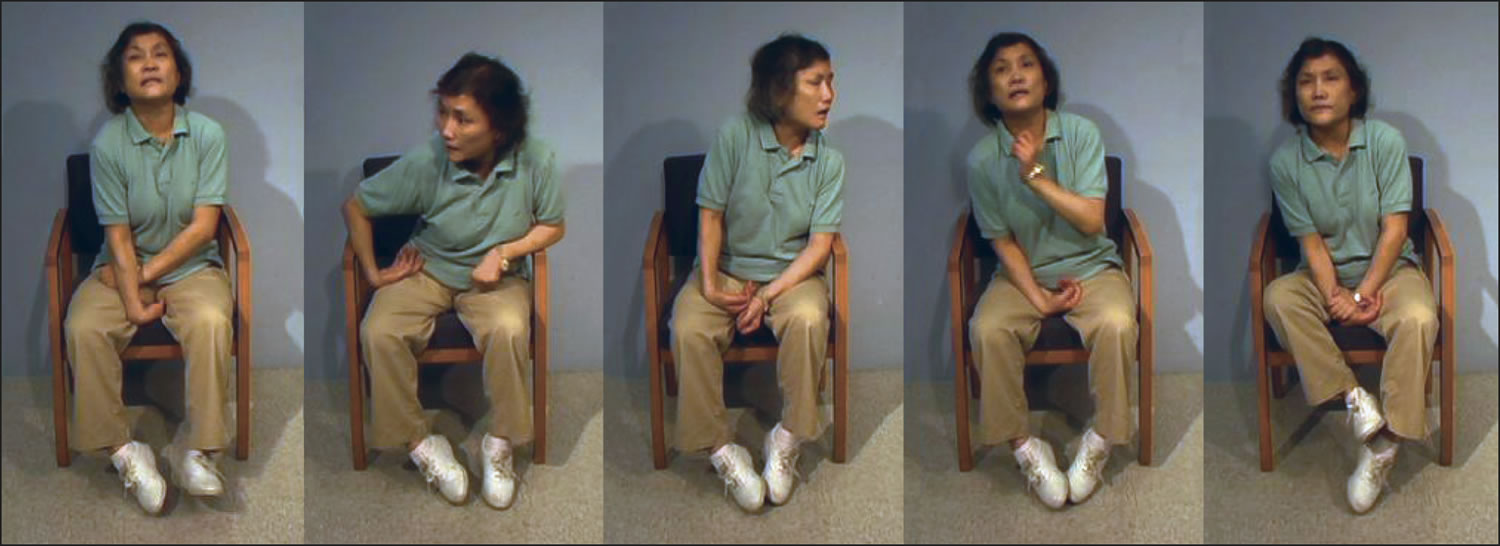
What is chorea
Chorea is a random-appearing, continuous (while awake), abnormal involuntary movement disorder, which can affect the entire body. Chorea often includes the face and tongue. Symptoms in arms and legs are often worse on one side of the body. Chorea is classed as one of a group of neurological disorders called dyskinesias, which are caused by overactivity of the neurotransmitter dopamine in the areas of the brain that control movement 1. Chorea is characterized by brief, irregular contractions that are not repetitive or rhythmic, but appear to flow from one muscle to the next. Chorea often occurs with athetosis, which adds twisting and writhing movements. Chorea is a primary feature of Huntington’s disease, a progressive, hereditary movement disorder that appears in adults, but it may also occur in a variety of other conditions. Sydenham’s chorea occurs in a small percentage (20 percent) of children and adolescents as a complication of rheumatic fever 2. Chorea can also be induced by drugs (levodopa, anti-convulsants, and anti-psychotics) metabolic and endocrine disorders, and vascular incidents.
Chorea can be due to a multitude of disorders with a wide variety of pathophysiologies. In addition to metabolic and structural causes, the list of potential investigations continues to grow as more potentially causative genes are discovered. Although treatments for genetic causes are not yet possible, correct diagnosis is essential for appropriate genetic counseling.
The prognosis for individuals with chorea varies depending on the type of chorea and the associated disease. Huntington’s disease is a progressive, and ultimately, fatal disease. Sydenham’s chorea is treatable and curable.
Types of chorea
Sydenham’s chorea
Sydenham chorea also called St. Vitus dance, is a rare neurological disorder of childhood characterized by sudden onset chorea, resulting from infection via Group A beta-hemolytic streptococcus, the bacterium that causes rheumatic fever 3. Sydenham chorea is characterized by rapid, irregular, and aimless involuntary movements of the arms and legs, trunk, and facial muscles. Additional symptoms of Sydenham chorea may include slurring of speech and difficulty maintaining steady hand grip. Anxiety, sadness, inattention, and obsessive compulsive thoughts and behaviors may also occur.
Sydenham chorea most often affects children over the age of 5 and 15 years of age. According to most studies, Sydenham chorea affects girls more often than boys. One distinct finding of Sydenham chorea is that it is almost never seen in children below 5 years of age. Rarely, the disorder has been reported in adults. Sydenham chorea affects individuals of all races and ethnicities.
Some children will have a sore throat several weeks before the symptoms begin, but the disorder can also strike up to 6 months after the fever or infection has cleared. Symptoms can appear gradually or all at once, and also may include uncoordinated movements, muscular weakness, stumbling and falling, slurred speech, difficulty concentrating and writing, and emotional instability. The symptoms of Sydenham chorea can vary from a halting gait and slight grimacing to involuntary movements that are frequent and severe enough to be incapacitating.
Sydenham chorea usually develops within weeks to months following group A beta-hemolytic streptococcal infection and may occur as an isolated finding or as a major complication of acute rheumatic fever. Rheumatic fever is considered an autoimmune disorder, meaning it occurs when the body’s immune system (which normally responds to foreign substances) mistakenly targets part of the body, disrupting normal function. The random, writhing movements of chorea are caused by an auto-immune reaction to the Group A beta-hemolytic streptococcus bacterium that interferes with the normal function of a part of the brain (the basal ganglia) that controls motor movements. Due to better sanitary conditions and the use of antibiotics to treat streptococcal infections, rheumatic fever, and consequently Sydenham chorea, are rare in North America and Europe. Rheumatic fever can still be found in developing countries.
Approximately, 25 percent of individuals with rheumatic fever develop Sydenham chorea. The incidence of rheumatic fever in North America declined steadily in the past 50 years, although there have been occasional outbreaks. Sydenham chorea is the most common cause of acute chorea during childhood in the United States. Sydenham chorea remains a major public health problem in many developing countries due primarily to cases where there is damage to heart valves.
Sydenham chorea symptoms
The severity of chorea and the presence of non-chorea symptoms of Sydenham chorea may vary greatly from one person to another. Most cases follow a streptococcal infection. Streptococcus is a group of bacteria that can cause several different infections, most commonly “strep throat” – often presenting with a sore throat (pharyngitis) or fever. Symptoms of Sydenham chorea may appear anywhere from 1 week to 6 months following streptococcal infection.
The onset of the abnormal movements (chorea) that characterize Sydenham chorea are most often sudden – appearing over hours and peaking within a few hours or days. Pediatricians and emergency physicians seldom see chorea and may not recognize it. Initially, doctors may misattribute the restless movements and involuntary facial expressions of Sydenham chorea to a child being extremely fidgety, hyperactive, clumsy and/or purposely uncooperative. Parents (and children) generally recognize however that these movements are a clear change from the child’s usual status.
The abnormal movements in Sydenham chorea range from mild symptoms, affecting coordination and tasks such as writing, to severe symptoms, disrupting walking, talking, and performing basic tasks such as dressing, eating, or simply holding objects. Choreic movements may fluctuate through the day. In most cases, chorea disappears during sleep.
In addition to choreic movements, individuals with Sydenham chorea may develop muscle weakness, slurred speech (dysarthria), diminished muscle tone (hypotonia), tics, obsessions, compulsions, inattention, anxiety, labile mood, and decreased verbal output. In some extremely rare cases (less than 2 percent), severe muscle weakness, irritability, or confusion may be profound and affected children may become bedridden, a condition sometimes referred to as paralytic chorea.
Because Sydenham chorea is a complication of rheumatic fever, some individuals will have additional symptoms of joint arthritis or arthralgia, inflammation of the heart valves causing permanent damage to the valves, and ongoing fever.
Sydenham chorea symptoms usually resolve within three weeks to six months. However, symptoms may last longer than one year. Occasionally, the symptoms of Sydenham chorea have recurred later during adult life, particularly in young women during the first trimester of pregnancy (so called chorea gravidarum, which may represent a recurrence of Sydenham chorea in some cases).
Sydenham chorea causes
Sydenham chorea is believed to be an autoimmune disorder. Most cases develop following a streptococcal infection or more severe rheumatic fever. An autoimmune disorder occurs when the body’s immune system mistakenly reacts against healthy tissue. In Sydenham chorea, streptococcal infection induces the body’s immune system to produce antibodies to combat the infection. For unknown reasons, these antibodies persist and subsequently target certain cells in the joints, kidneys, heart, and, in the brain, specifically cells of the basal ganglia (a key part of the brain for controlling motor movements). Researchers believe this ultimately leads to the characteristic symptoms of Sydenham chorea.
The exact underlying mechanisms that cause Sydenham chorea are not fully understood. Researchers believe that antigens (substances that are capable of stimulating an immune system response) on streptococcal bacterial cells are similar to antigens found on cells of the basal ganglia. When the immune system creates antibodies to combat the streptococcal infection, the antibodies also, in genetically predisposed individuals, mistakenly bind to healthy brain cells and disrupt their function.
Sydenham chorea diagnosis
A diagnosis of Sydenham chorea is made based upon identification of new onset choreic movements, a detailed patient history, and a thorough clinical evaluation.
If a streptococcus infection is suspected, tests will be done to confirm the infection. These include:
- Throat swab
- Anti-DNAse B blood test
- Antistreptolysin O (ASO) blood test
Further testing may include:
- Blood tests such as ESR, CBC
- MRI or CT scan of the brain
In the presence of new onset chorea, which is uncommon in childhood, the documentation of a prior streptococcal infection through throat swabs and/or the current presence of high blood titers of streptococcal antibodies (ASO, anti DNAseB) is useful, as are identification of co-occurring arthritis or cardiac valve abnormalities. In some cases, certain imaging techniques such as magnetic resonance imaging (MRI) may be recommended to exclude other causes. Usually, brain imaging is normal in Sydenham chorea. Of note, because the onset of Sydenham chorea usually occurs weeks after the infection, the characteristic signs of rheumatic fever or streptococcal infection are usually no longer present.
Individuals who are diagnosed with Sydenham chorea should receive an evaluation for inflammation of the heart (carditis).
Sydenham chorea treatment
A confirmed diagnosis of Sydenham chorea is nearly always an indication for long-term antibiotic treatment until adulthood. Antibiotics are used to kill the streptococcus bacteria. Your doctor may also prescribe antibiotics to prevent future rheumatic fever infections. This is called preventive antibiotics, or antibiotic prophylaxis. The purpose if this treatment is to prevent permanent heart valve damage which could result if the child experiences recurrent streptococcal infections. Most often penicillin is used. Physicians should consult current rheumatic fever guidelines.
Chorea-suppressing treatment
Chorea suppressing medications should be considered. Some mild cases may not require intervention because the disorder usually resolves on its own within weeks. When chorea symptoms are disabling, low doses of potent dopamine receptor blocking agents such as haloperidol, dopamine depleting agents such as tetrabenazine, anti-seizure medications such as valproic acid, or benzodiazepines may help. Because in most cases the treatment will only be needed for weeks to months and at low doses, side effects such as tardive dyskinesia are extremely unlikely. As is the case for any neurological medications, however, a careful discussion of potential benefits as well as risks is advised.
Immune system treatment
Additional short-term immune therapies have been used to treat individuals with impairing Sydenham chorea during the first weeks of symptoms, based on the idea that ongoing acute inflammation is contributing to symptoms. There is some scientific support for using oral steroids and intravenous immunoglobulins from small but rigorous clinical trials.
Huntington’s disease
Huntington disease is a genetic, progressive, neurodegenerative disorder that causes uncontrolled movements, emotional problems, and loss of thinking ability (cognition) 4. Huntington’s disease is characterized by the gradual development of involuntary muscle movements affecting the hands, feet, face, and trunk and progressive deterioration of cognitive processes and memory (dementia). Neurologic movement abnormalities may include uncontrolled, irregular, rapid, jerky movements (chorea) and athetosis, a condition characterized by relatively slow, writhing involuntary movements. Dementia is typically associated with progressive disorientation and confusion, personality disintegration, impairment of memory control, restlessness, agitation, and other symptoms and findings. In individuals with the disorder, disease duration may range from approximately 10 years up to 25 years or more. Life-threatening complications may result from pneumonia or other infections, injuries related to falls, or other associated developments.
Adult-onset Huntington disease, the most common form of this disorder, usually appears in a person’s thirties or forties. Early signs and symptoms can include irritability, depression, small involuntary movements, poor coordination, and trouble learning new information or making decisions. Many people with Huntington disease develop involuntary jerking or twitching movements known as chorea. As the disease progresses, these movements become more pronounced. Affected individuals may have trouble walking, speaking, and swallowing. People with this disorder also experience changes in personality and a decline in thinking and reasoning abilities. Individuals with the adult-onset form of Huntington disease usually live about 15 to 20 years after signs and symptoms begin.
A less common form of Huntington disease known as the juvenile form begins in childhood or adolescence. It also involves movement problems and mental and emotional changes. Additional signs of the juvenile form include slow movements, clumsiness, frequent falling, rigidity, slurred speech, and drooling. School performance declines as thinking and reasoning abilities become impaired. Seizures occur in 30 percent to 50 percent of children with this condition. Juvenile Huntington disease tends to progress more quickly than the adult-onset form; affected individuals usually live 10 to 15 years after signs and symptoms appear.
Huntington disease affects an estimated 3 to 7 per 100,000 people of European ancestry. Huntington’s disease appears to be less common in some other populations, including people of Japanese, Chinese, and African descent. About 30,000 people in the United States have Huntington’s disease and another 200,000 are at risk of developing the condition. Symptoms commonly develop between ages 30 and 50. Huntington’s disease progresses slowly and a person may live for another 15-20 years after the onset of symptoms.
Huntington disease signs and symptoms
Huntington’s disease is characterized by rapid uncontrollable muscle movements such as tics or muscle jerks (choreiform movements or chorea). This disorder causes a loss of coordination and personality changes. As the disease progresses, the ability to speak may be impaired, memory may fade, and the involuntary jerky muscle movements (chorea) become more severe.
Huntington’s disease runs a ten to 25 year progressive course. As the disorder progresses, the chorea may subside and there may be an absence of movement (akinesia). Dementia gradually develops. Patients with Huntington’s disease are at high risk of developing pneumonia as a result of being bedridden and undernourished.
Huntington disease causes
Huntington’s disease is inherited as an autosomal dominant trait. In dominant disorders, a single copy of the disease gene (received from either the mother or father) will be expressed “dominating” the other normal gene and resulting in the appearance of the disease. The risk of transmitting the disorder from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child.
Huntington’s disease is caused by changes (mutations) of a HTT gene (huntingtin gene) that is located on the short arm (p) of chromosome 4 (4p16.3). The HTT gene provides instructions for making a protein called huntingtin. Although the function of this protein is unknown, it appears to play an important role in nerve cells (neurons) in the brain. Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females. Each chromosome has a short arm designated as “p” and a long arm identified by the letter “q.” Chromosomes are further subdivided into bands that are numbered.
Known as huntingtin, the gene controls the production of a protein found in nerve cells (neurons) throughout the brain. However, the specific function of the huntingtin protein is not known. In individuals with the disorder, the huntingtin gene contains errors in the coded “building blocks” that make up its specific genetic instructions. The instructions within every gene consist of different arrangements of four basic chemicals (nucleotide bases) called adenine (A), cytosine (C), guanine (G), and thymine (T). In those with Huntington’s Disease, the huntingtin gene contains abnormally long repeats of coded instructions consisting of cytosine, adenine, and guanine (CAG trinucleotide repeat expansion). For example, individuals with the disease have over 35 CAG repeats within the huntingtin gene, with most having more than 39. However, individuals without the disorder tend to have about 20 repeats in the gene. Expanded CAG repeats are unstable and may expand further over time and with successive generations. This is thought to be responsible for genetic anticipation, a phenomenon in which an individual with Huntington’s disease may have symptom onset at a significantly earlier age than his or her affected parent. In addition, some researchers suggest that expanded CAG repeats of the huntingtin gene may become more unstable when the gene is transmitted from the father.
The specific symptoms associated with Huntington’s disease are caused by degenerative changes of nerve cells (neurons) within certain regions of the brain, including the basal ganglia and cerebral cortex. The basal ganglia are specialized nerve cells deep within the brain that play a role in regulating movements. The cerebral cortex, the outer region of the brain, is responsible for conscious thought and movement.
Huntington disease diagnosis
The diagnosis of Huntington’s disease may be confirmed by a thorough clinical evaluation, detailed patient history, and a variety of specialized tests. Specialized x-ray studies such as computerized tomography (CT) scanning, magnetic resonance imaging (MRI), or electroencephalography (EEG) may help confirm the diagnosis of Huntington’s Disease. During CT scanning, a computer and x-rays are used to create a file showing cross-sectional images of the brain. During MRI, a magnetic field and radio waves are used to create cross-sectional images of the brain. During an EEG, an instrument records electrical activity of the brain. Neuropsychological and/or genetic tests are also used to aid the diagnosis of Huntington’s disease.
Huntington disease treatment
In August 2008, the Food and Drug Administration (FDA) approved tetrabenazine (Xenazine) for the treatment of the repetitive, involuntary movements (chorea. associated with Huntington’s disease. This is the first and only FDA-approved treatment specifically for any symptom of Huntington’s disease. The drug has been available in Europe for several years.
Other treatment for Huntington’s disease is symptomatic and supportive. There are some treatments that may alleviate various symptoms temporarily. Neuroleptic medication such as haloperidon can partially suppress the involuntary movement, especially in the early stages. Other medication can often help depression and other emotional symptoms. Special high calorie food preparations may help an affected individual maintain weight and avoid choking during the later stages of Huntington’s disease.
Genetic counseling will be of benefit for affected individuals and their families. Family members of affected individuals should also receive clinical evaluations to detect any symptoms and physical characteristics that may be potentially associated with Huntington’s disease.
Chorea symptoms
Chorea is characterized by repetitive, brief, irregular, somewhat rapid, involuntary movements that typically involve the face, mouth, trunk and limbs. However, the appearance of chorea is not typically helpful in elucidating the diagnosis. The exceptions to this are when there is prominent orofacial involvement, or markedly asymmetric involvement of the limbs.
In neuroleptic-induced tardive dyskinesia facial muscles alone are most commonly affected. Prominent and severe orofacial and lingual involvement can be seen in chorea-acanthocytosis and pantothenate kinase-associated neurodegeneration. In chorea-acanthocytosis the tongue movements are dystonic, and specifically induced by eating.
Unilateral chorea in adults is often due to structural lesions. These are most commonly due to focal lesions of the basal ganglia, but may be seen with lesions in other locations, for reasons which are unclear.
Peripheral neuropathy
Peripheral neuropathy, as demonstrated by hyporeflexia and peripheral sensory changes is characteristic of the two core neuroacanthocytosis syndromes, chorea-acanthocytosis and McLeod syndrome, and of some of the inherited ataxias, both autosomal recessive (Friedreich’s ataxia, ataxias with oculomotor apraxia1and ataxias with oculomotor apraxia 2) and autosomal dominant (spinocerebellar ataxia 1, 2, 3).
Hyperreflexia
Upper motor neuron signs can be seen with disorders in which there is involvement of the cortex, e.g. Huntington’s disease, Huntington’s disease-like 2, and the spinocerebellar ataxias. Asymmetric hyperreflexia may indicate a focal lesion.
Ataxia
The inherited ataxias, both autosomal dominant and autosomal recessive, typically cause ataxia and eye movement abnormalities, however, occasionally these may be absent.
Cognitive and psychiatric symptoms
Changes in personality, frontal or subcortical dementia, and psychiatric disease can be seen in a number of the neurodegenerative causes of chorea, and are most likely due to involvement of the frontal cortex and caudate nucleus. (see accompanying article by Walterfang) Subtle changes in cognition can be detected by neuropsychological testing, for example, in pre-symptomatic carriers of the Huntington’s disease mutation.
Ophthalmologic findings
Analogous to the fragments of movements intruding into voluntary limb movements, intrusions of square-wave jerks can interfere with fixation of gaze. These are best documented in Huntington’s disease, but can be seen also in chorea of other etiologies. Impaired smooth pursuit and saccadic gaze, or ophthalmoplegia, may suggest a cerebellar disorder.
Pigmentary retinopathy is indicative of pantothenate kinase-associated neurodegeneration and is an invariable feature of typical (young-onset) disease.
The characteristic telangiectactic blood vessels seen in the conjunctivae of the eye may be helpful in making the diagnosis of ataxia-telangiectasia but may be absent in atypical, late-onset disease.
Other local ophthalmological findings may indicate autoimmune disease.
Seizures
Seizures are seen in approximately 50% of patients with a neuroacanthocytosis syndrome. These can predate the appearance of the movement disorder by several years. Seizures can also be present in young-onset Huntington’s disease, although these patients tend to have an akinetic-rigid phenotype rather than chorea. Younger onset patients with dentatorubropallidoluysian atrophy tend to present with seizures, although in these cases chorea is less prominent than ataxia. Older onset patients with chorea may develop seizures later in the disease course.
Myoclonus
Myoclonus is a frequent finding in prion diseases, and in younger patients with dentatorubropallidoluysian atrophy.
Chorea causes
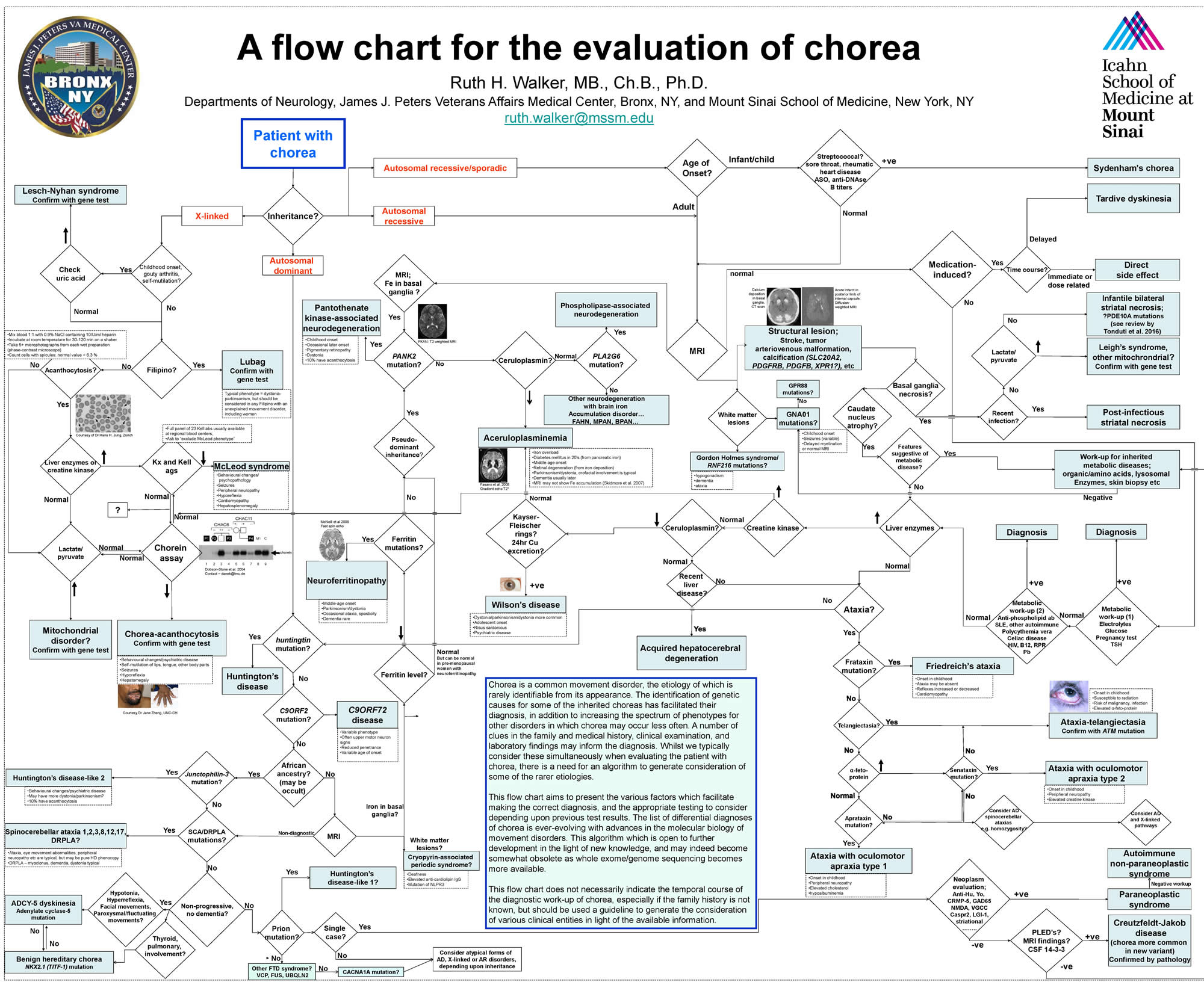
A large number of neurological disorders can cause chorea and diagnosis can be challenging (see the flowchart in Figure 1). While Sydenham chorea should be suspected as the most likely cause of acute chorea in children, there are other conditions doctors should consider as well.
Chorea can be present in a number of well-characterized genetic disorders, but also may occur as a rarer phenotypic variation in others. Despite extensive work-up some patients inevitably remain undiagnosed. Clues to the diagnosis of chorea may be found in features of the family and medical history and with careful clinical examination.
Many different disorders and conditions can be associated with acute chorea. In many developed countries, chorea is observed when psychiatric medications are started, increased, or stopped too abruptly. Specifically, abrupt withdrawal of dopamine receptor blocking medications (for example, haloperidol, risperidone, or aripiprazole) can cause chorea. These medications, also known as anti-psychotics or neuroleptics, are prescribed for conditions such as bipolar disorder, autism with irritability, schizophrenia, and Tourette syndrome. Other medications such as stimulants used to treat ADHD, or levodopa or anticholinergic medications can induce chorea when first started or if the dose is increased to a high level. Other acute choreas can be autoimmune, due to lupus or antiphospholipid antibody syndrome, or metabolic, due mitochondrial or other genetic metabolic diseases.
There are also many neurological diseases which are chronic or degenerative which may include chorea as one symptom. Typically, in these conditions neurologists recognize that the time course, other non-chorea symptoms, and neurological examination are different than Sydenham chorea. Such diseases include hereditary (genetic) disorders such as Huntington’s disease, neuroacanthocytosis, ataxia-telangiectasia, Wilson disease and dentatorubral-pallidoluysian atrophy.
Figure 1. Chorea disease causes
Age of onset
Infancy/childhood onset chorea
The commonest cause of acute chorea in childhood is Sydenham’s chorea, following beta-hemolytic streptococcal group A infection 3. A more insidious onset, especially where there are other neurological abnormalities, suggests a pediatric metabolic disorder. Dystonia appears to be more common than chorea for the disorders with early onset, probably related to the degree of long-tract myelination.
The commonest etiology of chronic movement disorders in this population is cerebral palsy. Spasticity may evolve to a hyperkinetic disorder with age. The history of the disorder should be diagnostic. Dystonia is more commonly seen, but chorea can also occur, and in some cases can be quite violent, being precipitated, for example by metabolic stress such as infection. Severe cases may require general anesthesia or neurosurgical intervention.
Adult onset chorea
Adult onset, often following the reproductive years, is typical for the autosomal dominant disorders. For the trinucleotide repeat expansion, the age of onset is inversely related to the size of the trinucleotide repeat disorders. However, young-onset forms are not uncommon, particularly with paternal inheritance.
Atypical forms of autosomal recessive disorders can present during adulthood, for example, when the causative mutations result in only partial enzyme deficiency. This can be seen with pantothenate kinase-associated neurodegeneration and ataxia-telangiectasia 6. Other autosomal recessive disorders, such as chorea-acanthocytosis 7, present later, for reasons which are unclear. In chorea-acanthocytosis subtle psychiatric signs of basal ganglia dysfunction may be present during adolescence. In other disorders, such as aceruloplasminemia, it is likely that symptoms emerge once a critical amount of substrate, e.g. iron in the case of aceruloplasminemia, has accumulated in the target organs 8.
The X-linked McLeod neuroacanthocytosis syndrome may be detected prior to neuropsychiatric manifestations if blood-typing is performed, or if serological tests demonstrate elevated levels of creatine kinase or liver enzymes; however, symptoms typically emerge in middle-age 9.
Late adult onset
The entity of “senile chorea” is obsolete as these cases are now recognized as most probably being due to late onset of genetic disorders, including Huntington’s disease. Elderly patients who develop chorea should be thoroughly evaluated for a paraneoplastic syndrome or for an autoimmune disorder, especially if female. Late onset of Huntington’s disease may be seen with CAG repeats in the borderline range. As the trinucleotide repeat will most likely expand with inheritance, genetic counseling issues in this age group may potentially involve several younger generations.
Disease progression
As with many other neurological and medical disorders, the characteristics of disease onset and progression can narrow the differential diagnosis. Sudden onset of chorea which does not progress indicates a stroke, either ischemic or hemorrhagic. Acute onset of chorea in childhood is most often due to Sydenham’s chorea.
Acute or subacute onset of chorea related to a medical illness suggests a common underlying metabolic, post-infectious, or autoimmune process. Examples of these include non-ketotic hyperglycemia in a non-insulin-dependent diabetic patient, and other metabolic disorders such as hyper-and hyponatremia, hyper- and hypocalcemia, hypomagnesemia, hyperthyroidism, hypo- and hyperparathyroidism. A variety of autoimmune diseases have been associated with movement disorders, including chorea; these include systemic lupus erythematosus (SLE), Sjögren’s syndrome, and anti-phospholipid antibody syndrome.
A sub-acute presentation may be seen with prion diseases, specifically new variant Creutzfeldt-Jakob disease 10 or with slowly growing mass lesions. Paraneoplastic syndromes can present similarly, and have been reported to occur with a variety of tumors, including renal, small cell lung, breast, Hodgkins and non-Hodgkins lymphoma. The causative auto-antibodies include anti-CRMP-5/CV2, anti-Hu, anti-Yo, and others. Bizarre movements can be seen in anti-NMDA-receptor antibody syndrome, which may be choreiform in nature, although are typically more complex 11.
A chronic, progressive disease course is likely to indicate a neurodegenerative process, for example due to Huntington’s disease, the autosomal dominant ataxias, a neuroacanthocytosis syndrome, or a neurodegeneration with iron accumulation syndrome. The appearance and progression of neurological, cognitive or psychiatric findings due to more widespread neurodegeneration can be informative.
Stable, chronic chorea which develops gradually, but does not progress, may be attributed to a medication, either as a direct side-effect, or as a tardive syndrome. A detailed medication history should reveal this diagnosis. Benign hereditary chorea should be considered in the setting of childhood onset and an autosomal dominant family history.
Episodic chorea may be due to one of the paroxysmal hyperkinetic disorders, although these are more typically dystonic and are classified with the genetic dystonias. The diagnosis of these disorders was previously based upon their phenomenology and precipitating factors, but this is being replaced by molecular identification 12. These include paroxysmal non-kinesigenic dyskinesia (paroxysmal dystonic choreoathetosis; DYT8), which is precipitated by alcohol, caffeine, stress, extremes of temperature, fatigue, or fasting; paroxysmal choreoathetosis with spasticity (DYT9), which is brought on by exercise, stress, alcohol consumption and sleep deprivation; paroxysmal kinesigenic dyskinesia (paroxysmal kinesigenic dystonia; DYT10), characterized by very frequent, brief, episodes of limb dystonia following movement; paroxysmal exertional dyskinesia (DYT18) develops following more prolonged exertion than paroxysmal kinesigenic dystonia, and is due to mutations of the glucose transporter GLUT1. Many of these disorders appear to be due to mutations of ion channels and respond to anticonvulsants. Rarely, episodic chorea may be psychogenic in etiology.
Medication and drug use
Chorea can be caused by a variety of medications, and can be either as a direct, and often predictable, effect, as with dopaminergic agents in Parkinson’s disease.
The movements of tardive dyskinesia (tardive dyskinesia) are more accurately described as chorea, although dystonia can also be seen, and is more disabling. Although much less common than with typical neuroleptics, tardive dyskinesia has been reported with many of the newer atypical antipsychotics. Other dopaminergic antagonists, such as those used for nausea, specifically compazine and metoclopromide, may also cause tardive dyskinesia.
It may be hard to confirm or refute the association of other medications with tardive dyskinesia, as the movements may get worse, rather than resolving, when the offending agent is stopped. Other classes of medications have also been implicated including selective serotonin re-uptake inhibitors (SSRIs), lithium, and anticonvulsant medications.
Chorea may be seen as a direct side-effect of other medications, including dilantin, gabapentin, lamotrigine, and lithium. This may be seen particularly when there is an underlying structural brain lesion, as in cerebral palsy. Stimulants which release catecholamines can be causative, whether used therapeutically, or recreationally, such as amphetamine and cocaine. The timing and the resolution of the movements with discontinuation of the offending agent makes the diagnosis and treatment straightforward.
Practitioners should be alert to the appearance of atypical neurologic and cognitive features in patients who have been diagnosed with tardive dyskinesia following neuroleptic use for psychiatric disease, as these may be indicative of a neurodegenerative basal ganglia disorder.
Chorea disease diagnosis
Diagnostic tests
Neuroimaging
Neuroimaging should be performed in all patients with undiagnosed chorea. Ideally this should be brain MRI with contrast to exclude a space-occupying lesion, and with sequences sensitive to iron deposition. In addition to tumors, benign or malignant, causative lesions may be vascular, most commonly stroke, but also vasculitis, moya-moya disease, cavernous angioma, or arteriovenous malformation. Multiple sclerosis is a rare cause of chorea.
In the neurodegenerative disorders the head of the caudate nucleus is typically atrophic, and may be interpreted as being consistent with a diagnosis of Huntington’s disease.
Mineral deposition can be seen particularly in the basal ganglia, especially the globus pallidus, probably due to high metabolic demands. CT scan may be necessary to distinguish calcium from iron deposition. “Fahr’s disease” refers to the non-specific neuroradiological finding of intra-cranial deposition of calcium, which may be associated with a variety of neurologic features, including chorea, dystonia, parkinsonism. The structural lesion due to calcium deposition in the putamen may be the direct cause of chorea, but this could also be attributable to effects upon neurotransmission. Intra-cranial calcium deposition may be due to a number of different disorders, including those involving calcium and mitochondrial metabolism.
Neurodegeneration with brain iron accumulation refers to several distinct genetic disorders. These include neuroferritinopathy, aceruloplasminemia, and mutations of PLA2G6. The classical MRI finding is the “eye-of-the-tiger”, due to basal ganglia iron deposition and central edema, although there may be some variation in the different disorders.
Cases of reversible chorea associated with herniated cervical discs have been reported, although the pathophysiology of chorea originating outside the basal ganglia is obscure.
Serological tests
Sydenham’s chorea can be diagnosed by the presence of anti-streptolysin O and anti-DNAse B antibodies 3. Testing for appropriate autoantibodies should be performed to evaluate for autoimmune diseases including SLE, antiphospholipid antibody syndrome and Sjögren’s syndrome.
If indicated, serum and cerebrospinal fluid can be tested for the presence of autoantibodies due to a paraneoplastic syndrome. If these are found, or if there is a high degree of suspicion for a neoplasm, patients should undergo neuroimaging of the thorax, abdomen and pelvis. Sometimes, for example with ovarian teratomas, neoplasms are only detected following exploratory surgery.
The finding of elevated levels of serum liver enzymes raises the suspicion of Wilson’s disease 13 or a neuroacanthocytosis syndrome 14. Elevated creatine kinase is typical of either of the NA syndromes due to myopathy, especially in McLeod syndrome.
In addition to Wilson’s disease, markedly reduced ceruloplasmin may be found in aceruloplasminemia. Low ferritin can be indicative of neuroferritinopathy.
Serum alpha-fetoprotein (AFP) may be elevated in ataxia-telangiectasia and help distinguish this disorder from other childhood-onset ataxias. However, atypical forms have recently been reported, thus this may be a useful screening test for adults with unusual movement disorders.
Hypoalbuminemia and elevated cholesterol can be seen in ataxias with oculomotor apraxiaI. Findings of elevated cholesterol, creatine kinase, and alpha-fetoprotein support the diagnosis, although alpha-fetoprotein levels are typically lower than those seen in ataxia-telangiectasia 15.
Peripheral blood smear
Acanthocytosis refers to the finding of contracted thorny erythrocytes on peripheral blood smear. The presence of acanthocytes suggests, but is not necessary for the diagnosis of, one of the core neuroacanthocytosis syndromes. Acanthocytes can be found in approximately 10% of cases of pantothenate kinase-associated neurodegeneration and Huntington’s disease-like 2. The presence of acanthocytosis may vary over time for reasons which are not understood, and may not be detected even with repeated testing, despite utilization of a sensitive protocol 16. In this circumstance the serum enzymes mentioned above may be more informative.
Erythrocyte phenotyping
Erythrocyte antigen phenotyping should be performed if the diagnosis of McLeod syndrome is suspected 17. Testing for these antigens requires a panel of anti-Kx and anti-Kell antibodies, and is usually performed at regional blood banks, which should be asked explicitly to exclude McLeod phenotype.
Electroencephalography
EEG may be informative in identifying disorders in which myoclonus and seizures co-occur with chorea. These include prion diseases, dentatorubropallidoluysian atrophy, and the core neuroacanthocytosis syndromes.
Chorea treatment
There is no standard course of treatment for chorea. Treatment depends on the type of chorea and the associated disease. Treatment for Huntington’s disease is supportive, while treatment for Sydenham’s chorea usually involves antibiotic drugs to treat the infection, followed by drug therapy to prevent recurrence. Adjusting medication dosages can treat drug-induced chorea. Metabolic and endocrine-related choreas are treated according to the cause(s) of symptoms.
References- Chorea Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Chorea-Information-Page
- Sydenham Chorea Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Sydenham-Chorea-Information-Page
- Gilbert DL. (2009). Acute and chronic chorea in childhood. Semin.Pediatr.Neurol. 16:71-76.
- Huntington disease. https://ghr.nlm.nih.gov/condition/huntington-disease
- An approach to the patient with chorea. https://www.movementdisorders.org/MDS-Files1/News/Archived-Content/choreaposter12.pdf
- Verhagen MMM, Abdo WF, Willemsen MAAP et al. (2009). Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology 73:430-437.
- Danek A, Walker RH. (2005). Neuroacanthocytosis. Curr.Opin.Neurol. 18:386-392.
- McNeill A, Pandolfo M, Kuhn J et al. (2008). The neurological presentation of ceruloplasmin gene mutations. Eur.Neurol. 60:200-205.
- Danek A, Rubio JP, Rampoldi L et al. (2001). McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol 50:755-764.
- Bowen J, Mitchell T, Pearce R et al. (2000). Chorea in new variant Creutzfeldt-Jacob disease. Mov Disord. 15:1284-1285.
- Dalmau J, Tuzun E, Wu HY et al. (2007). Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann.Neurol. 61:25-36.
- Mink JW. (2007). Paroxysmal dyskinesias. Curr.Opin.Pediatr. 19:652-656.
- Machado A, Fen CH, Mitiko DM et al. (2006). Neurological manifestations in Wilson’s disease: Report of 119 cases. Mov Disord. 21:2192-2196.
- Rampoldi L, Danek A, Monaco AP. (2002). Clinical features and molecular bases of neuroacanthocytosis. J.Mol.Med. 80:475-491.
- Le Ber I, Bouslam N, Rivaud-Pechoux S et al. (2004). Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 127:759-767.
- Storch A, Kornhass M, Schwarz J. (2005). Testing for acanthocytosis – A prospective reader-blinded study in movement disorder patients. J.Neurol. 252:84-90.
- Russo D, Redman C, Lee S. (1998). Association of XK and Kell blood group proteins. J.Biol.Chem. 273:13950-13956.





{kind=link}