Alstrom syndrome
Alstrom syndrome also called Alstrom-Hallgren syndrome, is a ultra-rare complex genetic disorder with a wide variety of symptoms affecting multiple organ systems of the body 1, where survival beyond the age of 50 is rare 2. Many of the signs and symptoms of Alstrom syndrome begin in infancy or early childhood, although some appear later in life. Symptoms develop gradually, beginning in infancy, and can be variable. In childhood, Alstrom syndrome is generally characterized by vision and hearing abnormalities, childhood obesity, and heart disease (dilated cardiomyopathy). Over time, diabetes mellitus, liver problems, and slowly progressive kidney dysfunction which can lead to kidney failure may develop 3. Some individuals with Alström syndrome have a skin condition called acanthosis nigricans, which causes the skin in body folds and creases to become thick, dark, and velvety. Additional symptoms including lung (pulmonary), liver (hepatic), kidney (renal), and endocrine dysfunction can also occur. Although some children may experience delays in attaining developmental milestones, intelligence is usually unaffected. Alström syndrome is caused by mutations in the ALMS1 gene. The protein encoded by this gene has been implicated in ciliary function, cell cycle control, and intracellular transport. Alstrom syndrome is inherited in an autosomal recessive manner 1.
Alstrom syndrome affects males and females in equal numbers. The exact incidence is unknown. Estimates have ranged from 1 in 10,000 to less than 1 in 1,000,000 individuals in the general population. Approximately 1200 affected individuals have been identified worldwide 3. Because some cases of Alstrom syndrome may go unrecognized or misdiagnosed, the disorder may be under-diagnosed, making it difficult to determine its true frequency in the general population.
While there is no specific treatment for Alström syndrome, symptoms can be managed by a team of specialists with the goal of improving the quality of life and increasing the lifespan 4.
Alstrom syndrome treatment is focused on managing the symptoms present in each individual. This may involve a team of specialists including but not limited to: pediatricians, cardiologists, audiologists (hearing specialists), ophthalmologists, endocrinologists, and orthopaedists 3.
Treatment may include 5:
- Specially-tinted, prescription glasses and vision aids to assist with vision loss
- Hearing aids and cochlear implants for hearing loss
- Dietary measures, exercise programs, and oral medications and/or insulin to control diabetes
- ACE inhibitors and other medications to manage heart and kidney problems with some individuals requiring a kidney or heart transplant
- Hormone therapy if the male testes or female ovaries produce lower than average levels.
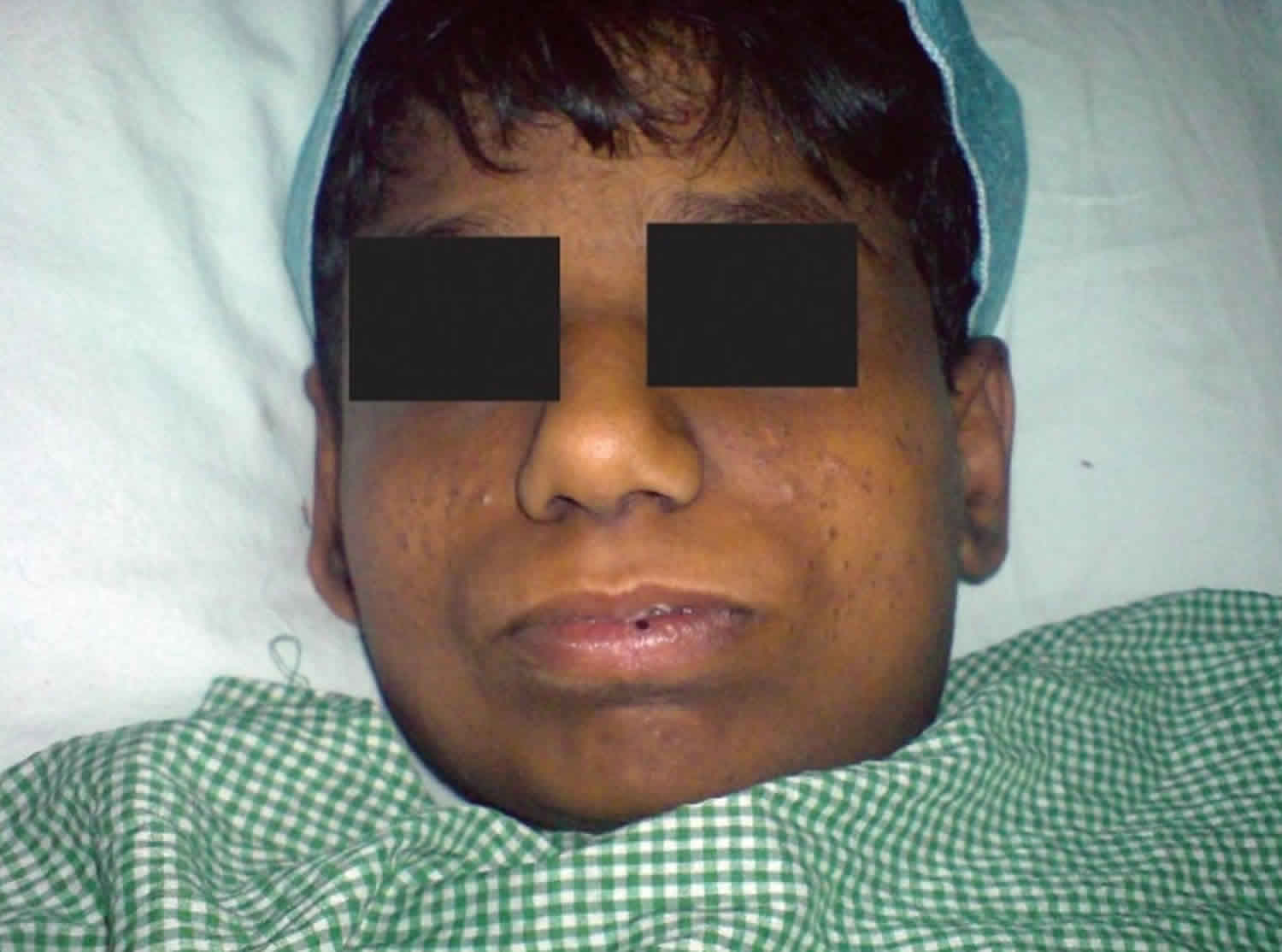
Figure 1. Alstrom syndrome
Footnote: Obesity is an early and consistent feature observed in most children with Alstrom syndrome. A, B. Clinical pictures of a male with Alstrom syndrome at the age of 6 years, 8 months presenting characteristic truncal obesity. C. Note characteristic face and prominent ears.
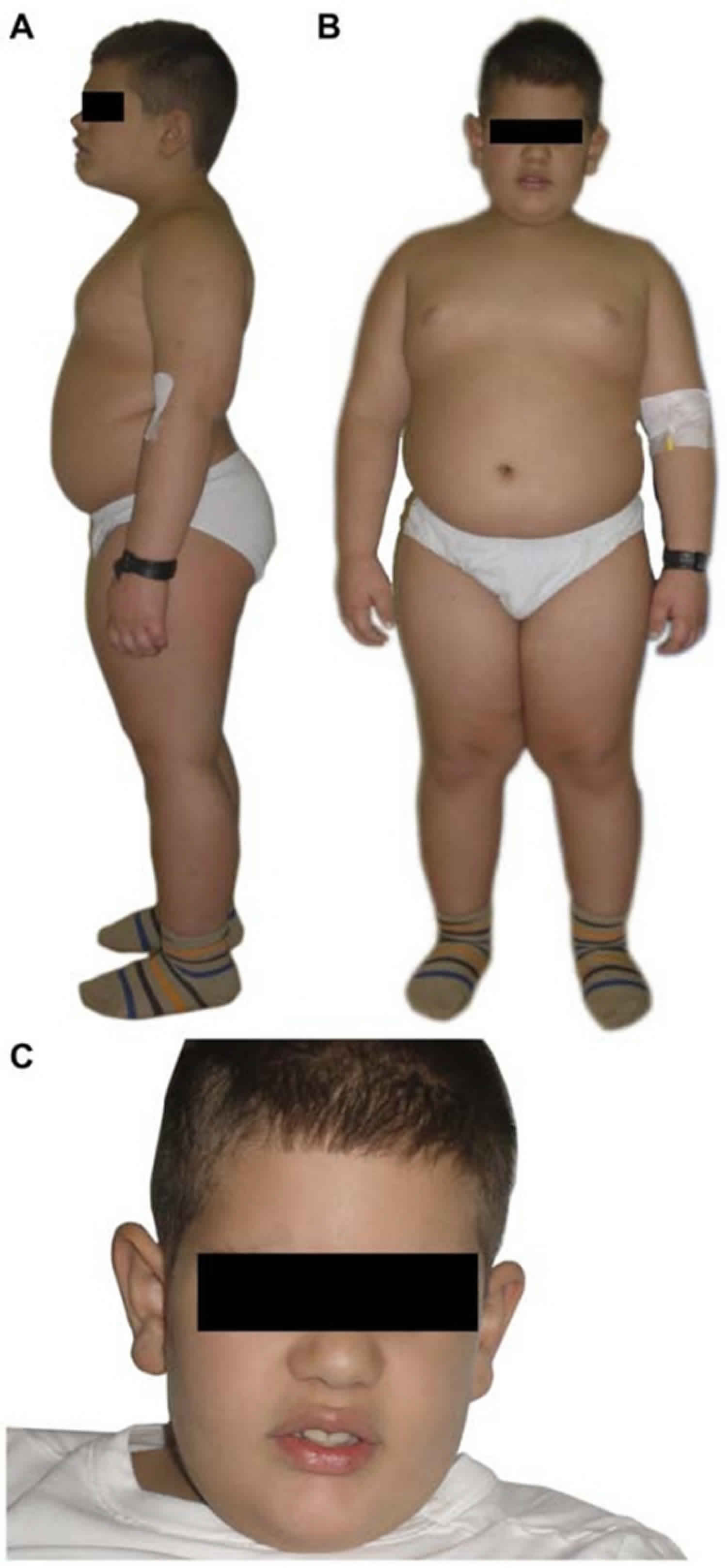
[Source 6 ]Figure 2. Alstrom syndrome

Footnote: A. Close-up of broad hands with stubby fingers and brachydactyly from a 3 year, 3 month old child with Alstrom syndrome. B. Pes planus typically seen in Alstrom syndrome.
[Source 6 ]What percentage of patients with Alstrom syndrome become deaf?
Progressive sensorineural hearing loss is found in 88% of individuals with Alström syndrome 5. About 10% of affected indivduals progress to profound deafness and must rely on sign language for communication 7.
What is the average age of deafness in patients with Alstrom syndrome?
In up to 70% of individuals with Alström syndrome, hearing loss becomes evident between ages one and ten years. The average age of onset is 9 years old but can range from 2 years to 25 years old 5. Hearing loss may progress to be more severe by the end of the first to second decade 5.
Alstrom syndrome causes
Mutations in the ALMS1 gene cause Alström syndrome. The ALMS1 gene provides instructions for making a protein whose function is not fully understood, but believed to be involved in ciliary function, cell cycle control, and intracellular transport 3. Mutations in ALMS1 gene probably lead to the production of an abnormally short, nonfunctional version of the ALMS1 protein. This protein is normally present at low levels in most tissues, so a loss of the protein’s normal function may help explain why the signs and symptoms of Alström syndrome affect many parts of the body.
Some research has indicated that the protein encoded by the ALMS1 gene has a role in the proper function, formation, and/or maintenance of cilia, the hair-like structures that can be found in almost all types of cells in the body, and possibly some related structures such as the basal body (which “anchors” the cilia to a cell). A related disorder known as Bardet-Biedl syndrome has also been linked to ciliary dysfunction. Several disorders, referred to as ciliopathies, have been linked to ciliary dysfunction. More research is necessary to determine what role, if any, that cilia and related structures play in the development of Alstrom syndrome.
Investigators have determined that the ALMS1 gene is located on the short arm (p) of chromosome 2 (2p13) 3. Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 2q13” refers to band 13 on the long arm of chromosome 2. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
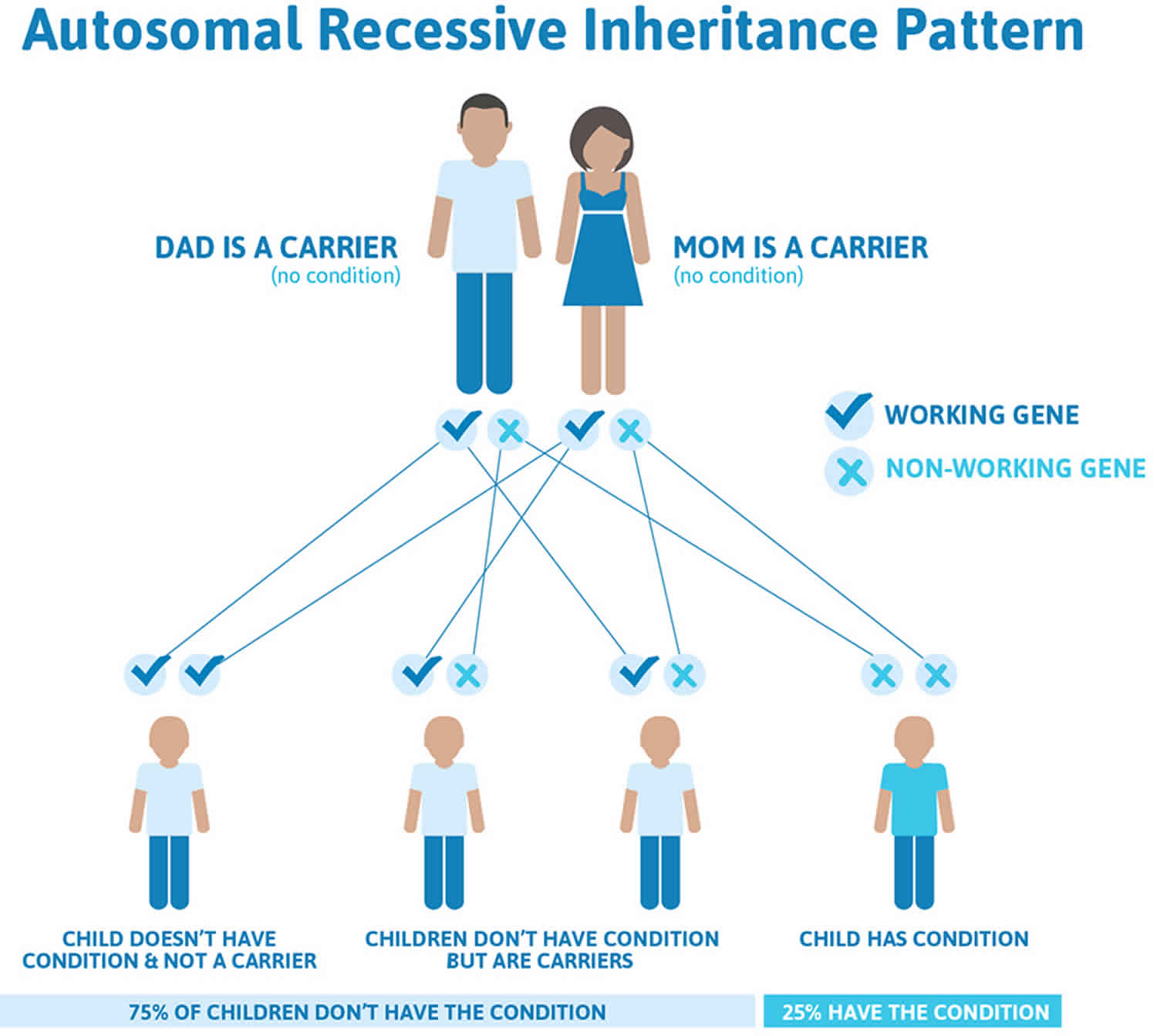
Alstrom syndrome inheritance pattern
Alstrom syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Affected people inherit one mutated copy of the gene from each parent, who is referred to as a carrier. Carriers of an autosomal recessive condition typically do not have any signs or symptoms (they are unaffected). When 2 carriers of an autosomal recessive condition have children, each child has a:
- 25% (1 in 4) chance to be affected
- 50% (1 in 2) chance to be an unaffected carrier like each parent
- 25% (1 in 4) chance to be unaffected and not be a carrier
Figure 3. Alstrom syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Alstrom syndrome symptoms
Alstrom syndrome may potentially affect several different organ systems of the body. The signs and symptoms of Alstrom syndrome vary among affected individuals, even among members of the same family. The age that symptoms begin also varies. Symptoms may first appear anywhere from infancy to early adulthood 3.
Alstrom syndrome signs and symptoms may include 3:
- Vision abnormalities, specifically cone-rod dystrophy and cataracts
- Progressive sensorineural hearing loss in both ears and chronic infection or inflammation of the middle ear
- Heart disease that enlarges and weakens the heart muscle (dilated cardiomyopathy)
- Excessive eating (hyperphagia) and rapid weight gain leading to obesity
- Insulin resistance leading to high levels of insulin in the blood (hyperinsulinemia) and type 2 diabetes mellitus
- Elevated levels of fats (lipids) in the blood (hyperlipidemia)
- Fatty liver that may progress to significant liver disease
- Short stature
- Skin findings including abnormally increased coloration and “velvety” thickening of the skin in certain areas of the body (acanthosis nigricans)
- Lower hormone levels produced by the male testes or the female ovaries (hypogonadism)
Alstrom syndrome can also cause serious or life-threatening medical problems involving the liver, kidneys, bladder, and lungs 3.
It is important to note that affected individuals will not have all of the symptoms discussed above and individual cases may be dramatically different. Some symptoms may present in the first weeks of life, others symptoms may not develop until adolescence or early adulthood.
Individuals with Alstrom syndrome often develop vision abnormalities, specifically cone-rod dystrophy, between birth and 15 months of age. Cone-rod dystrophy is a form of retinal dysfunction. The retina is the light-sensitive membrane upon which images are focused at the back of the eye. In affected individuals, the cells in the retina (cones and rods [photoreceptors]) that convert light into nerve impulses gradually deteriorate (cone-rod dystrophy), causing vision loss. In addition to visual impairment, affected individuals may develop severe sensitivity of the eyes to light (photophobia) and rapid, involuntary eye movements (nystagmus). The progression and degree of visual impairment varies among affected individuals. Some individuals may also develop clouding of the lenses of the eyes (cataracts). In most cases, vision becomes progressively worse through the first and second decade and may result in blindness by the mid-teens. Some individual are able to read large print into their third decade.
Hearing may also be affected in Alstrom syndrome. Hearing is usually normal at birth, but sometime during the first decade of life, progressive sensorineural hearing loss may affect both ears (bilateral) in approximately 70% of patients. Sensorineural hearing loss is caused by an impaired ability of the auditory nerves to transmit sensory input to the brain. Hearing loss may be mild to moderate in degree or may progress to severe or moderately severe by the end of the first or second decade of life. Chronic infection or inflammation of the middle ear (otitis media) may also occur. Some individuals may develop the accumulation of thick, sticky fluid behind the eardrum (glue ear). Long-standing glue ear can cause conductive hearing loss in some cases. Conductive hearing loss is cause by the blockage of sound waves.
While vision and hearing are affected in individuals with Alstrom syndrome, intelligence is usually unaffected. Some infants and children may experience delays in reaching developmental milestones such as crawling or walking. Some children may have delays in developing certain language skills or develop learning disabilities.
Birth weight is normal in infants with Alstrom syndrome, but excessive eating beyond the normal need to satisfy hunger (hyperphagia) and rapid weight gain may occur during the first year of life. Some affected children develop childhood truncal obesity, a condition in which fat is disproportionately distributed on the abdomen and chest rather than the arms and legs. As affected individuals age, some may see their body weight fall, often regaining normal or slightly above-average weight for their size.
In childhood, height is typically normal or above normal. As the child grows into adolescence and adulthood, growth slows and final adult height is below the 50th percentile.
More than 60 percent of children with Alstrom syndrome develop a condition known as dilated cardiomyopathy, in which weakening of the myocardium–the heart muscle forming the walls of the heart chambers–leads to enlargement (dilatation) of the heart’s lower chambers (ventricles). Dilated cardiomyopathy may not be associated with any symptoms initially, but eventually leads to weakening of the heart’s pumping action, which impairs the circulation of blood through the lungs and the rest of the body resulting in fluid buildup in the heart, lung and various body tissues (congestive heart failure).
Associated symptoms and findings may depend upon the degree of heart failure, the affected child’s age, and other factors. For example, in some infants, signs of heart failure may include feeding difficulties and poor weight gain, irritability, excessive sweating; labored, rapid breathing (tachypnea); bluish discoloration of the skin and mucous membranes due to abnormally low levels of circulating oxygen (cyanosis), among other findings. Children with heart failure may develop fatigue; shortness of breath (dyspnea), coughing, lack of appetite (anorexia); or abdominal pain.
The onset, severity and progression of dilated cardiomyopathy vary greatly even among members of the same family. Dilated cardiomyopathy can develop during infancy or in early adulthood. In some cases, it has preceded the development of the characteristic eye abnormalities of Alstrom syndrome. In older children and adults, restrictive cardiomyopathy can develop.
Affected children often experience insulin resistance, a condition in which the body fails to react to insulin. Insulin is a hormone secreted by the pancreas that regulates blood glucose levels by promoting the movement of glucose into cells for energy production or into the liver and fat cells for storage. Glucose is a simple sugar that is the body’s primary source of energy for cell metabolism. In response to insulin resistance, the pancreas secretes more insulin, resulting in abnormally high levels of insulin in the blood (hyperinsulinemia).
Individuals with Alstrom syndrome eventually develop type 2 diabetes mellitus, although the age of onset varies. Children as young as five have developed type 2 diabetes mellitus. In this form of diabetes, the pancreas produces insulin but the body becomes resistant to its effects, leading to insufficient absorption of glucose and abnormally increased glucose levels in the blood (hyperglycemia) and urine. As a result, there may be a gradual onset of certain symptoms, including excessive urination (polyuria) and increased thirst (polydipsia), and the development of particular complications without appropriate treatment.
Individuals with Alstrom syndrome often develop a condition known as acanthosis nigricans, a skin disorder characterized by abnormally increased coloration (hyperpigmentation) and “velvety” thickening (hyperkeratosis) of the skin, particularly of skin fold regions, such as of the neck and groin and under the arms (axillae). Acanthosis nigricans may be a skin manifestation of insulin resistance.
Affected individuals may also have elevated levels of certain fats (lipids) in the blood (hyperlipidemia). Hyperlipidemia is usually characterized by elevated triglycerides in the blood (hypertryglyceridemia). Some affected individuals are at risk of a rapid increase in triglycerides, which can cause inflammation of the pancreas (pancreatitis). Pancreatitis can be associated with abdominal pain, chills, jaundice, weakness, sweating, vomiting, and weight loss.
Some males with Alstrom syndrome may experience diminished hormone production by the testes (hypogonadotrophic hypogonadism). The onset of puberty may be delayed. Some affected males may develop abnormally enlarged breasts (gynecomastia).
Hypogonadism also occurs in affected females, but may not be apparent until puberty. Affected females may develop polycystic ovarian syndrome (PCOS). PCOS can result in irregular menstrual periods or a lack of menstruation, oily skin that is prone to acne, cysts on the ovaries and mild hirsutism (a male pattern of hair growth). Hair may develop on the upper lip and chin. PCOS may occur as a symptom of insulin resistance. In some cases, females enter puberty early (before the age of 8), a condition called precocious puberty.
Some individuals with Alstrom syndrome develop various urological abnormalities. As with other symptoms, the severity of urological abnormalities can vary greatly. Affected individuals may be unable to coordinate the muscles of the bladder and the tube that carries urine from the bladder out of the body (urethral dysynergia). Additional abnormalities include difficulty beginning urination, reduced flow, increased time between urinating, inability to control bladder movements (incontinence), and urinary retention. Urinary abnormalities may alternate between underactivity and over activity of the bladder. Many individuals with Alstrom syndrome also have recurrent urinary tract infections.
Affected individuals often experience slowly progressive dysfunction of the kidneys. Onset of kidney dysfunction may be during adolescence or adulthood. In many individuals, early kidney dysfunction may not cause symptoms (asymptomatic). Two common signs of kidney disease are excessive urination (polyuria) and excessive thirst (polydipsia). Eventually, symptoms including swelling of the ankles or a general feeling of ill health (malaise) may develop. Kidney dysfunction may progressively worsen eventually causing end stage renal failure, which can occur as early as the mid or late teen-aged years.
Some individuals may develop breathing (respiratory) or lung (pulmonary) problems such as chronic respiratory infections beginning early during childhood. These chronic infections can contribute to the development of asthma, chronic inflammation of the sinuses (sinusitis), a dry cough, and repeated episodes of inflammation of the bronchial tubes (bronchitis) or pneumonia. More serious pulmonary complications can occur including high blood pressure of the main artery of the lungs (pulmonary hypertension), chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome, and emphysema.
The liver may be involved in some cases resulting in abnormal enlargement of the liver (hepatomegaly). The severity of liver involvement can range from elevated liver enzymes, which are common in childhood to fatty liver disease (steatohepatitis). Steatohepatitis is characterized by the accumulation of fatty material in the liver and is often associated with diabetes or obesity. Liver (hepatic) dysfunction may occur and can progress to cause scarring (cirrhosis) within the liver, high blood pressure of the main vein of the liver (portal hypertension), abnormal enlargement of the spleen (splenomegaly), the abnormal accumulation of fluid in the abdominal cavity (ascites) and, eventually, liver failure by the second or third decade.
Additional serious complications associated with liver disease including esophageal varices and hepatic encephalopathy can develop. Esophageal varices are damaged, swollen blood vessels in the throat that are prone to bleeding and can rupture potentially causing life-threatening bleeding complications. Hepatic encephalopathy is a brain disorder that occurs in some individuals with chronic liver disease. It is a complex disorder that encompasses a spectrum of disease ranging from a subtle condition with no outward signs to a severe form that can cause life-threatening neurological complications.
Additional symptoms may be associated with Alstrom syndrome including low levels of growth hormone, which may result in short stature in adulthood; high blood pressure (hypertension), abnormally decreased activity of the thyroid gland and underproduction of thyroid hormones (hypothyroidism), advanced bone age; patchy areas of hair loss (alopecia); gastroesophageal reflux; pain in the middle of the abdomen just below the sternum (epigastric pain); and abnormal side-to-side (scoliosis) or front-to-back (kyphosis) curvature of the spine.
A variety of neurobehavioral findings have been reported including seizures, decreased reflexes (hyporeflexia), exaggerated response when startled, unexplained joint or muscle pain and dystonia, a group of neurological conditions generally characterized by involuntary muscle contractions that force the body into abnormal, sometimes painful movements and positions.
Alstrom syndrome diagnosis
Alstrom syndrome is diagnosed based on clinical findings (e.g., cone-rod dystrophy, sensorineural hearing impairment, cardiomyopathy, obesity, kidney dysfunction, diabetes), medical history, and family history and a variety of specialized tests. Making a diagnosis can be complicated by the variation in age of symptom onset from one individual to another. The absence of certain findings (e.g., polydactyly, intellectual disability) distinguishes Alstrom syndrome from similar syndromes such as Bardet-Biedl syndrome or Laurence-Moon syndrome.
Molecular genetic testing is not necessary to make the diagnosis of Alstrom syndrome, although it can be useful to confirm a diagnosis 5. Genetic testing of the ALMS1 gene is available for Alstrom syndrome. Although genetic testing is not necessary to make a diagnosis of Alstrom syndrome, it can be helpful to confirm a diagnosis. Molecular genetic testing can detect mutations in the ALMS1 gene known to cause the disorder. Molecular genetic testing detects ALMS1 mutations in approximately 70%-80% of individuals of Northern European descent and in approximately 40% of individuals worldwide. If a mutation is not identified in both copies of the ALMS1 gene of an individual suspected to have Alstrom syndrome, it does not rule out the diagnosis 5.
Clinical testing and work-up
An eye specialist (ophthalmologist) using specialized tests can make diagnosis of disorders affecting the retina of the eye such as Alstrom syndrome. An electroretinogram (ERG) may be used to detect abnormalities in the retina, and an electro-oculogram (EOG) may be used to measure retinal function. Sensorineural deafness may be confirmed through a variety of specialized hearing (auditory) tests.
Individuals with a suspected diagnosis of Alstrom syndrome should receive a thorough physical examination to detect the potential presence of additional heart, endocrinological, and kidney abnormalities often associated with Alstrom syndrome.
Alstrom syndrome treatment
There is no specific treatment for individuals with Alstrom syndrome. Treatment is directed toward the specific symptoms that are apparent in each individual. Treatment will require the coordinated efforts of a team of specialists. Pediatricians, cardiologists, specialists who asses and treat hearing problems (audiologists), specialists who asses and treat vision problems (ophthalmologists), specialists who deal with the system of glands that secrete hormones into the bloodstream (endocrinologists), specialists who assess and treat skeletal problems (orthopedists), and other healthcare professionals may need to systematically and comprehensively plan an affect child’s treatment.
Photophobia may be treated with specially-tinted, prescription glasses. In cases where cataracts significantly interfere with vision, they may be removed surgically. Whether or not surgery helps to improve vision often depends on how far the retinal changes have advanced.
Various vision aids may help people with Alstrom syndrome to make the maximum use of their remaining vision. These include optical aids such as Corning and NOIR glasses, the Fresnel Prising telescope, microscopes, and night vision devices. Non-optical aids that may also be useful include the Apollo Laser, Visualtek closed circuit television, the Wide Angle Mobility Light, paper guides, large-print typewriters, and adjustable stands. There are also reading machines and talking computers that can enhance the quality of life for individuals whose vision is severely impaired by Alstrom syndrome.
Children with Alstrom syndrome should receive instruction while they retain sight. Physicians have recommended that, in anticipation of total blindness, children learn mobility training, adaptive living skills, computing skills such as voice recognition and transcription software, and the use of Braille before sight is lost.
There is no specific treatment for sensorineural hearing loss. Hearing aids may help to maximize remaining hearing, and speech therapy may enhance the ability of a child to communicate orally. In the case of deafness associated with Alstrom syndrome, teaching a child sign language may not be an option as vision loss may also occur. Therefore, educational methods and options should be chosen carefully. A surgical procedure called a myringotomy, in which a thin incision is made in the eardrum to release fluid, may be performed for individuals with glue ear. Cochlear implants, which improve hearing by stimulating nerve fibers within the inner ear, have been beneficial in some cases. Tactile sign language could be of help for some deaf-blind individuals.
Strict dietary measures and exercise programs may help to control obesity, as well as aid in the management of diabetes mellitus and/or glucose intolerance associated with Alstrom syndrome. In most of the diagnosed patients of Alstrom syndrome, diabetes is controlled by diet and exercise alone. However, it is necessary in some cases to treat the diabetes with oral anti-diabetic agents or insulin. Patients may control diabetes for many years with insulin-sensitizing agents such as metformin. In several individuals, the group of incretins agents such as the Glucagon-like peptide 1 (GLP-1) analogues and the dipeptidyl peptidase-4 (DPP-4) inhibitors have been used with success.
If insulin therapy should become necessary for diabetes associated with Alstrom syndrome, a daily routine of insulin-injection, a controlled diet, exercise to burn off glucose, and testing for blood sugar level is vital in achieving and maintaining good blood sugar control. Self-monitoring of blood glucose levels uses a single drop of blood which is obtained with a finger stick, and placed on a chemically treated pad on a plastic strip; the color change of the chemically treated pad is compared to a color chart or “read” by a battery-operated portable meter. For blind individuals, talking glucose meters are available so that testing can be done independently.
In many individuals with Alstrom Syndrome, insulin is ineffective. Insulin must be given by injection, usually two or more times each day. Portable “insulin pumps” have been developed that permit continuous administration of insulin, as well as additional amounts of insulin when needed to control the changes in blood sugar level that occur after meals.
If excess levels of protein are detected in the urine (proteinuria), drugs known as angiotensinogen-converting enzyme (ACE) inhibitors may be recommended. In the event of kidney failure a procedure to remove toxins from the blood (dialysis) may be necessary. Kidney transplantation has been successfully performed in a growing number of individuals with Alstrom syndrome. However, the procedure can be contraindicated by other potential complications of the disorder such as morbid obesity, uncontrolled diabetes or cardiomyopathy.
Hypertriglyceridemia may be treated with a low-fat diet or lipid lowering medications.
Cardiac abnormalities may be treated with a variety of drugs including ACE inhibitors, which relax blood vessels, thereby lowering the blood pressure and minimizing the effort needed by the heart to pump blood throughout the body; drugs that reduce abnormal fluid retention by promoting the production and excretion of water and sodium from the body through the urine (diuretics); drugs such as digoxin that increase the efficiency of heart muscle contractions and produce a more regular heartbeat; and, in some cases, drugs that reduce the workload of the heart by blocking certain substances from binding to structures within the heart (beta blockers).Rarely, a heart (cardiac) transplant has been successfully performed on individuals with Alstrom syndrome.
As children attain puberty, an evaluation should be performed to see whether hormonal adjustment therapy is necessary. For example, male hypogonadism should be treated with testosterone to preserve sexuality, muscle strength and bone health. Individuals who have hypothyroidism may be treated with L-thyroxine, a type of thyroid hormone. Pediatric patients may be treated with recombinant growth hormone to promote their linear growth. Although controversial for Alstrom patients growth hormone therapy may benefit body composition (the balance between the fat mass and fat free mass) and bone health. Some female disorders such as irregular menses, polycystic ovary syndrome (PCOS) and hyperandrogenism may be treated with estrogen and progestin. Female individuals with PCOS and who are overweight and have diabetes may benefit from treatment with diet, exercise and insulin-sensitizing agents such as metformin.
A variety of techniques may be used to treat the complications of liver involvement. Portal hypertension may be treated with beta-blocker drugs. Sclerotherapy may be used to treat esophageal varices. Sclerotherapy is a procedure in which a solution, called a sclerosant or sclerosing agent, is injected directly into the affected veins of the throat and the areas adjacent to the affected veins. The solution injected into the veins causes blood clots to form in the veins stopping bleeding. The solution injected into the surrounding areas causes stops bleeding by thickening and swelling the vein to compress the blood vessel.
Banding (the application of rubber bands at the bleeding site) may be done to prevent bleeding in the upper gastrointestinal tract. Individuals who fail to respond to other methods may be candidates for transjugular intrahepatic portosystemic shunt (TIPS). During this procedure, a small metal device called a stent is placed into the liver creating an artificial passage from the portal vein to the hepatic vein, thereby improving blood flow. It can decrease the risk of variceal bleeding associated with portal hypertension. Some individuals with severe liver complications may be evaluated for a liver transplantation.
Genetic counseling may be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Alstrom syndrome prognosis
The prognosis for Alstrom syndrome varies depending on the progression of symptoms, specifically heart and kidney disease. The lifespan and overall quality of life for individuals with Alstrom syndrome can be improved by early diagnosis, treatment, surveillance, and proper management of symptoms 8.
References- Alström syndrome. https://ghr.nlm.nih.gov/condition/alstrom-syndrome
- Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675–83. doi: 10.1001/archinte.165.6.675
- Alström Syndrome. https://rarediseases.org/rare-diseases/alstrom-syndrome
- Alström syndrome. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=64
- Paisey RB, Steeds R, Barrett T, et al. Alström Syndrome. 2003 Feb 7 [Updated 2019 Jun 13]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1267
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alström syndrome: genetics and clinical overview. Curr Genomics. 2011;12(3):225-235. doi:10.2174/138920211795677912 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3137007
- Girard, D., Petrovsky, N. Alström syndrome: insights into the pathogenesis of metabolic disorders. Nat Rev Endocrinol 7, 77–88 (2011). https://doi.org/10.1038/nrendo.2010.210
- Van Groenendael S, Giacovazzi L, Davison F, et al. High quality, patient centred and coordinated care for Alstrom syndrome: a model of care for an ultra-rare disease. Orphanet J Rare Dis. 2015;10:149. Published 2015 Nov 24. doi:10.1186/s13023-015-0366-y https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4657378





{kind=link}