near vision is often better than distance vision. Generally, those who have the least amount of pigment (i.e. most severely affected) have the poorest vision.
- Foveal hypoplasia (absence of a foveal pit) also known as macular hypoplasia: In albinism, the retina does not develop normally before birth and in infancy because of inappropriate retinal pigment epithelium (RPE) pigmentation that is required for macular development. Optical coherence tomography (OCT) can demonstrate an absence of the foveal pit and the loss of normal thinning of the retina. Also, the foveal avascular zone is small or nonexistent with vessels crossing the area 2 disc diameter temporal to the optic disc margin. Foveal hypoplasia is the single most important contributor to poor vision in albino people 14.
- Pendular nystagmus: Nystagmus refers to rhythmic, involuntary, flicker of the eyes (back-and-forth movement of the eyes). Affected infants may have large amplitude with low frequency pattern of eye movement starting at 2-3 months of age, later changing to a pendular form without distinct fast or slow phases. Head movements, such as bobbing or tilting the head, to try to reduce the involuntary eye movements and see better. Eye muscle surgery may be considered to reduce nystagmus.
- Sensitivity to light (photophobia) – an intolerance or sensitivity to bright light due to reduced or absent pigment in the iris. The iris in albinism has little to no pigment to screen out stray light coming into the eye resulting in scattering of light within the eye. On slit lamp exam, the examiner may detect speckled or diffuse transillumination defect. This finding, while common with albinism, is not specific as iris transillumination occurs in diseases unrelated to albinism such as pseudoexfoliation, pigment dispersion syndrome, megalocornea, iris atrophy, and Axenfeld-Rieger spectrum 14. When present in an otherwise normal individual, this finding may indicate carrier status of a hypomelanotic gene mutation. The iris may be translucent and the margin of the crystalline lens may be visible on transillumunation during slit lamp examination. Patients may prefer to wear sunglasses to reduce their sensitivity to light.
- Refractive errors: Both myopia (nearsightedness) and hyperopia (farsightedness) can occur, and astigmatism (abnormal curvature of the front surface of the eye or the lens inside the eye) is very common.
- Strabismus (‘squint’. ‘lazy eye’ or ‘turned eye’): Misalignment of the eyes and related anomalous head tilt can occur in association with albinism. Kumar et al 51 reported that the strabismus is seen in higher proportions of those with albinism compared to those with idiopathic infantile nystagmus, suggesting different mechanisms underlying the cause of strabismus in the two disorders.
- Visual pathway anomalies (misrouting of the optic nerve) may also be present, particularly problems with depth perception. Nerve signals from the retina to the brain that don’t follow the usual nerve pathways. Normally about half of optic nerve fibers from each eye decussates at the optic chiasm to the contralateral side, contributing to stereopsis. Albinism is associated with fiber over-decussation, resulting in crossing of up to 90% of fibers to the contralateral side and thus strabismus and loss of stereopsis. Evidence of this abnormality can be detected by 3-lead visual evoked potential (VEP) for proper counseling regarding visual potential of a patient.
Eye color
Eyelashes and eyebrows are often pale. Eye color can range from very light blue to brown and may change with age.
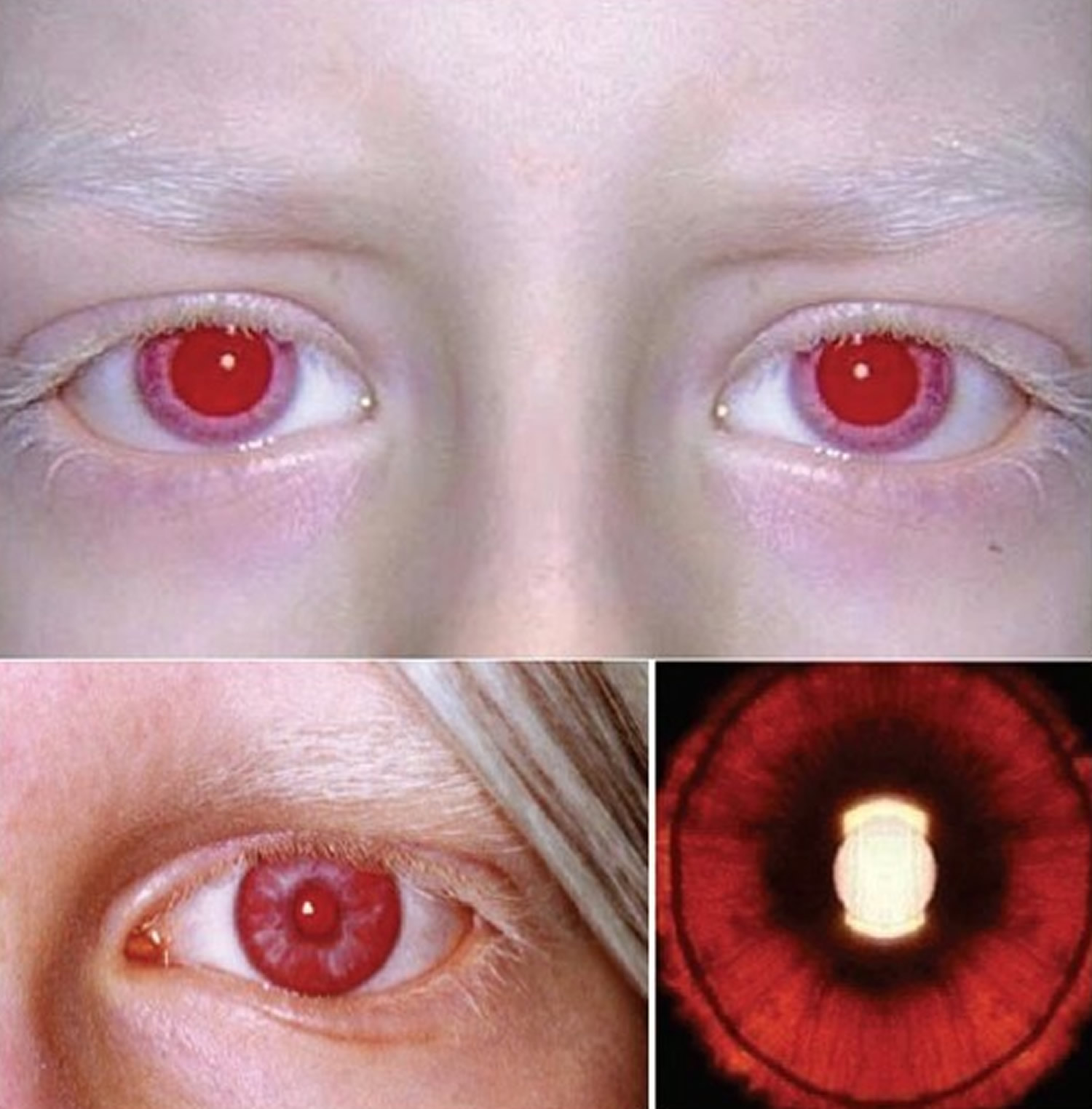
The lack of pigment in the colored part of the eyes (irises) makes the irises somewhat translucent. This means that the irises can’t completely block light from entering the eye. Because of this, very light-colored eyes may appear red in some lighting.
Figure 4. Albino person eyes
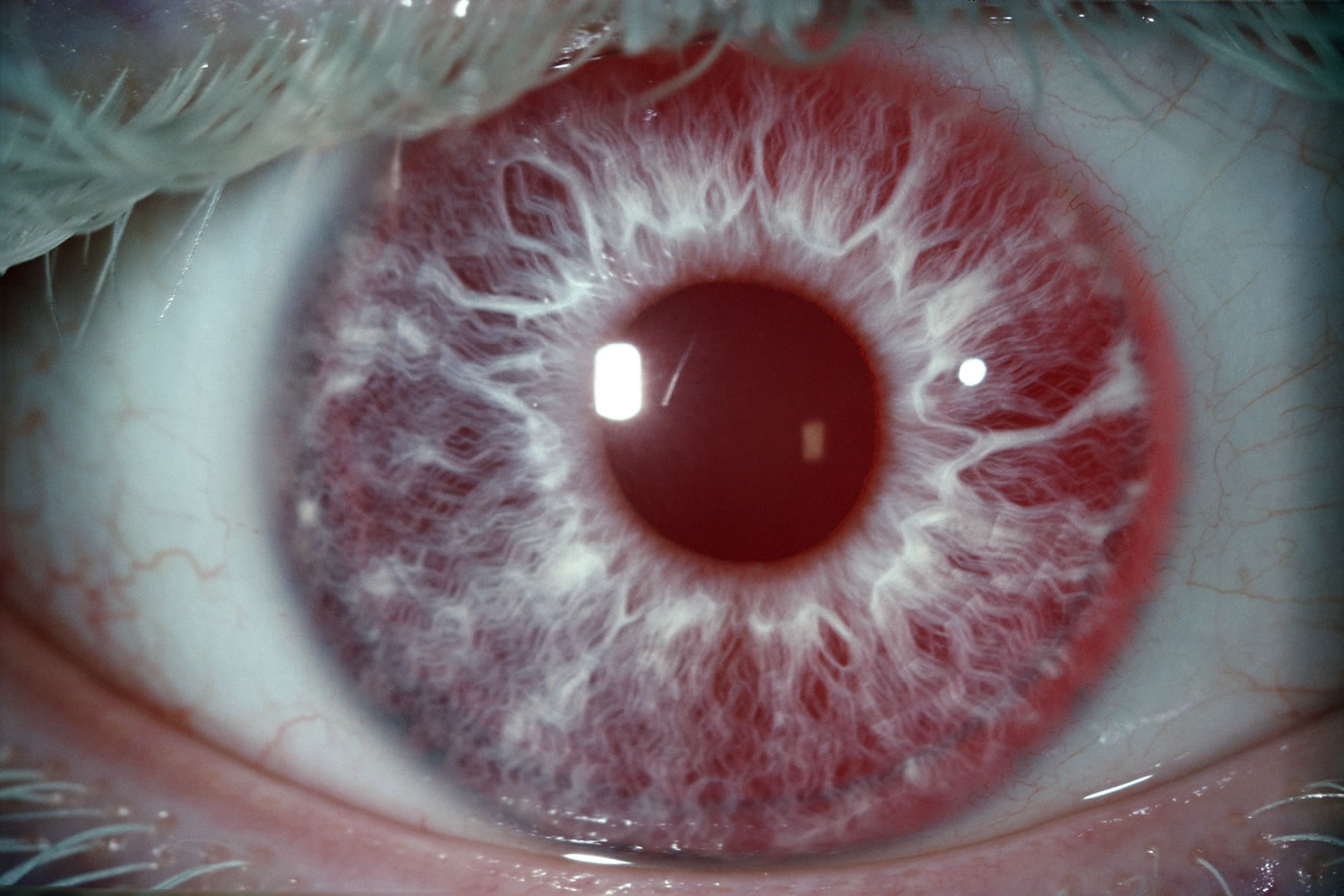
Albinism complications
Albinism can include skin and eye complications as well as social and emotional challenges.
Eye complications
People with albinism may be legally blind, but they can learn to use their vision over time. Problems with vision can impact learning, employment and the ability to drive. Some people may be able to correct problems with astigmatism, farsightedness and nearsightedness with eyeglasses or contacts.
Skin complications
People with albinism have skin that is very sensitive to light and sun exposure. Sunburn is one of the most serious complications associated with albinism because it can increase the risk of developing skin cancer and sun damage-related thickening of the skin.
Social and emotional challenges
Some people with albinism may experience discrimination. People with albinism are at an increased risk of isolation due to the social stigma behind the condition. The reactions of other people to those with albinism can often have a negative impact on people with the condition.
People with albinism may experience bullying, teasing or probing questions about their appearance, eyewear or visual aid devices. They usually look very different from members of their own families or ethnic groups, so they may feel like outsiders or be treated like outsiders. These experiences may contribute to social isolation, poor self-esteem and stress.
Using the term “person with albinism” is preferred to avoid the stigma of other terms.
Albinism diagnosis
Diagnosis of albinism is based on:
- A physical exam that includes checking skin and hair pigmentation
- A thorough eye exam
- Comparison of your child’s pigmentation to that of other family members
- Review of your child’s medical history, including whether there has been bleeding that doesn’t stop, excessive bruising or unexpected infections
A medical doctor specializing in vision and eye disorders (ophthalmologist) should conduct your child’s eye exam. The exam includes an assessment of potential nystagmus, strabismus and photophobia. The doctor also uses a device to visually inspect the retina and determine if there are signs of abnormal development.
Genetic consultation can help determine the type of albinism and the inheritance.
Researchers have identified some, but not all of the DNA defects in ocular albinism. Therefore blood tests to identify genes for various types of albinism are not conclusive enough to be used for genetic counseling. Most of the time an ophthalmologist can identify a carrier of the X-linked albinism gene by seeing mottling of pigment in her retinas. If this mottling is not apparent, research labs can examine a hair bulb or skin biopsy from a child. This will show unusually large granules of pigment in X-linked ocular albinism, but not in autosomal recessive ocular albinism.
Albinism treatment
Because albinism is a genetic disorder, albinism itself has no treatment. But some conditions that albino people have are treatable. Your care team may involve your primary care doctor and doctors specializing in eye care (ophthalmologist), skin care (dermatologist) and genetics. The goal of albinism treatment is to address the symptoms present in each individual. Treatment focuses on getting proper eye care and monitoring skin for signs of abnormalities.
Albinism treatment generally includes:
- Eye care. This includes receiving an annual eye exam by an ophthalmologist and most likely wearing prescription corrective lenses. Use low vision aids, such as a hand-held magnifying glass, a monocular or a magnifier that attaches to glasses, and a tablet synced to a smart board (an interactive electronic board with a touch screen) in the classroom. Although surgery is rarely part of treatment for eye problems related to albinism, your ophthalmologist may recommend surgery on optical muscles to minimize nystagmus. Surgery to correct strabismus may make the condition less noticeable.
- Skin care and prevention of skin cancer. This includes receiving an annual skin assessment to screen for skin cancer or lesions that can lead to cancer. An aggressive form of skin cancer called melanoma can appear as pink skin lesions.
People with Hermansky-Pudlak or Chediak-Higashi syndromes usually require regular specialized care to address medical needs and prevent complications.
People with albinism should protect their skin and eyes from the sun. This can be done by 5:
- strictly avoiding high-risk or prolonged sun exposure, such as being outside for long periods of time or in the middle of the day, at high altitudes, and on sunny days with thin cloud cover.
- using broad spectrum sunscreen that protects against both UVA and UVB light with a high sun protection factor (SPF) rating (50 or higher),
- wearing protective clothing, including clothes with color, such as long-sleeve, collared shirts, long pants and socks; broad-brimmed hats; and special UV-protection clothing.
- protecting eyes wearing dark, UV-blocking sunglasses or transition lenses (photochromic lenses) that darken in bright light.
Individuals with vision problems may need corrective lenses. For example, strabismus can be treated with glasses or surgery. Glasses can also help improve vision and reduce light sensitivity. For children with low vision, low vision aids such as hand-held magnifiers can help. Glasses with small telescopes attached are helpful for older children and adults. These lenses can help with both close and distant vision. They should also have regular follow-up exams with an ophthalmologist.
Patients with albinism are at higher risk for squamous cell and basal cell cancers in sun-exposed areas, and thus should be strongly encouraged to limit their sun exposure through avoiding outdoor occupations and through using sun-protective products such as appropriate clothing, large-brim hats, sunglasses, and sunscreen agents. Individuals with albinism should also have regular skin assessments to screen for skin cancer or lesions that can lead to cancer 53.
As with other heritable conditions, genetic testing and counseling support may offer invaluable support for families faced with albinism.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Ocular albinism treatment
There is currently no definitive treatment regimen; however ongoing ophthalmology care is needed. An early eye examination is required to detect high refractive errors and to facilitate early diagnosis and management of amblyopia. Patients with strabismus or nystagmus may benefit from eye muscle surgery, so identification of such patients early may help optimize visual function. Also, photochromic lenses are useful to alleviate photosensitivity and ultraviolet protection is important.
Low Vision Aids
There are dozens of different low vision aids to help improve the lives of people with albinism. Each aid has its own advantages and limitations. To get the most from a low vision aid, it’s important to see and understand what that device can and cannot do. Since different aids help with different tasks, the person with low vision will likely choose a mix of different low vision aids to accomplish all of his or her goals.
An optometrist or ophthalmologist who specializes in low vision evaluates the individual’s vision, and then recommends specific aids based on the patient’s needs and goals.
Tasks, goals, and technology change over time. Whether an individual has used low vision aids for years or just for a short time, the individual should periodically reevaluate the low vision aids he or she uses.
- Glasses: Glasses cannot fix low vision. Eyeglasses are not considered a low vision device unless they have a high reading “add” or contrast enhancement tint, but they deserve special mention here since glasses ensure that the person’s eyes see the clearest image possible and can easily focus at close distances. It is important for babies and young children with albinism to wear glasses to correct refractive error if they are nearsighted, farsighted, or have astigmatism. If a child with albinism who needs glasses doesn’t wear them, that child may tire easily and quickly give up doing certain tasks, and may even affect their development.
- Short viewing distances: Beginning in infancy, people with albinism instinctively hold things closer to their eyes to see them better. Up until the age of five or six, this technique sufficiently compensates for low vision because books for young children already feature large print, or they can sit closer to the TV. As a result of using this desire of moving closer to what needs to be seen, children this age generally do not need low vision aids. Looking closely at objects does not hurt the individual’s eyes, and this strategy continues throughout life.
- Near vision aids: Dome magnifiers, reading glasses, hand-held and stand magnifiers, and microscopes are near vision aids that help people with albinism read, look at pictures, diagrams, and maps, and accomplish other tasks that require seeing small details up close.Dome magnifiers are one of the easiest aids to use and arguably the most important for those with albinism [Figure 1]. Despite their seemingly modest ability to magnify print (1.7 to 2.2 times), in combination with a short viewing distance many reading tasks can be easily accomplished even in the primary grades of school. These magnifiers are also known as bright field magnifiers, paper weight magnifiers, and Visolett magnifiers.
- Reading glasses help the user focus on text or other objects while holding the object close to the user’s eyes. Reading glasses allow the widest field of view for reading compared to other aids.
- Hand-held and stand magnifiers enlarge close-up images, allowing the user to see small print and images at greater distances from the user’s eyes. There are many different styles and sizes of magnifiers useful for people with albinism.
- Microscopes help people see smaller details than magnifiers produce. Microscopes enlarge close-up objects the same way telescopes enlarge far-away objects. Some telescopes and bioptic telescopes, designed for distance vision, also allow the user to refocus the scope for up-close use.
- Distance vision aids: Telescopes can help people with low vision improve distance vision. Distance vision includes seeing a chalkboard in a classroom, seeing a menu board at a fast-food restaurant, or seeing the stage at a concert or the action at a sporting event. The two specifications that differentiate telescopes from one another are the magnification power and the field of view. Magnification power indicates how much larger an image appears through a telescope compared to how large it appears to the naked eye. Typical magnification powers for low vision telescopes range from 2x to 8x. When a person with 20/100 vision uses a 4x telescope, that person theoretically sees an image 4 times bigger than normal and, therefore, sees the same details a person with 20/25 vision sees. In general, the greater the magnification power of a telescope, the more fine detail the person with low vision will see using the telescope. However, as magnifying power increases, the field of view, or the size of the area enlarged by the telescope, generally decreases.
- One of the most common low vision aids is the hand-held telescope, also called a monocular. Hand-held telescopes come in a wide variety of sizes, magnification powers and prices. Hand held telescopes work best to quickly view a distant object, such as reading a sign or locating an object. Clip-on telescopes allow the user to slip the telescope over his or her glasses for hands-free use [Figure 4]. The clip-on telescope works well for the person who needs to use the telescope for extended time periods, such as for watching TV, a movie, or a live stage performance. The user can remove the clip-on telescope and use it as a hand-held telescope for quick viewing tasks.clip on telescope
- A bioptic is a special pair of glasses with a telescope permanently mounted in the glasses’ lens. While looking straight ahead, a bioptic user sees a normal, unmagnified image through the glasses. Then by dipping one’s head slightly, the bioptic user instantly sees a magnified image through the telescope. This “bi-optical” system allows the user to rapidly switch between a normal view and a magnified view without ever using his or her hands. In some areas, some people with low vision can use bioptics to drive. A person with albinism can also use bioptics in the same situations he or she might use a hand-held telescope, such as seeing a classroom chalkboard. Hands-free use can help older students take notes in class more quickly and easily. Some bioptic users prefer telescopes in both eyes, while many users with albinism choose to have only one mounted telescope for their dominant eye.
- One of the most common and simplest bioptic designs is from Designs for Vision. The Designs for Vision bioptic uses a telescope that extends about 3/4 to 1-1/2 inches from the front of the glasses’ lens and is 1/2 to 1 inch wide. Designs for Vision offers both fixed focus telescopes in a black metal housing and slightly larger focusable telescopes in a black plastic housing. They also make a bioptic telescope with a clear plastic housing to improve the bioptic’s appearance.
- To make using a bioptic more discreet, Edwards Optical created the BITA system. This bioptic uses telescopes that are only 1/2 to 3/4 inches long and about as wide as a pencil. The telescopes also extend behind the glasses lens, instead of in front of it. The BITA design is much less noticeable than traditional bioptic designs, but the small telescopes also produce a smaller field of view.
- Ocutech manufactures a bioptic system that mounts the telescope across the glasses’ bridge. This approach creates a wider field of view at higher magnification powers compared to conventional bioptic designs, plus offers a different appearance. Ocutech also manufactures an auto-focus bioptic, which automatically changes the focus as the user looks at different objects. The auto-focus bioptic is bulkier and more expensive than other bioptics.
- Electronic aids: When traditional optical low vision aids don’t help accomplish a task, electronic aids might help. Closed-Circuit Television (CCTV) systems help people who need greater magnification than reading glasses, magnifiers, and microscopes provide. CCTV systems also allow the user to adjust the size, brightness, and contrast of the magnified image to best match the user’s vision. The user can even read white letters on a black background to decrease glare. CCTV systems have historically been far more expensive and far less portable than other near vision aids. Newer CCTV designs, however, such as the MagniCam®, are now smaller, more versatile, more portable, and less expensive then previous CCTV systems. Other electronic aids help users see far away objects.
- Computers and Software: Desktop and laptop computers have advanced the ability of those with albinism to learn, communicate, and follow their chosen careers. Monitors are available in various sizes to suit an individual’s magnification needs. Magnification can be changed in common software such as Microsoft Outlook, Excel, and Word. This can be done in a variety of ways such as directly changing it on the toolbar, or by going to the toolbar clicking on the “View” option. Another method that is to hold down the Ctrl key on the keyboard and then turn the small wheel in the middle of the mouse away from you or towards you to change the print size on many word processing and HTML screens. Specialized software is also available such as Zoomer, ZoomText, Kurzweil, or Big Shot. These have many advanced features such as magnification of specific areas on a page, or a split screen option.
- Contrast enhancement aids: For people with albinism, bigger isn’t always better. Increasing the contrast of text is often more effective than increasing the size of text. Black felt-tipped pens and dark lined paper can make writing easier for some people with low vision. #1 pencils (as opposed to traditional #2 pencils) and bright-colored chalk can also help students with albinism.
- Writing guides, which are templates with open areas where one writes, can help people write in straight lines, or with tasks like writing checks. Colored filters can make it easier to see certain colors. For example, a yellow filter can make light blue letters appear darker and easier to read. Lighting plays a major role in how well a person with low vision sees. Experimenting with different types and brightness of light, as well as the location of the light, can make tasks a lot easier. Keep in mind that too much light and too little light can both cause problems.
Investigative treatments
- Nitisinone is an FDA-approved inhibitor of tyrosine degradation for hereditary tyrosinemia. Brooks and colleagues 54 hypothesized that the relative deficiency of tyrosinase in OCA type 1 may be reduced by increasing the concentration of tyrosine with nitisinone and thus improving pigmentation with OCA 1. In mice studies, nitisinone indeed improved pigmentation in fur and irides, suggesting potential for benefit in humans affected by OCA 1.
- A group by Martinez-Garcia et al 27 attempted to supplement L-DOPA, an intermediate metabolite of melanin synthesis pathway in mouse models. Various experiments suggested that L-DOPA supplementation may help overcome visual abnormalities associated with albinism 27. Phase 2 clinical trial (link to trial) is underway in the US to study the effects of L-Dopa supplementation in human subjects 55.
- A Japanese group suggested the use of aminoglycosides that can read through non-sense mutations for a potential treatment measure for common mutations found in albinism 56.
Coping and support
Making school or work adjustments
If your child has albinism, begin early to work with teachers and school administrators to take measures to help your child adapt to classroom learning. If necessary, start with educating the school professionals about albinism and how it affects your child. Also ask about services that the school or workplace offers to assess and meet needs.
Adjustments to the classroom or work environment that may help include:
- A seat near the front of the classroom
- Large-print textbooks or a tablet computer
- A tablet computer that can be synced to an interactive whiteboard (SMART board) at the front of the room, allowing the child to sit farther back in the classroom
- Handouts of the content written on boards or overhead screens
- High-contrast printed documents, such as black type on white paper, rather than using colored print or paper
- Enlarging font size on a computer screen
- Avoiding bright light in the learning or work setting
- Allowing extra time for taking tests or reading material
Help your child develop skills to deal with other people’s reactions to albinism. For example:
- Encourage your child to talk to you about experiences and feelings.
- Practice responses to teasing or embarrassing questions.
- Find a peer support group or online community through agencies such as the National Organization for Albinism and Hypopigmentation (NOAH).
- Talk to a mental health professional who can help you and your child develop healthy communication and coping skills, if needed.
Albinism prognosis
Most people with albinism live a normal lifespan and have the same types of medical problems as the rest of the population 57. Although the risk to develop skin cancer is increased, with careful surveillance, UV protection and prompt treatment, this is usually curable 58.
Patients may have children with no complications. Whether or not their children have albinism depends on the genetic makeup of their partner.
Albinism does not cause a delay in development or mental retardation.
Hermansky-Pudlak syndrome (HPS) can shorten a person’s lifespan due to lung disease or bleeding problems.
References- Cruz-Inigo AE, Ladizinski B, Sethi A. Albinism in Africa: stigma, slaughter and awareness campaigns. Dermatol Clin. 2011 Jan;29(1):79-87
- Schlessinger DI, Anoruo MD, Schlessinger J. Biochemistry, Melanin. [Updated 2021 May 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459156
- D’Alba L, Shawkey MD. Melanosomes: Biogenesis, Properties, and Evolution of an Ancient Organelle. Physiol Rev. 2019 Jan 1;99(1):1-19. doi: 10.1152/physrev.00059.2017
- Federico JR, Krishnamurthy K. Albinism. [Updated 2021 Aug 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519018
- Albinism. https://medlineplus.gov/ency/article/001479.htm
- What is albinism? https://aapos.org/glossary/albinism
- What is Albinism? https://www.albinism.org/information-bulletin-what-is-albinism/
- Del Bino, S., Duval, C., & Bernerd, F. (2018). Clinical and Biological Characterization of Skin Pigmentation Diversity and Its Consequences on UV Impact. International journal of molecular sciences, 19(9), 2668. https://doi.org/10.3390/ijms19092668
- Ismail, Rosnah & Ahmad, Salmiah & Aotd,. (2004). {PAGE } Sodium lactates in skin lightening formulations: its synergy with other skin lightening agents. https://www.researchgate.net/publication/307863602_PAGE_Sodium_lactates_in_skin_lightening_formulations_its_synergy_with_other_skin_lightening_agents
- Cruz-Inigo AE, Ladizinski B, Sethi A. Albinism in Africa: stigma, slaughter and awareness campaigns. Dermatol Clin. 2011 Jan;29(1):79-87.
- Boissy RE, Nordlund JJ. Molecular basis of congenital hypopigmentary disorders in humans: a review. Pigment Cell Res. 1997 Feb-Apr;10(1-2):12-24.
- RGiebel LB, Musarella MA, Spritz RA. A nonsense mutation in the tyrosinase gene of Afghan patients with tyrosinase negative (type IA) oculocutaneous albinism. J Med Genet. 1991 Jul;28(7):464-7.
- Grønskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007 Nov 2;2:43.
- Levin AV, Stroh E. Albinism for the busy clinician. J AAPOS. 2011 Feb;15(1):59-66. doi: 10.1016/j.jaapos.2010.10.012
- King RA, Wirtschafter JD, Olds DP, Brumbaugh J. Minimal pigment: a new type of oculocutaneous albinism. Clin Genet. 1986 Jan;29(1):42-50.
- Kono M, Kondo T, Ito S, Suzuki T, Wakamatsu K, Ito S, Tomita Y. Genotype analysis in a patient with oculocutaneous albinism 1 minimal pigment type. Br J Dermatol. 2012 Apr;166(4):896-8.
- Lund PM. Oculocutaneous albinism in southern Africa: population structure, health and genetic care. Ann Hum Biol. 2005 Mar-Apr;32(2):168-73. Review
- Spritz RA, Oh J, Fukai K, Holmes SA, Ho L, Chitayat D, France TD, Musarella MA, Orlow SJ, Schnur RE, Weleber RG, Levin AV. Novel mutations of the tyrosinase (TYR) gene in type I oculocutaneous albinism (OCA1). Hum Mutat. 1997;10(2):171-4.
- Fridman C, Hosomi N, Varela MC, Souza AH, Fukai K, Koiffmann CP. Angelman syndrome associated with oculocutaneous albinism due to an intragenic deletion of the P gene.Am J Med Genet A. 2003 Jun 1;119A(2):180-3.
- King RA, Lewis RA, Townsend D, Zelickson A, Olds DP, Brumbaugh J. Brown oculocutaneous albinism. Clinical, ophthalmological, and biochemical characterization. Ophthalmology. 1985 Nov;92(11):1496-505.
- Manga P, Kromberg J, Turner A, Jenkins T, Ramsay M. In Southern Africa, brown oculocutaneous albinism (BOCA) maps to the OCA2 locus on chromosome 15q: P-gene mutations identified. Am J Hum Genet. 2001;68:782–7.
- Zhang KH, Li Z, Lei J, Pang T, Xu B, Jiang WY, Li HY. Oculocutaneous albinism type 3 (OCA3): analysis of two novel mutations in TYRP1 gene in two Chinese patients. Cell Biochem Biophys. 2011 Dec;61(3):523-9.
- Kromberg JG, Castle DJ, Zwane EM, Bothwell J, Kidson S, Bartel P, Phillips JI, Jenkins T. Red or rufous albinism in southern Africa. Ophthalmic Paediatr Genet. 1990 Sep;11(3):229-35.
- Manga P, Kromberg JG, Box NF, Sturm RA, Jenkins T, Ramsay M. Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. Am J Hum Genet. 1997 Nov;61(5):1095-101.
- Jimbow K. Biological role of tyrosinase-related protein and its relevance to pigmentary disorders (vitiligo vulgaris). J Dermatol. 1999 Nov;26(11):734-7.
- Newton JM, Cohen-Barak O, Hagiwara N, Gardner JM, Davisson MT, King RA, Brilliant MH. Mutations in the human orthologue of the mouse underwhite gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am J Hum Genet. 2001 Nov;69(5):981-8.
- Mártinez-García M, Montoliu L. Albinism in Europe. J Dermatol. 2013 May;40(5):319-24.
- Kausar T, Bhatti MA, Ali M, Shaikh RS, Ahmed ZM. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet. 2013 Jul;84(1):91-3.
- Wei AH, Zang DJ, Zhang Z, Liu XZ, He X, Yang L, Wang Y, Zhou ZY, Zhang MR, Dai LL, Yang XM, Li W. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J Invest Dermatol. 2013 Jul;133(7):1834-40.
- Grønskov K, Dooley CM, Østergaard E, Kelsh RN, Hansen L, Levesque MP, Vilhelmsen K, Møllgård K, Stemple DL, Rosenberg T. Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism.Am J Hum Genet. 2013 Mar 7;92(3):415-21.
- Morice-Picard F, Lasseaux E, François S, Simon D, Rooryck C, Bieth E, Colin E, Bonneau D, Journel H, Walraedt S, Leroy BP, Meire F, Lacombe D, Arveiler B. SLC24A5 Mutations Are Associated with Non-Syndromic Oculocutaneous Albinism. J Invest Dermatol. 2013 Aug 28.
- Montoliu L, Grønskov K, Wei AH, Martínez-García M, Fernández A, Arveiler B, Morice-Picard F, Riazuddin S, Suzuki T, Ahmed ZM, Rosenberg T, Li W. Increasing the complexity: new genes and new types of albinism. Pigment Cell Melanoma Res. 2013 Sep 21.
- Rosenberg T, Schwartz M. X-linked ocular albinism: prevalence and mutations–a national study. Eur J Hum Genet. 1998;6(6):570.
- Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, Bruttini M, Gebbia M, Bergen AA, Lewis RA, Ballabio A. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet. 1995 May;10(1):13-9.
- Shiono T, Tsunoda M, Chida Y, Nakazawa M, Tamai M. X linked ocular albinism in Japanese patients. Br J Ophthalmol. 1995;79(2):139.
- Lang GE, Rott HD, Pfeiffer RA. X-linked ocular albinism. Characteristic pattern of affection in female carriers. Ophthalmic Paediatr Genet. 1990 Dec;11(4):265-71.
- Schnur RE, Gao M, Wick PA, Keller M, Benke PJ, Edwards MJ, Grix AW, Hockey A, Jung JH, Kidd KK, Kistenmacher M, Levin AV, Lewis RA, Musarella MA, Nowakowski RW, Orlow SJ, Pagon RS, Pillers DA, Punnett HH, Quinn GE, Tezcan K, Wagstaff J, Weleber RG. OA1 mutations and deletions in X-linked ocular albinism. Am J Hum Genet. 1998 Apr;62(4):800-9.
- Winship IM, Babaya M, Ramesar RS. X-linked ocular albinism and sensorineural deafness: linkage to Xp22.3. Genomics. 1993 Nov;18(2):444-5.
- Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol. 1999;38(9):656.
- O’Donnell FE, Green WR, McKusick VA, Forsius H, Eriksson AW. Forsius-Eriksson syndrome: its relation to the Nettleship-Falls X-linked ocular albinism. Clin Genet. 1980 Jun;17(6):403-8.
- Pillers DA, Towbin JA, Chamberlain JS, Wu D, Ranier J, Powell BR, McCabe ER. Deletion mapping of Aland Island eye disease to Xp21 between DXS67 (B24) and Duchenne muscular dystrophy. Am J Hum Genet. 1990 Nov;47(5):795-801.
- Hawksworth NR, Headland S, Good P, Thomas NS, Clarke A. Aland island eye disease: clinical and electrophysiological studies of a Welsh family. Br J Ophthalmol. 1995 May;79(5):424-30.
- Seward SL Jr, Gahl WA. Hermansky-Pudlak syndrome: health care throughout life. Pediatrics. 2013 Jul;132(1):153-60.
- Wildenberg SC, Oetting WS, Almodóvar C, Krumwiede M, White JG, King RA. A gene causing Hermansky-Pudlak syndrome in a Puerto Rican population maps to chromosome 10q2. Am J Hum Genet. 1995;57(4):755–765.
- Kaplan J, De Domenico I, Ward DM. Chediak-Higashi syndrome. Curr Opin Hematol. 2008 Jan;15(1):22-9.
- Reddy RR, Babu BM, Venkateshwaramma B, Hymavathi Ch. Silvery hair syndrome in two cousins: Chediak-Higashi syndrome vs Griscelli syndrome, with rare associations. Int J Trichology. 2011 Jul;3(2):107-11.
- Antunes H, Pereira A, Cunha I. Chediak-Higashi syndrome: pathognomonic feature. Lancet. 2013 Nov 2;382(9903):1514.
- Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, Barbosa E, Falik-Borenstein T, Filipovich A, Ishida Y, Kivrikko S, Klein C, Kreuz F, Levin A, Miyajima H, Regueiro J, Russo C, Uyama E, Vierimaa O, Spritz RA. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak-Higashi syndrome. Am J Med Genet. 2002 Feb 15;108(1):16-22.
- Cooper DN, Ball EV, Krawczak M. The human gene mutation database. Nucleic Acids Res. 1998 Jan 1;26(1):285-7.
- Ocular albinism. https://medlineplus.gov/genetics/condition/ocular-albinism/
- Kumar A, Gottlob I, McLean RJ, Thomas S, Thomas MG, Proudlock FA. Clinical and oculomotor characteristics of albinism compared to FRMD7 associated infantile nystagmus. Invest Ophthalmol Vis Sci. 2011 Apr 8;52(5):2306-13.
- Hertle, Richard. (2013). Albinism: Particular Attention to the Ocular Motor System. Middle East African journal of ophthalmology. 20. 248-255. 10.4103/0974-9233.114804
- Albinism. https://www.mayoclinic.org/diseases-conditions/albinism/symptoms-causes/syc-20369184
- Onojafe IF, Adams DR, Simeonov DR, Zhang J, Chan CC, Bernardini IM, Sergeev YV, Dolinska MB, Alur RP, Brilliant MH, Gahl WA, Brooks BP. Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism. J Clin Invest. 2011 Oct;121(10):3914-23.
- Lavado A, Jeffery G, Tovar V, de la Villa P, Montoliu L. Ectopic expression of tyrosine hydroxylase in the pigmented epithelium rescues the retinal abnormalities and visual function common in albinos in the absence of melanin. J Neurochem. 2006 Feb;96(4):1201-11.
- K. Fukai, H. Kunimoto, K. Nakajima, T. Suzuki, M. Ishii. In vitro analysis of read-through effect of aminoglycosides to tyrosinase R278X nonsense mutation in melan-c cells. The 24th Annual Meeting of the Japanese Society for Pigment Cell Research (JSPCR-2012) 19 OCT 2012.
- Albinism. https://emedicine.medscape.com/article/1200472-overview#a2
- Chalya PL, Gilyoma JM, Kanumba ES, Mawala B, Masalu N, Kahima JK, Rambau P: Dermatological malignancies at a University Teaching Hospital in north-western Tanzania: a retrospective review of 154 cases. Tanzania Journal of Health Research 2021, 14(1):1-7.





{kind=link}