What is Epstein Barr virus
Epstein-Barr virus, also known as human herpesvirus 4, is a gamma herpes virus that occurs only in humans 1. Epstein-Barr virus is one of the most common human viruses. Epstein Barr virus is found all over the world. According to the Centers for Disease Control and Prevention (CDC), most people in the United States are infected by Epstein Barr virus at some point in their lives 2. Epstein Barr virus is very contagious and spreads most commonly from person to person through bodily fluids, primarily saliva, which is why Epstein-Barr virus infection is sometimes called “kissing disease.” Epstein-Barr virus is present in the saliva of infected individuals and can be spread through close contact such as kissing and through sharing utensils or cups. Epstein Barr virus can cause infectious mononucleosis, also called mono, and other illnesses.
Epstein Barr virus replicates primarily in β-lymphocytes but also may replicate in the epithelial cells of the pharynx and parotid duct 3. The infection is spread primarily by saliva, and the incubation period is four to eight weeks. In an acute infection, heterophile antibodies that agglutinate sheep erythrocytes are produced. This process forms the basis for the Monospot rapid latex agglutination test. Antibodies to viral capsid antigen (i.e., VCA-IgG and VCA-IgM) are produced slightly earlier than the heterophile antibody and are more specific for Epstein Barr virus infection. The VCA-IgG antibody persists past the stage of acute infection and signals the development of immunity 3.
After initial exposure to Epstein Barr virus, there is a period of several weeks before associated symptoms may appear, called the incubation period. During the acute primary infection, the virus multiplies in number. This is followed by a decrease in viral numbers and resolution of symptoms, but Epstein-Barr virus never completely goes away. Latent Epstein Barr virus remains in the person’s body for the rest of that person’s life and may reactivate but usually causes few problems unless the person’s immune system is significantly weakened.
Most people are infected by Epstein Barr virus in childhood and experience few or no symptoms. However, when the initial infection occurs in adolescence, it can cause infectious mononucleosis, commonly called mono, a condition associated with fatigue, fever, sore throat, swollen lymph nodes, an enlarged spleen, and sometimes an enlarged liver. These symptoms occur in about 25% of infected teens and young adults and usually resolve within a month or two.
People with infectious mononucleosis (mono) are typically diagnosed by their symptoms and the findings from a complete blood count (CBC) and a mono test (which tests for a heterophile antibody). About 25% of those with infectious mononucleosis (mono) do not produce heterophile antibodies and will have a negative mono test; this is especially true with children. Tests for Epstein Barr virus antibodies can be used to determine whether or not the symptoms these people are experiencing are due to a current infection with the Epstein Barr virus virus.
Epstein Barr virus is the most common cause of infectious mononucleosis (mono). According to the CDC, examples of other causes of infectious mononucleosis (mono) include cytomegalovirus (CMV), hepatitis A, hepatitis B or hepatitis C, rubella, and toxoplasmosis. Sometimes, it can be important to distinguish Epstein Barr virus from these other illnesses. For instance, it may be important to diagnose the cause of symptoms of a viral illness in a pregnant woman. Testing can help to distinguish a primary Epstein Barr virus infection, which has not been shown to affect a developing baby, from a cytomegalovirus (CMV), herpes simplex virus, or toxoplasmosis infection, as these illnesses can cause complications during the pregnancy and may harm the fetus.
Epstein Barr virus transmission
Epstein Barr virus spreads most commonly through bodily fluids, especially saliva. However, Epstein Barr virus can also spread through blood and semen during sexual contact, blood transfusions, and organ transplantations.
Epstein Barr virus can be spread by using objects, such as a toothbrush or drinking glass, that an infected person recently used. The virus probably survives on an object at least as long as the object remains moist.
The first time you get infected with Epstein Barr virus (primary Epstein Barr virus infection) you can spread the virus for weeks and even before you have symptoms. Once the virus is in your body, it stays there in a latent (inactive) state. If the virus reactivates, you can potentially spread Epstein Barr virus to others no matter how much time has passed since the initial infection.
Health experts aren’t sure how long people with Epstein Barr virus stay contagious after symptoms are gone. They believe that people can spread the infection for many months after that — some studies show as long as 18 months. Then, the Epstein Barr virus stays dormant (inactive) in the body for the rest of a person’s life.
Sometimes the dormant Epstein Barr virus can “wake up” and find its way into a person’s saliva (spit). That person might not feel ill or show any infectious mononucleosis symptoms, but can spread the virus to other people. So there’s a very small chance that people who have had Epstein Barr virus infection in the past can pass it to others, even when they feel OK.
Epstein Barr virus infection
Infectious mononucleosis, or “mono”, is an infection usually caused by the Epstein-Barr virus. Other viruses can also cause infectious mononucleosis are cytomegalovirus (CMV), hepatitis A, hepatitis B or hepatitis C, rubella, and toxoplasmosis. Infectious mononucleosis is usually suspected in patients 10 to 30 years of age who present with sore throat and significant fatigue, palatal petechiae, posterior cervical or auricular adenopathy, marked adenopathy, or inguinal adenopathy 4. Some of the symptoms of infectious mononucleosis are similar to those of a cold or flu and serious problems are rare.
Symptoms of infectious mononucleosis include:
- Fever
- Sore throat
- Swollen lymph glands in the neck and armpits
- Extreme fatigue
- Head and body aches
- Swollen liver or spleen or both
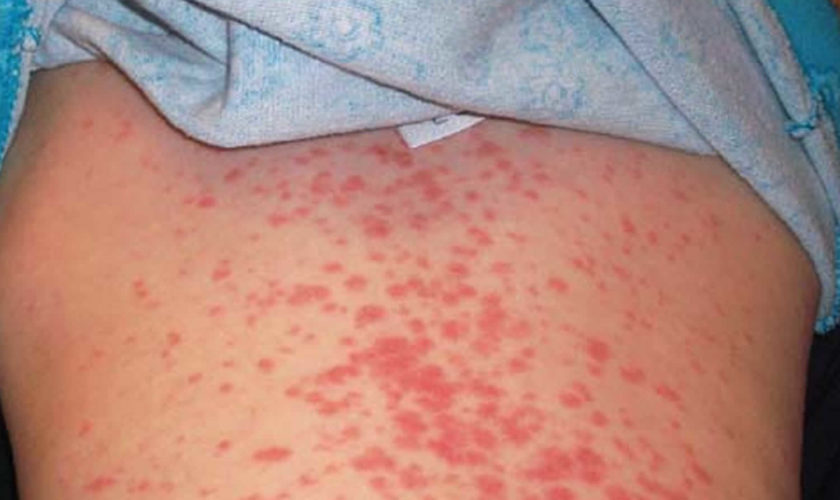
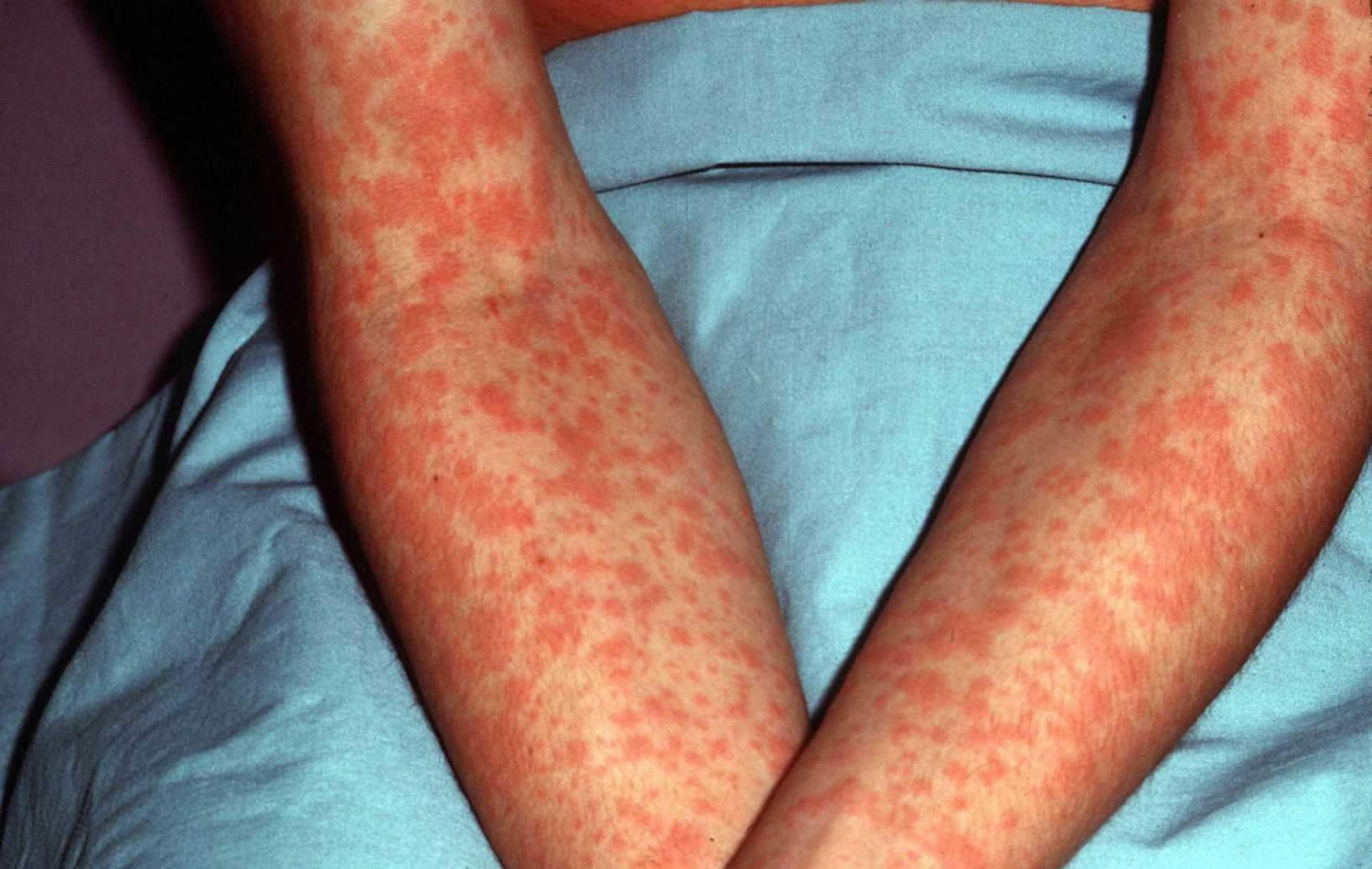
- Rash
Enlarged spleen and a swollen liver are less common symptoms. For some people, their liver or spleen or both may remain enlarged even after their fatigue ends. Jaundice and splenic rupture (spleen rupture) can occur in patients with infectious mononucleosis, but these complications are rare 5.
A ruptured spleen is rare in people who have mono. But it’s wise to be aware of the signs:
- sharp pain in the left upper part of your abdomen (under the left chest)
- feeling lightheaded
- feeling confused
- blurred vision
- fainting.
Other complications can include anemia, nervous system problems, or hepatitis with jaundice. They could cause symptoms including:
- breathing difficulty
- persistent high fevers (101.5 °F or higher)
- severe headache
- weakness in arms or legs
- yellow color in you eyes or skin (jaundice)
- trouble swallowing.
Call your doctor right away if you notice any of the above symptoms.
A blood test can show if you have infectious mononucleosis (mono).
Most people get better in two to four weeks. However, you may feel tired for a few months afterward. Occasionally, the symptoms of infectious mononucleosis can last for six months or longer. Treatment focuses on helping symptoms and includes medicines for pain and fever, warm salt water gargles and plenty of rest and fluids.
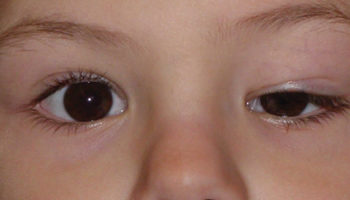
Figure 1. Epstein Barr virus rash
Can mononucleosis recur?
Most people who have infectious mononucleosis, or mono, get it only once. Rarely, however, mononucleosis symptoms may recur months or even years later.
Most cases of mononucleosis are caused by infection with the Epstein-Barr virus (Epstein Barr virus). Once you’re infected with Epstein Barr virus, you carry the virus — usually in a dormant state — for the rest of your life. Periodically, however, the virus may reactivate. When this happens, the virus can be detected in your saliva — but you’re not likely to become ill. Rarely, reactivated Epstein Barr virus may cause illness in people who have weak immune systems, such as those who have AIDS.
Mononucleosis rarely leads to a serious condition called chronic active Epstein Barr virus infection, which is characterized by persistent illness more than six months after the initial mononucleosis diagnosis.
If you’re experiencing signs or symptoms of mononucleosis — such as fatigue, weakness, fever, sore throat and swollen lymph nodes — and you’ve had mono before, consult your doctor to determine the cause of your current symptoms. Keep in mind that many other conditions, such as hepatitis and toxoplasmosis, can mimic the symptoms of mononucleosis.
Epstein Barr virus infection complications
Epstein-Barr virus can cause illnesses and complications aside from infectious mononucleosis. People with weakened immune systems may develop more severe symptoms and complications from Epstein Barr virus infection. They may also have more severe illness caused by Epstein Barr virus infection.
Complications of Epstein Barr virus infection include:
- Peritonsillar abscesses (pus-filled tissue near the tonsils)
- Acute bacterial sinusitis (bacterial infection of the sinus cavities)
- Suppurative lymph nodes (swelling of lymph nodes)
- Mastoiditis (bacterial infection of the mastoid bone of the skull)
- Sialadenitis (swelling and injury of salivary glands)
- Blockage of the air passages in the nose and throat
- Acute interstitial nephritis
- Hemolytic anemia
- Myocarditis and cardiac conduction abnormalities
- Neurologic abnormalities
- Cranial nerve palsies
- Encephalitis
- Meningitis
- Mononeuropathies
- Retrobulbar neuritis
- Thrombocytopenia
- Upper airway obstruction
Patients with infectious mononucleosis are likely to have splenomegaly. Although most patients do not have a palpable spleen on physical examination, a study of 29 patients who were hospitalized with infectious mononucleosis (and who therefore may have had more severe disease) found that all patients had splenomegaly on ultrasound examination and that one half of them had hepatomegaly 6. Only 17 percent of the enlarged spleens and 8 percent of the enlarged livers were palpable on physical examination, a finding that is consistent with other studies 7. Because an enlarged spleen is at risk for rupture, athletes should not compete in contact or collision sports for a minimum of three to four weeks after the onset of symptoms 8.
Nervous System
Epstein Barr virus infection can affect a person’s brain, spinal cord, and nerves.
Epstein Barr virus infection can cause conditions such as:
- Viral meningitis (swelling of the tissues that cover the brain and spinal cord)
- Encephalitis (swelling of the brain)
- Optic neuritis (swelling of the eye nerve)
- Transverse myelitis (swelling of the spinal cord)
- Facial nerve palsies (paralysis of facial muscles)
- Guillain-Barré syndrome (an immune system disease)
- Acute cerebellar ataxia (sudden uncoordinated muscle movement)
- Hemiplegia (paralysis on one side of the body)
- Sleep disorders
- Psychoses
Hematological System
Epstein Barr virus infection can affect a person’s blood and bone marrow. The virus can cause the body to produce an excessive number of white blood cells called lymphocytes (lymphocytosis).
Epstein Barr virus can also weaken the immune system, making it more difficult for the body to fight infection.
Examples of some of these conditions include:
- Neutropenia with secondary infections
- Hemophagocytic syndrome (hemophagocytic lymphohistiocytosis)
- Acquired hypogammaglobulinemia
- X-linked lymphoproliferative disease
Other Conditions
Epstein Barr virus infection can also cause:
- Pneumonia (injury of the lungs)
- Interstitial lung disease (a large group of disorders, most of which cause scarring of lung tissue)
- Pancreatitis (swelling of the pancreas)
- Myocarditis (swelling of the heart muscle)
- Oral cavity-oral hairy leukoplakia (raised, white patches on the tongue), which is usually seen in people infected with HIV
Cancers
Cancers associated with Epstein Barr virus infection include:
- Burkitt’s lymphoma (cancer of the lymphatic system)
- Nasopharyngeal carcinoma (cancer of the upper throat)
- Hodgkin’s disease and non-Hodgkin’s lymphoma (cancers of the lymphatic system)
- Post-transplant lymphoproliferative disorder (white blood cells are produced in excess)
- Other tumors including leiomyosarcomas (cancer in the soft tissue) and T-cell lymphomas
Chronic Epstein Barr virus
Several cohort studies have examined the long-term outcomes of infectious mononucleosis. Between 9 and 22 percent of patients reported persistent fatigue or hypersomnia six months after clinical infectious mononucleosis, compared with zero to 6 percent of patients following uncomplicated upper respiratory infection 3. The best study of the natural history of infectious mononucleosis followed 150 patients for six months 9. In this study, sore throat, fever, headache, rash, cough, and nausea largely had resolved one month after the onset of symptoms. Fatigue resolved more slowly (77 percent initially, 28 percent at one month, 21 percent at two months, and 13 percent at six months), as did sleeping too much (45 percent initially, 18 percent at one month, 14 percent at two months, and 9 percent at six months) and sore joints (23 percent initially, 15 percent at one month, 6 percent at two months, and 9 percent at six months) 9. This study also followed functional status over the same six-month period and found that patients required about two months to achieve a stable level of recovery. The association between Epstein Barr virus infection and chronic fatigue syndrome remains uncertain, and a positive IgG test for Epstein Barr virus does not imply a causal relationship. In addition, evidence of Epstein Barr virus infection is not part of the definition of chronic fatigue syndrome or myalgic encephalomyelitis 10.
Infectious mononucleosis prevention and treatment
There is no vaccine to protect against infectious mononucleosis. You can help protect yourself by not kissing or sharing drinks, food, or personal items, like toothbrushes, with people who have infectious mononucleosis.
You can help relieve symptoms of infectious mononucleosis by:
- drinking fluids to stay hydrated
- getting plenty of rest
- taking over-the-counter medications for pain and fever
If you have infectious mononucleosis, you should not take penicillin antibiotics like ampicillin or amoxicillin. Based on the severity of the symptoms, a healthcare provider may recommend treatment of specific organ systems affected by infectious mononucleosis.
Because your spleen may become enlarged as a result of infectious mononucleosis, you should avoid contact sports until you fully recover. Participating in contact sports can be strenuous and may cause the spleen to rupture.
Epstein Barr virus symptoms
Many people become infected with Epstein Barr virus in childhood. Epstein Barr virus infections in children usually do not cause symptoms, or the symptoms are not distinguishable from other mild, brief childhood illnesses. People who get symptoms from Epstein Barr virus infection, usually teenagers or adults, get better in two to four weeks. However, some people may feel fatigued for several weeks or even months.
Symptoms of Epstein Barr virus infection can include:
- fatigue
- fever
- inflamed throat
- swollen lymph nodes in the neck
- enlarged spleen
- swollen liver
- rash
After you get an Epstein Barr virus infection, the virus becomes latent (inactive) in your body. In some cases, the virus may reactivate. This does not always cause symptoms, but people with weakened immune systems are more likely to develop symptoms if Epstein Barr virus reactivates.
Epstein Barr virus causes
Epstein Barr virus is a herpes virus that replicates primarily in β-lymphocytes but also may replicate in the epithelial cells of the pharynx and parotid duct.7
Epstein Barr virus is spread by saliva through:
- kissing
- sharing drinks and food
- using the same cups, eating utensils, or toothbrushes
- having contact with toys that children have drooled on
Data collected more than 30 years ago on the incidence of infectious mononucleosis show the highest rates in persons 10 to 19 years of age (six to eight cases per 1,000 persons per year) 11. The incidence in persons younger than 10 years and older than 30 years is less than one case per 1,000 persons per year 11, but mild infections in younger children often may be undiagnosed. The infection is most common in populations with many young adults, such as active-duty military personnel and college students, in whom the annual incidence for infectious mononucleosis ranges from 11 to 48 cases per 1,000 persons 12.
Infectious mononucleosis is relatively uncommon in adults, accounting for less than 2 percent of all adults presenting with sore throat 7. Family physicians should expect to diagnose one to four patients with infectious mononucleosis per year, depending on the number of adolescents in their practice 12. The incidence of infectious mononucleosis shows no consistent seasonal peak 13.
Epstein Barr virus diagnosis
Diagnosing Epstein Barr virus infection can be challenging because the symptoms are similar to other illnesses. Epstein Barr virus infection can be confirmed with a blood test that detects antibodies. About nine out of ten of adults have antibodies that show that they have a current or past Epstein Barr virus infection.
Epstein Barr virus test
Epstein-Barr virus antibody test is a blood test to detect for antibodies to Epstein Barr virus in the blood and help establish a diagnosis of Epstein Barr virus infection. Epstein-Barr virus antibody test can also be important to rule out Epstein Barr virus infection and to look for other causes of the symptoms. Those with strep throat, an infection caused by group A streptococcus, for instance, need to be identified and treated with antibiotics. A person may have strep throat instead of mono or may have both conditions at the same time.
Several tests for different types and classes of Epstein Barr virus antibodies are available. The antibodies are proteins produced by the body in an immune response to several different Epstein-Barr virus antigens. During a primary Epstein Barr virus infection, the level of each of these Epstein Barr virus antibodies rises and falls at various times as the infection progresses. Measurement of these antibodies in the blood can aid in diagnosis and typically provides the healthcare practitioner with information about the stage of infection and whether it is a current, recent, or past infection.
Healthcare practitioners can test for antibodies to the following Epstein Barr virus-associated antigens:
Viral capsid antigen (VCA)
- Anti-VCA IgM appears early in Epstein Barr virus infection and usually disappears within four to six weeks.
- Anti-VCA IgG appears in the acute phase of Epstein Barr virus infection, peaks at two to four weeks after onset, declines slightly then persists for the rest of a person’s life.
Early antigen (EA)
- Anti-EA IgG appears in the acute phase of illness and generally falls to undetectable levels after three to six months. In many people, detection of antibody to EA is a sign of active infection. However, 20% of healthy people may have antibodies against EA for years.
Epstein Barr virus nuclear antigen (EBNA)
Antibody to EBNA, determined by the standard immunofluorescent test, is not seen in the acute phase of Epstein Barr virus infection but slowly appears two to four months after onset of symptoms and persists for the rest of a person’s life. Other EBNA enzyme immunoassays may report false positive results.
Monospot test
The Monospot test is not recommended for general use. The antibodies detected by Monospot can be caused by conditions other than infectious mononucleosis. Moreover, studies have shown that the Monospot produces both false positive and false negative results. For example, the heterophile antibodies detected by Monospot are often not present in children with infectious mononucleosis. At best, the Monospot test may indicate that a person has a typical case of infectious mononucleosis, but does not confirm the presence of Epstein Barr virus infection.
Interpretation of Epstein Barr virus Antibody Tests
Epstein Barr virus antibody tests are not usually needed to diagnose infectious mononucleosis. However, specific antibody tests may be needed to identify the cause of illness in people who do not have a typical case of infectious mononucleosis or have other illnesses that can be caused by Epstein Barr virus infection. Symptoms of infectious mononucleosis generally resolve within four weeks. If a person is ill for more than six months and does not have a laboratory-confirmed diagnosis of Epstein Barr virus infection, other causes of chronic illness or chronic fatigue syndrome should be considered.
The interpretation of Epstein Barr virus antibody tests requires familiarity with these tests and access to the patient’s clinical information.
Interpretation of Epstein Barr virus antibody tests and diagnosis of Epstein Barr virus infection is summarized as follows:
- Susceptibility to infection: People are considered susceptible to Epstein Barr virus infection if they do not have antibodies to the viral capsid antigen (VCA).
- Primary (new or recent) infection: People are considered to have a primary Epstein Barr virus infection if they have anti-VCA IgM but do not have antibody to Epstein Barr virus nuclear antigen (EBNA). Other results that strongly suggest a primary infection are a high or rising level of anti-VCA IgG and no antibody to Epstein Barr virus nuclear antigen (EBNA) after at least four weeks of illness. Resolution of the illness may occur before the diagnostic antibody levels appear. In rare cases, people with active Epstein Barr virus infections may not have detectable Epstein Barr virus-specific antibodies.
- Past infection: The presence of antibodies to both viral capsid antigen (VCA) and Epstein Barr virus nuclear antigen (EBNA) suggests past infection (from several months to years earlier). Since over 90% of adults have been infected with Epstein Barr virus, most adults will show antibodies to Epstein Barr virus from infection years earlier. High or elevated antibody levels may be present for years and are not diagnostic of recent infection.
Testing paired acute- and convalescent-phase serum samples is not useful to distinguish between recent and past Epstein Barr virus infections. In most cases, the antibody response occurs rapidly during primary Epstein Barr virus infection. The clinical findings of infectious mononucleosis occur in conjunction with the appearance of IgG and IgM anti-VCA antibodies. However, the antibody pattern is not stable before symptoms appear.
Epstein Barr virus treatment
There isn’t a cure for Epstein Barr virus infection. The virus will go away on its own. Symptoms usually last about 4 weeks. The main goal of treatment is to relieve your symptoms. Here are some steps you can take:
- Rest. Sleep helps your body fight infection.
- Drink plenty of fluids. They help prevent dehydration.
- Soothe a sore throat. Gargle with salt water or suck on throat lozenges, hard candy, or flavored frozen desserts (such as Popsicles).
- Relieve the pain. Take acetaminophen (Tylenol) or ibuprofen (Advil, Motrin) to relieve pain and fever. Do not give aspirin to children. Aspirin has been associated with a rare disease in children called Reye’s syndrome. Reye’s syndrome is a serious illness that can lead to death.
Antibiotics are not effective against mono. Infectious mononucleosis is caused by Epstein Barr virus. Antibiotics don’t work against viruses. If you have infectious mononucleosis, you should not take penicillin antibiotics like ampicillin or amoxicillin because they may cause a morbilliform rash in patients with infectious mononucleosis. If you have a bacterial infection (such as strep throat) in addition to having infectious mononucleosis, your doctor may give you an antibiotic to treat that infection.
Based on the severity of the symptoms, a healthcare provider may recommend treatment of specific organ systems affected by infectious mononucleosis.
Because your spleen may become enlarged as a result of infectious mononucleosis, you should avoid contact sports until you fully recover. Participating in contact sports can be strenuous and may cause the spleen to rupture. The risk of spleen rupture is estimated at 0.1 percent, based on a retrospective series of 8,116 patients 14. In a review of 55 athletes with splenic rupture, almost all ruptures occurred in the first three weeks of illness 15. Interestingly, in the same series, one half of ruptures were atraumatic. These data suggest that patients should be kept out of athletics for at least three to four weeks and until they are asymptomatic. Some experts suggest a longer duration of restricted activity of five to six weeks 16 or even six months 17, although data from natural history studies do not necessarily support these recommendations. Because the physical examination is so insensitive, ultrasound imaging to assess the size of the spleen at three weeks may be a better guide for determining whether a patient should return to athletics 18. The cost to prevent one traumatic spleen rupture probably is well over $1 million when this strategy is used, given that the risk of rupture spleen overall is one in 1,000, the risk of rupture beyond four weeks is considerably less, about one half of ruptures are atraumatic, and the cost of an ultrasound scan is several hundred dollars. Although the strategy may make sense when used selectively (e.g., when an athlete would like to return to competition in less than four weeks), it cannot be endorsed as routine practice.
A meta-analysis 19 of five randomized controlled trials involving 339 patients found that patients who took acyclovir (Zovirax) had less oropharyngeal shedding at the end of therapy, but this treatment provided no significant or consistent clinical benefit and is therefore not recommended. Another trial found no significant benefit from the use of ranitidine (Zantac) in patients with infectious mononucleosis 20.
Corticosteroids have been advocated for the treatment of patients with infectious mononucleosis 21 and some early studies 22 seemed to show a benefit from these agents with regard to normalization of temperature and laboratory values. However, these studies had significant methodologic limitations. A more recent and better-designed study 23 found no benefit from a combination of acyclovir and prednisone. In a small, double-blind, randomized trial of 40 children with suspected infectious mononucleosis (33 of whom had confirmed infectious mononucleosis), those who were given oral dexamethasone (0.3 mg per kg) had less pain at 12 hours but not at 24, 48, and 72 hours 24. This finding indicates that repeated doses may be needed.
- Epstein-Barr Virus and Infectious Mononucleosis. Laboratory Testing. https://www.cdc.gov/epstein-barr/laboratory-testing.html[↩]
- About Epstein-Barr Virus (EBV). Epstein-Barr Virus and Infectious Mononucleosis. https://www.cdc.gov/epstein-barr/about-ebv.html[↩]
- Andersson JP. Clinical aspects of Epstein-Barr virus infection. Scand J Infect Dis Suppl. 1991;80:94–104.[↩][↩][↩]
- Epstein-Barr Virus Infectious Mononucleosis. Am Fam Physician. 2004 Oct 1;70(7):1279-1287. https://www.aafp.org/afp/2004/1001/p1279.html[↩]
- Bailey RE. Diagnosis and treatment of infectious mononucleosis. Am Fam Physician. 1994;49:879–88.[↩]
- Dommerby H, Stangerup SE, Stangerup M, Hancke S. Hepatosplenomegaly in infectious mononucleosis, assessed by ultrasonic scanning. J Laryngol Otol. 1986;100:573–9[↩]
- Aronson MD, Komaroff AL, Pass TM, Ervin CT, Branch WT. Heterophil antibody in adults with sore throat: frequency and clinical presentation. Ann Intern Med. 1982;96:505–8.[↩][↩]
- Burroughs KE. Athletes resuming activity after infectious mononucleosis. Arch Fam Med. 2000;9:1122–3.[↩]
- Rea TD, Russo JE, Katon W, Ashley RL, Buckwald DS. Prospective study of the natural history of infectious mononucleosis caused by Epstein-Barr virus. J Am Board Fam Pract. 2001;14:234–42.[↩][↩]
- Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Possible Causes. https://www.cdc.gov/me-cfs/about/possible-causes.html[↩]
- Fry J. Infectious mononucleosis: some new observations from a 15-year study. J Fam Pract. 1980;10:1087–9.[↩][↩]
- Candy B, Chalder T, Cleare AJ, Wessley S, White PD, Hotopf M. Recovery from infectious mononucleosis: a case for more than symptomatic therapy? A systematic review. Br J Gen Pract. 2002;52:844–51.[↩][↩]
- Henke CE, Kurland LT, Elveback LR. Infectious mononucleosis in Rochester, Minnesota, 1950 through 1969. Am J Epidemiol. 1973;98:483–90.[↩]
- Farley DR, Zietlow SP, Bannon MP, Farnell MB. Spontaneous rupture of the spleen due to infectious mononucleosis. Mayo Clin Proc. 1992;67:846–53.[↩]
- Maki DG, Reich RM. Infectious mononucleosis in the athlete. Diagnosis, complications, and management. Am J Sports Med. 1982;10:162–73[↩]
- Eichner ER. Infectious mononucleosis. Phys Sportsmed. 1996;24:49–54[↩]
- Rutkow IM. Rupture of the spleen in infectious mononucleosis: a critical review. Arch Surg. 1978;113:718–20.[↩]
- Sevier TL. Infectious disease in athletes. Med Clin North Am. 1994;78:389–412.[↩]
- Torre D, Tambini R. Acyclovir for treatment of infectious mononucleosis: a meta-analysis. Scand J Infect Dis. 1999;31:543–7.[↩]
- Vendelbo Johansen L, Lildholdt T, Bende M, Toft A, Brahe Pedersen C, Danielsson GP. Infectious mononucleosis treated by an antihistamine: a comparison of the efficacy of ranitidine (Zantac) vs placebo in the treatment of infectious mononucleosis. Clin Otolaryngol. 1997;22:123–5.[↩]
- Hoagland RJ. Infectious mononucleosis. Prim Care. 1975;2:295–307.[↩]
- Bolden KJ. Corticosteroids in the treatment of infectious mononucleosis. An assessment using a double blind trial. J R Coll Gen Pract. 1972;22:87–95.[↩]
- Tynell E, Aurelius E, Brandell A, Julander I, Wood M, Yao QY, et al. Acyclovir and prednisolone treatment of acute infectious mononucleosis: a multicenter, double-blind, placebo-controlled study. J Infect Dis. 1996;174:324–31.[↩]
- Roy M, Bailey B, Amre DK, Girodias JB, Bussieres JF, Gaudreault P. Dexamethasone for the treatment of sore throat in children with suspected infectious mononucleosis: a randomized, double-blind, placebo-controlled, clinical trial. Arch Pediatr Adolesc Med. 2004;158:250–4.[↩]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}