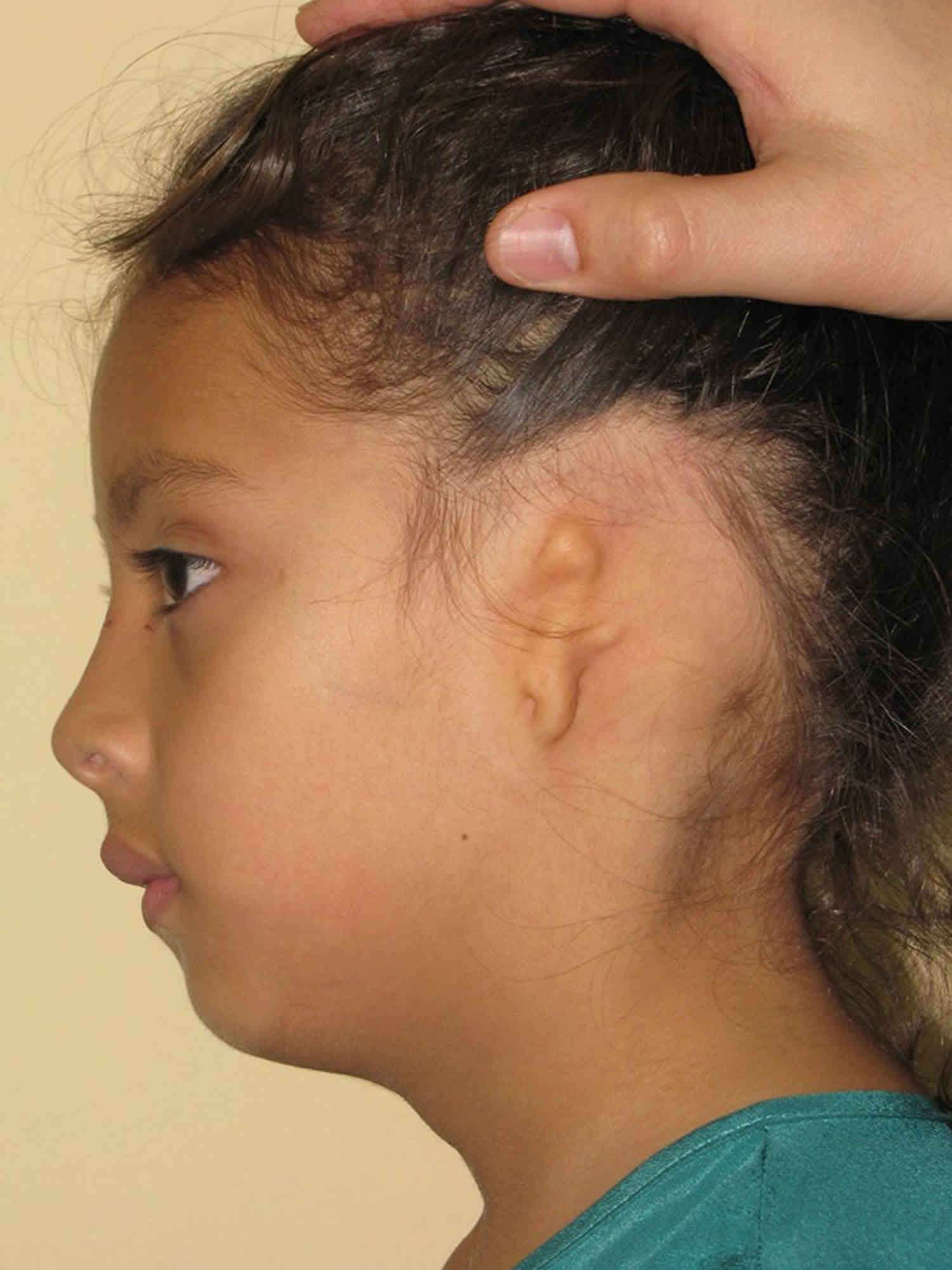
Aural atresia
Aural atresia is the absence of the ear canal. Aural atresia can be in one ear (unilateral) or both (bilateral). It is often part of microtia, a condition when the outside of the ear (pinna) does not form correctly. Microtia refers to a small, abnormally shaped ear, which can range from a minimally deformed small ear to a complete absence of the ear. In congenital aural atresia, the external auditory canal and structures in the middle ear fail to develop completely, which means that microtia and atresia are commonly found together. This causes a hearing loss (usually a “maximal conductive loss”) because the sound waves cannot get through to the eardrum/inner ear. Usually, the nerve for hearing and inner ear is normal, and this can be determined with a hearing test (audiogram).
Microtia is classically graded 1, 2, and 3:
- Grade 1 is simply a smaller than normal ear with all the normal hills and valleys of the delicate cartilage.
- Grade 2 is smaller than normal but not all the hills and valleys of the cartilage developed.
- Grade 3 microtia is a “peanut” ear with a small cartilage or soft tissue remnant.
Embryologic development of the ear canal and middle ear may be arrested at any point in the process. Therefore, the clinician may encounter varying degrees of severity of this malformation. In the severe form of the disorder, no identifiable ear canal exists (complete atresia), and the middle ear and its structures (ossicles, or ear bones) may be absent or show significant underdevelopment. If a semblance of an external auditory meatus is present, the ear canal may end in a shallow blind pouch. In less severe forms of the disorder, the ear canal may be stenotic (narrow) with a pinpoint aperture leading into the medial ear canal and, possibly, a rudimentary tympanic membrane. The tympanic membrane in these cases may or may not be attached to the ear bones (the ossicular chain).
There are two main problems in children with aural atresia and microtia: hearing loss and cosmetic deformity. Hearing loss is the top priority – studies have shown that children with hearing loss even just on one side can have increased difficulties in school. This can also affect interactions with friends and family. Some children with hearing loss on one side do very well without intervention, but we strive to achieve the highest potential for each child and monitor their hearing status very closely.
The cosmetic deformity of having an abnormally shaped ear can be surprising for parents, but children do not usually notice that they are “different” until around age 4-6. This can cause some shyness or self-consciousness with friends, but children can be counseled to refer to their microtic ear as their “little ear” and it becomes less of an issue. Children with microtia are usually normal kids who just happen to be missing an ear.
A child with unilateral aural atresia should:
- Not rely solely on a newborn hearing screening. An audiologist should perform a complete hearing assessment for your child at birth.
- Have regular follow up evaluations with an audiologist to
- Monitor communication skills
- Determine if a speech and language evaluation is required
- Be watched closely for ear infections. These may cause delays in speech and language skills
A child with bilateral aural atresia should:
- Have a complete audiological evaluation by a pediatric audiologist
- Be enrolled in early intervention services for speech and language therapy
All children with aural atresia undergo a formal hearing test (audiogram), usually between 6 and 12 months of age. Then, they should have their hearing checked at least yearly, or anytime there is a major change. Children with bilateral atresia are typically fitted with a headband which has hearing aids attached, so that they may hear sounds and develop speech in a normal fashion.
One option for treatment is an “osseointegrated” hearing device, either the BAHA Attract or Sophono Alpha 2. These are hearing processors that attach to a magnetic device implanted under the skin. The device sends the hearing signal from the bone directly to the hearing nerve to transmit sound. The device is safe and implanted with one surgery. Activation is done only one month later. When children reach age 5, they are candidates for implantation and are tested with the audiologists to see if this will benefit them.
Surgery can often be performed on children who have unilateral or bilateral atresia. Your child must be at least 5 years old to undergo surgery.
First, a computed tomography (CT) scan will be performed to make sure that the middle and inner portions of the ear are developed enough to allow surgery to be performed.
Next, a surgeon first repairs the microtia, if necessary. After that heals, an ear surgeon (otologist) creates an ear canal and eardrum to improve hearing. This is called an atresia repair. A small skin graft from the thigh or hip is usually needed to line the new ear canal.
Follow-up visits to the ear surgeon are required every six to 12 months to clean out the ear canal. Earplugs may be needed while bathing or swimming to prevent infections.
Hearing results for children with auricular atresia are good. Many children can hear on the telephone and notice an improvement in identifying the direction from which sounds are coming. While many children have near-normal hearing, completely normal hearing should not be expected.
Some children may need to wear a hearing aid in the operated ear.
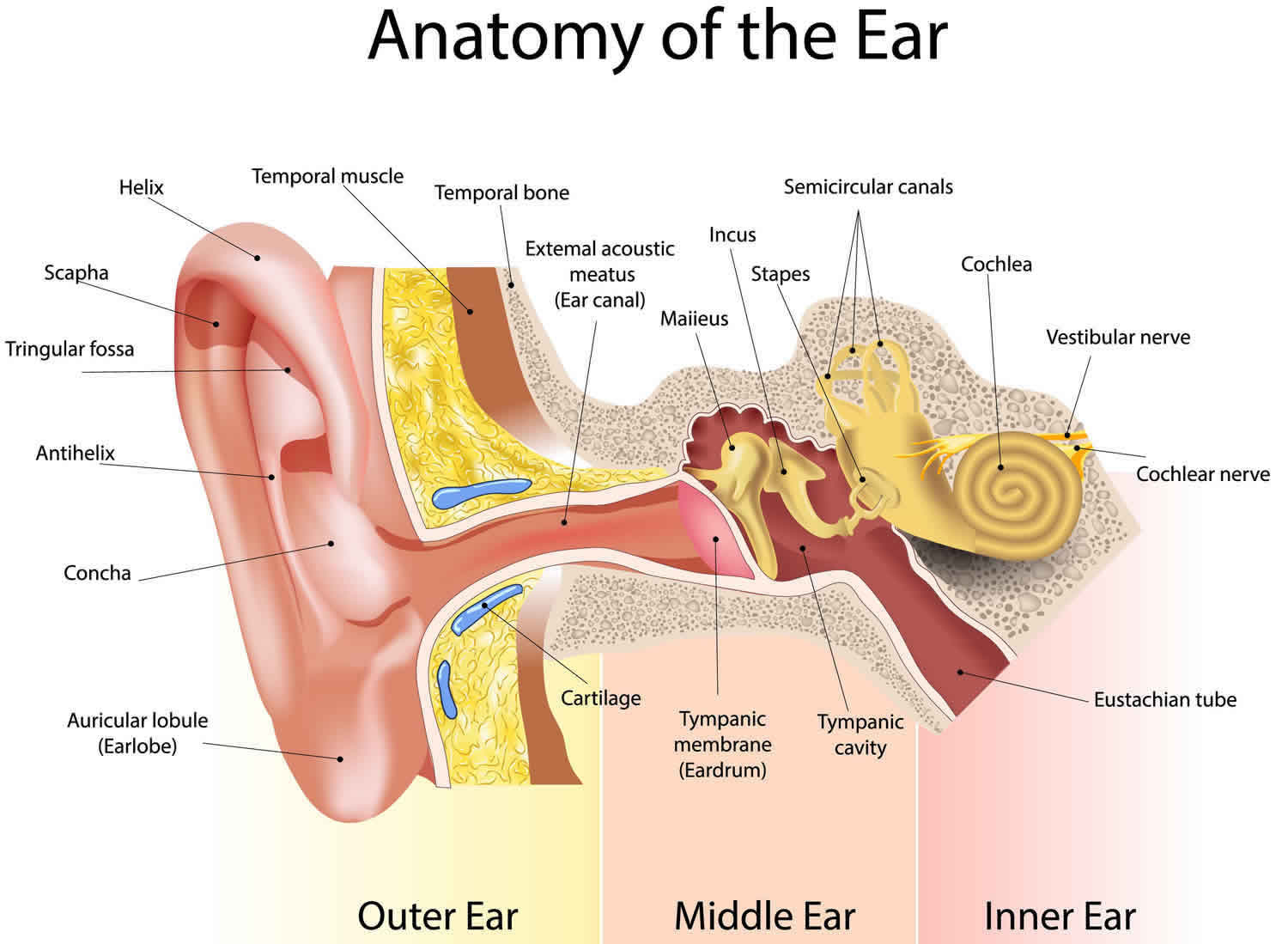
Figure 1. Ear anatomy
Is there an age that is too young to repair congenital aural atresia?
Certainly, the child who cannot sit still in the office for the necessary packing removal and ear canal cleaning is not old enough to undergo the surgery, unless the surgeon returns the child to the operating room for packing removal and debridement. In addition, younger children are more susceptible to “ear infections” and middle ear fluid, negating the hearing gains of atresia surgery. Waiting until the child is aged 5-7 years mitigates this potential complication.
The auditory system, unlike the visual system, sends signals from both ears to both sides of the brain. Central auditory pathways cross the midline very early in the auditory pathway so that both sides of the brain receive stimulation from both ears. This means that both sides of the brain are being stimulated by the one good ear in unilateral aural atresia. The argument that the earlier the surgeon opens the atretic ear, the less chance the brain will “close down” (and not be able to respond) to input from the reconstructed ear has yet to be supported. In fact, studies have shown that children with unilateral aural atresia do not have the same grade retention rates as their peers with unilateral sensorineural hearing loss with the same resource utilization 1.
Even still scientists are just beginning to understand whether a patient can “use” their new ear. Certainly in some cases yes, but sound localization and some tasks of hearing in noise remain elusive for many postoperative atresia patients, even with excellent hearing outcomes 2. In general, hearing in noise appears to be improved by atresia surgery 3.
The difficulty patients have with one good ear and one atretic ear is difficulty hearing in background noise and difficulty locating a sound source. Successful surgery to open the atretic ear canal and restore hearing to the ear may eliminate (or greatly reduce) these 2 problems, whether the patient is 5 or 55. Ongoing research is attempting to quantify and qualify the difficulties children with unilateral hearing loss have, both in school and in their day-to-day activities.
Time and ongoing analysis with longitudinal studies will determine if atresia surgery is an advantage in the classroom for children with aural atresia.
Classification of congenital aural atresia
Congenital aural atresia is a birth defect that is characterized by hypoplasia or aplasia of the external auditory canal, often in association with dysmorphic features of the auricle, middle ear, and, occasionally, inner ear structures. The classification of congenital aural atresia recommended here is based on both clinical and surgical observations 4.
- Type A. Stenosis: Stenosis is narrowing of the fibrocartilaginous or bony parts of the EAC, in which the tympanic membrane is present, but it is smaller than normal, with slight deformity. The ossicular chain generally develops normally but is often fixed; most cases have a mild to moderate conductive hearing loss.
- Type B. Partial atresia: Some part of the fibrocartilaginous or bony external auditory canal is present. There is a bony atretic plate, and the tympanic membrane is missing or may be rudimentary. The tympanic membrane is often not attached to the ossicular chain, which may also be underdeveloped. The patient generally has a moderate or moderate–severe conductive hearing loss.
Type C. Total atresia: There is complete aplasia of the external auditory canal with a bony atretic plate. Both fibrocartilaginous and bony parts of the external auditory canal are absent. The tympanic membrane is also absent. There is some degree of underdevelopment of the middle ear and its associated structures. The patient has a moderate or moderate–severe conductive hearing loss.
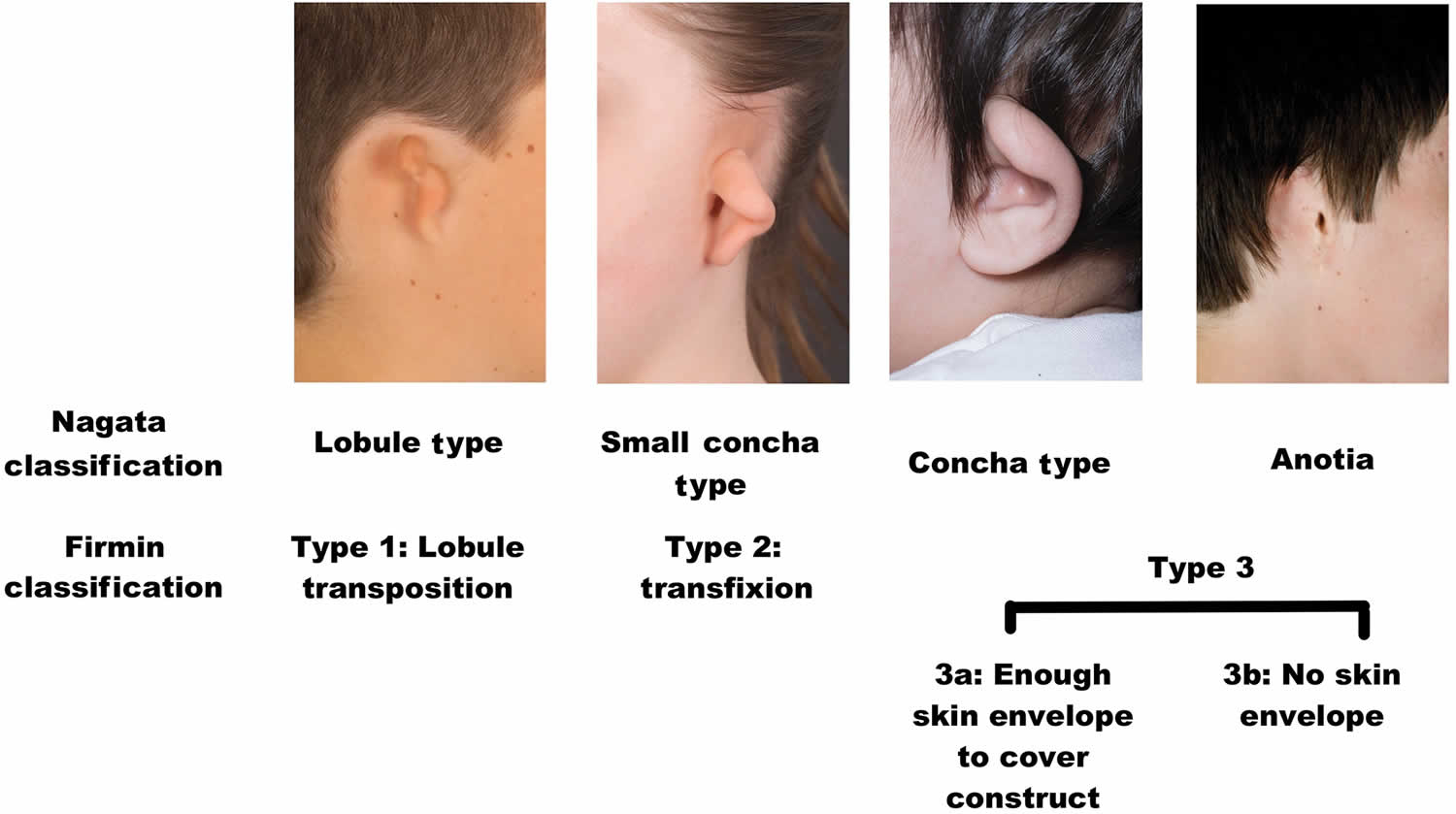
Classification of microtia
Microtia is a term used to describe a smaller and usually malformed auricle. One widely adopted classification system, originally described by Weerda and simplified by Aguilar, assigns a grade from 1 to 3 based on the severity of the deformity (Figure 2).
- Grade 1 depicts a slightly smaller than normal ear with essentially normal features.
- Grade 2 represents an auricle that is rudimentary and malformed but contains some recognizable components.
- Grade 3 includes the classic “peanut” ear, which is severely attenuated with a small lump of deformed tissue often containing some cartilage.
- Grade 4 includes anotia.
Another commonly cited classification system, proposed by Nagata, categorizes microtia according to the vestigial structures present. Lobule-type microtic ears have a remnant ear and lobule but lack a concha, acoustic meatus, and tragus; these usually correspond to Grade 3. Concha-type microtic ears possess some degree of a lobule, concha, acoustic meatus, tragus, and incisura tragica; these usually correspond to Grade 2, according to the previously described system. Small concha-type microtic ears contain the remnant ear and lobule with a small indentation for a concha.
Figure 2. Microtia classification

Aural atresia causes
Factors that cause the developmental sequence of the ear canal and middle ear to cease in congenital aural atresia are not known. Regions adjacent to the ear canal, including the jaw bone (mandible) may be affected as well. In general, the inner ear (cochlea and hearing nerve) is not affected because its development is from a completely separate set of structures from the ear canal and middle ear, and the development of the inner ear is complete by the time the ear canal begins to form. In general, the middle ear, ossicles, and the auricle are affected because their development is concurrent with that of the ear canal. Recent studies have linked mutations in chromosome 18 to some patients with aural atresia 5.
Embryology of the ear
The development of the ear consists of a complex series of events. The inner ear, middle ear space, ossicles, ear canal, and auricle each are derived from the 3 embryonic germ cell layers (ie, ectoderm, mesoderm, endoderm). These structures assume individual paths of growth and maturation.
The ear canal and middle ear develop from the branchial apparatus, a series of ectodermal outpouchings (arches) and indentations (clefts/grooves) on the lateral surface of the embryo that are first clearly seen at 24 days gestation. The formation of the ear canal begins with the invagination of the first branchial groove (ie, primary meatus). This area is located between the first branchial arch rostrally (toward the head) and the second branchial arch caudally (toward the tail).
The branchial groove invaginates and advances medially as an epithelial plate as early as the second month of fetal life. Its ingrowth temporarily meets the lateral growth of the first pharyngeal pouch. The first pharyngeal pouch is derived from endoderm and subsequently develops into the middle ear cleft and Eustachian tube. The union between the ingrowth of the first branchial groove and first pharyngeal pouch forms the meatal plate, which is the precursor to the tympanic membrane. At 6 months of fetal life, the epithelial plate begins to canalize (open) from a medial to lateral direction (from the inside out) to meet the primary meatus.
At birth, the ear canal comprises the bony tympanic ring medially and a membranous cartilaginous portion laterally. Postnatally, the bony tympanic ring lengthens and transforms from a ring into a bony cylinder. Thus, the ear canal increases in length and reaches adult proportions by the age of 4-5 years.
In congenital aural atresia, development of the ear may be interrupted at any point. If the process is halted before canalization of the ear canal, total atresia occurs. If development is halted during canalization, the patency of the external ear canal varies depending on how far canalization has progressed.
Development of the middle ear space and ear bones (ossicles) is closely aligned with the development of the ear canal, from the branchial apparatus. The middle ear space arises from lateral growth of the first pharyngeal pouch. The middle ear eventually envelops the ossicles and incorporates them into the middle ear space. The ossicles are derived from both first and second branchial arches, specifically Meckel and Reichert cartilages, respectively. Arrest in the development of the ear canal affects the middle ear to the extent that the growth and maturation of the middle ear will also be incomplete, and the middle ear space is constricted. The ossicular chain is usually deformed, typically featuring a fused malleus-incus complex, shortened malleus with absent manubrium, and occasionally, hypoplastic stapes suprastructure. The malleus neck may be fused to the bony atretic plate. In cases of complete atresia, the tympanic membrane is absent.
The inner ear begins development as early as the third week of fetal life with the formation of the otic placode, a local thickening of the ectoderm. The otic placode invaginates to form the otic pit. The epithelium of the otic pit fuses to become the otic vesicle, which forms the membranous labyrinth of the inner ear. A series of infoldings of the otic vesicle compartmentalizes the membranous labyrinth into the vestibule, cochlea, and endolymphatic regions. The inner ear completes its development by the 20th week of fetal life, which predates the formation of the ear canal and explains why patients with congenital aural atresia generally have a cochlea that is functional and able to be stimulated.
Congenital aural atresia commonly coexists with microtia. However, rarely, congenital aural atresia may occur alone with an auricle that appears healthy. The embryologic precursors of the auricle are the axonal hillocks, a series of ectodermal elevations derived from the first and second branchial arches located along the rostral end of the branchial apparatus. The axonal hillocks fuse with each other around the primitive ear canal. Each of the 6 hillocks is responsible for forming a distinctive part of the auricle. Hillock 1 forms the tragus; hillock 2 forms the crus helicis; hillock 3 forms the helix; hillock 4 forms the antihelix; hillock 5 forms the antitragus; and hillock 6 forms the ear lobule. The auricle assumes adult shape (but not size) by the 20th week of fetal life. Microtia (“small ear”) results when the development of these hillocks is arrested. In general, the middle ear is less developed when microtia is severe than when it is not 6.
Aural atresia symptoms
The patient with congenital aural atresia may present with unilateral or bilateral atresia. If the deformity is present in both ears, the severity often varies between the ears. Hearing evaluation through auditory-evoked brainstem response (ABR; BAER) testing and hearing rehabilitation, generally with a bone conducting hearing aid, are critical early in life for normal language development in children with bilateral congenital aural atresia.
A study by Montino et al 7 found that out of 68 children with congenital aural atresia, 94.3% had age-adequate feeding abilities, 44.1% had language development delays, and, in patients with bilateral, right, or left congenital aural atresia, 57.2%, 53.9%, and 53.4%, respectively, had orofacial development delays.
Colman graded the severity of congenital aural atresia into 3 categories: minor, moderate, and severe. Patients with severe congenital aural atresia have total canal atresia and usually present with unfavorable middle ear anatomy and temporal bone development for reconstruction. In comparison, patients with moderate congenital aural atresia have more favorable anatomy (ie, an identifiable ear canal with deformed ossicles and an aerated middle ear space). Most patients with congenital aural atresia present with this moderate form. In minor cases, the ear canal is present but narrowed, and the middle ear is better developed.
Congenital aural atresia may coexist with syndromes that feature first and second branchial arch deformities (eg, Treacher-Collins syndrome, hemifacial microsomia, Goldenhar syndrome, and other craniofacial abnormalities).
A child with canal stenosis (mild atresia) may present to the clinician with or without associated hearing loss in the ear. These patients with narrowed canals (generally less than 2 mm are at risk of canal cholesteatoma 8. Cholesteatoma is a pocket or cyst of skin that grows and expands as dead skin cells fill the cyst. Careful observation, follow-up, and microscopic examination and cleaning may ensure the patient does not develop a canal cholesteatoma. However, when the opening to the ear canal is too small to permit examination or when the patient’s history (eg, otorrhea, otalgia) suggests a cholesteatoma, radiographic imaging with CT scanning is used to evaluate the possibility of a canal cholesteatoma.
Children with canals 2 mm or smaller in diameter develop canal cholesteatoma more frequently than do other children. In addition, canal cholesteatomas are rarely, if ever, observed to occur in children younger than 3 years. Cole and Jahrsdoerfer reported these findings and noted that 50% of 54 stenotic ears with canals smaller than 4 mm developed canal cholesteatoma 8. Clinicians should readily incorporate CT scanning into their diagnostic workup to ensure that no canal cholesteatoma, medial to the stenosis, is present.
Detection at birth
If the anatomic abnormality is detected at birth, the otologist examines the newborn and sees the parents in consultation. The parents are usually anxious and eager to have their many questions answered. Reassurance and parent education are critical during this time. Regardless of whether this condition is present in 1 or both ears, cochlear function must be assessed in both ears by means of specialized audiologic testing, such as auditory-evoked brainstem responses (ABR or BAER testing) elicited from both air-conducted and bone-conducted signals. If bilateral congenital aural atresia and good cochlear function are present, the infant needs amplification with a bone-conducting hearing aid (eg, BAHA Softband) until a decision is made to pursue surgical correction around the age of 5-7.
The decision to amplify, or place a bone-conducting hearing device, on the child with unilateral congenital aural atresia is one made individually by the family. There are advantages and disadvantages to using a bone conductor as well as not using one. Physicians should never discourage families from trying a bone conductor on their child with unilateral congenital aural atresia, but families should not be discouraged if their child refuses to wear it. While researchers are discovering some of the more subtle problems associated with unilateral hearing loss, especially in school-age children, these studies have been conducted in children with unilateral sensorineural hearing loss, not conductive hearing loss 9. A more recent study has shown that children with unilateral congenital aural atresia did not require repeating a grade in elementary education but did utilize resources such as an individual education program (IEP) to a greater degree 1. In general, normal hearing in one ear is enough hearing to support normal speech and language development.
That said, it is critical in the early years to monitor the hearing in the good ear to ensure that the ear is hearing well. Follow-up ABR testing is recommended every 3-4, then every 6 months in the first 1-2 years of life, followed by behavioral testing every 6 months in the second 2 years of life and, if the hearing is stable, annually thereafter.
Once in the elementary school years, the child should be monitored for academic success. Resources such as an individual education program (IEP), speech therapy, preferential seating in class, and FM systems should be instituted early to set the child up for success in the academic environment.
Detection in the older child just before microtia repair
The otologist may encounter the older child with unilateral congenital aural atresia before a plastic surgeon repairs microtia. In these patients, coordination of care between the plastic surgeon and otologist is important to delineate a clear surgical plan and schedule to prevent confusion and unrealistic expectations 10. Using the patient’s own rib (“autologous”), the plastic surgeon sculpts an auricle and pockets it under the skin, moves the lobule, elevates the sculpted rib off the skull with a skin graft, and creates a tragus in a multistage (3-4 operations over a one year period) procedure. The otologist opens the new ear canal, frees the ossicular chain so the ear bones can vibrate, constructs a tympanic membrane, lines the ear canal with a skin graft, and opens the meatus in continuity with the new ear canal in a single stage procedure.
A porous polyethylene implant (Medpor) has recently been established as a viable means of reconstructing the auricle. Long-term (12 years) data show a complication rate of exposure of the implant at 7% 11. The implant in the setting of atresia can be problematic if the atresia repair is performed after the implant has been placed. In this instance, the implant is at risk for exposure, infection, and extrusion. Patients have undergone atresia repair before Medpor implantation with short-term success 12. Revision atresia repair in these patients again puts the Medpor implant at risk.
Adult with congenital aural atresia
Adults with congenital aural atresia who present to the otologist usually have a unilateral condition that was not corrected in childhood. In some patients, the condition was unnoticed, especially in those with stenotic ears. If it was noticed, the patients may have been told that their condition was inoperable or too risky to repair in light of possible facial nerve injury. This is especially true if the contralateral ear was morphologically and functionally normal. In this situation, the patient may make an informed decision to proceed with an evaluation for surgery with dedicated CT scan and audiometry with the hopes of obtaining binaural hearing.
Aural atresia diagnosis
Imaging studies
Thin section (≤1-mm sections), high-resolution, axial and coronal computed tomography (CT) of the temporal bone is the study of choice in the surgical workup and evaluation of patients with congenital aural atresia (congenital aural atresia). Given the radiation exposure of this imaging modality, the CT scan is not recommended until the child is being considered for surgery, around the age of 5-6 years.
- Perform the study by obtaining contiguous images with a thickness of 1 mm or less (submillimeter thickness) in both axial and coronal planes (coronal reconstruction is adequate with the appropriate software).
- Imaging provides information on whether surgical repair is feasible based on the anatomy present.
- Obtain imaging when surgical correction is contemplated or just before surgery.
- As long as audiologic evidence reveals that the inner ear is functional, imaging is not necessary during early infancy and childhood.
In 1992, Jahrsdoerfer et al 13 developed a 10-point grading system to determine surgical candidacy based on key features from the CT scan and the appearance of the external ear. This scheme is also used to indirectly estimate the likelihood of success if surgical correction is performed 14.
Jahrsdoerfer 13 recommends operating on unilateral patients with scores of 7 or higher; bilateral patients with a grade 5-6 or higher. This system bases surgical success on a postoperative speech-reception threshold (SRT) of less than or equal to 30 dB HL, which, depending on the anatomy, is attainable in over 80% of patients 14. As a reflection of the conservative nature of this grading system, a score of 10 is never given.
The following is a summary of Jahrsdoerfer’s 10-point grading system 14:
- Stapes present = 2 points
- Oval window open = 1 point
- Middle ear space = 1 point
- Facial nerve = 1 point
- Malleus-incus complex = 1 point
- Mastoid pneumatization = 1 point
- Incus-stapes connection = 1 point
- Round window = 1 point
- External ear appearance = 1 point
Each ear is given a grade based on the CT anatomy and this scale. The anatomy predicts the candidacy of that ear for atresia surgery. Of patients with scores of 7 or greater, 80-90% have been shown to achieve normal or near-normal hearing (≤30 dB HL speech reception threshold) 14.
Audiometry
Audiometry is equally as important as imaging in the evaluation of the patient with congenital aural atresia. Air and bone conduction pure-tone thresholds at 250 Hz, 500 Hz, 1000 Hz, 2000 Hz, 4000 Hz, 6000 Hz, and 8000 Hz, speech reception thresholds, and speech discrimination scores comprise the routine audiological assessment. Air conduction thresholds in the atretic ear tend to run in the 60-70 dB HL range with similar speech reception thresholds. Bone conduction thresholds in the atretic ear are typically in the normal range (and if they are not, the patient may not be a candidate for surgery), with excellent speech discrimination scores.
The masking dilemma arises when testing the patient with bilateral congenital aural atresia.
How does the audiologist establish bone conduction thresholds for each ear in bilateral congenital aural atresia?
The sensorineural acuity level test (SAL) allows the establishment of individual bone conduction thresholds in bilateral congenital aural atresia, as follows:
- Determine patient’s thresholds through earphones to 500, 1000, 2000, and 4000 Hz.
- Place a bone oscillator on the forehead and present masking through it at the following intensities:
- 500 Hz – 50 db HL
- 1000 Hz – 50 db HL
- 2000 Hz – 60 db HL
- 4000 Hz – 60 db HL
- With masking present, reestablish air conduction thresholds.
- At each frequency, determine the difference between the masked air conduction threshold and the unmasked air conduction threshold
- Norms of threshold shift with bone conduction masking are as follows:
- 500 Hz – 40 db HL
- 1000 Hz – 50 db HL
- 2000 Hz – 45 db HL
- 4000 Hz – 50 db HL
If the masked threshold shifts more than the norm, the bone conduction threshold is 0 dB HL. If the masked threshold shifts less than the norm, then the bone conduction threshold is the difference between the norm and the amount of shift (eg, if the norm is 50 and the amount of shift is 30, the masked bone conduction is 20).
Laboratory studies
In a healthy patient with no other abnormalities, laboratory investigation is generally not necessary. A small subset of patients with aural atresia will have 18q chromosome deletion syndrome. These children typically have normal auricles with complete atresia of the ear canal. Genetic testing can identify this rare condition 15.
Aural atresia treatment
Surgery to repair congenital aural atresia is one of the most challenging operations the ear specialist faces. The goals of surgery are to provide the patient with a clean, dry, skin-lined external auditory canal with long-term restoration or improvement in hearing.
An important point to remember: While most all patients with grade 3 microtia are candidates for reconstruction, not all patients with congenital aural atresia are candidates for surgical correction. Among patients with associated syndromes such as Treacher-Collins syndrome or hemifacial microsomia or Goldenhar syndrome, approximately 50% of patients are not surgical candidates because of the existing anatomy. In isolated unilateral cases of congenital aural atresia (most patients), approximately 65-75% are surgical candidates.
For surgical candidates with favorable anatomy, the otolaryngologist/otologist/pediatric otolaryngologist must rely on the skills, techniques, and experience from middle ear and mastoid surgery to optimize the surgical result. Surgical success is based on restoration of useful hearing, long-term stability of hearing, and maintenance of a patent, skin-lined ear canal. Otologists agree that when these goals are achieved, few accomplishments are as gratifying as successfully treating a patient with congenital aural atresia.
International Microtia and Atresia Workgroup Guidelines
Consensus recommendations on the treatment of total congenital aural atresia, published in 2019 by the International Microtia and Atresia Workgroup, include the following 16:
- Bone conduction technology for children with bilateral aural atresia is very strongly recommended to support speech and language development
- Evaluation of the temporal bones using careful computed tomography (CT) scanning is recommended, as is precise assessment of the type of hearing loss and of preoperative motivation with the softband
- To prevent the processor from interfering with microtia reconstruction, a discussion between the otolaryngologist/otologist/pediatric otolaryngologist and microtia surgeon regarding placement of the osseointegrated bone conduction hearing device is strongly recommended
- It is recommended that children over age 6 years whose hearing and anatomy support surgical reconstruction (ie, those with normal inner ear function and a Jahrsdoerfer score of 7 or higher) undergo atresia surgery with the understanding that revision surgery may be required if the canal and hearing are compromised by new bone growth during puberty
- It is strongly recommended that atresia repair be performed in combination with or subsequent to autologous rib graft microtia repair and prior to porous polyethylene microtia repair
Medical therapy
Alternatives to surgical correction include amplification devices, such as a bone-conducting hearing aid or bone-anchored hearing device/BAHA System (Cochlear Corp., Englewood, CO) 17. Conventional hearing aids in patients with canal stenosis may also be used if the hearing device is able to provide adequate amplification. A distinction should be made between the BAHA System (Cochlear Corp., Englewood, CO), which is the osseointegrated titanium implant and speech processor, and the BAHA Softband (Cochlear Corp., Englewood, CO), which is a bone conducting hearing device that is worn on a soft headband around the head. The BAHA system is FDA-approved for children 5 and older in the United States. Oticon Medical Corp. (Somerset, NJ) also has a commercially available bone anchored hearing aid called the Ponto.
As noted above, 2 semi-implantable alternative bone conduction devices can also be considered: the Sophono and the BAHA Attract. These devices require surgical placement of a metallic plate into the bone behind the ear. Once healed, the sound processor, affixed to a magnetic plate, is placed over the implanted metallic plate to stimulate bone conduction of sound energy.
Experience using the BAHA device is considerable, especially in Europe. Long-term results of the US experience with the BAHA are also favorable 18. This device features a surgically implanted, percutaneous titanium fixture-abutment that osseointegrates into the temporal bone. A sound transducer attaches to this titanium implant and delivers the sound energy directly to the cochlea via bone conduction. Therefore, one does not need an ear canal or middle ear to hear 19.
This type of technology is more efficient than the bone-conducting hearing aids because the sound energy is not attenuated by the skin and intervening soft tissues. Speech reception thresholds (SRTs) less than 30 dB are obtainable with the use of these devices. Bilateral BAHA devices have even been used for patients with bilateral atresia 20. Bilateral implants may impart better sound localization ability and possibly better hearing in noisy environments, but this has not been demonstrated consistently experimentally. Furthermore, the use of the BAHA device does not preclude future reconstructive surgery. Potential complications of BAHA placement include failure of osseointegration with extrusion of the fixture/abutment, skin overgrowth over the abutment, and flap necrosis with secondary healing.
Important: When implanting the BAHA implant in a patient who has not had microtia repair, it is critically important to place the implant far enough away from the virginal skin and soft tissue in the postauricular area so that the reconstructive microtia surgeon has room to place the autologous rib cartilage graft without scarring.
These alternatives may be considered in patients with unilateral or bilateral congenital aural atresia who do not desire surgical correction or in whom the existing anatomy is not favorable (a score of 6 or less on the Jahrsdoerfer CT grading scale) but the cochlea is still functional and can be stimulated.
Some audiologists have also tried conventional hearing amplification in patients with canal stenosis. The amplification provided may overcome a mild-moderate conductive hearing loss associated with canal stenosis or minor middle ear malformation.
A study by Attaway et al 21 indicated that in children with aural atresia who are provided with an amplification aid, the development of speech and language abilities are influenced by the age of first amplification, the degree of compliance in aid use, and which ear has atresia. For example, children fitted with the aid before age 1 year and those with left-sided atresia demonstrated better results.
Aural atresia surgical repair
Surgical repair of congenital aural atresia is offered if the patient is deemed a good candidate from both the radiological (Imaging Studies) and audiological evaluations. Patients with unilateral atresia are offered surgery if the anatomy is favorable. Surgery to correct congenital aural atresia is usually performed after the child undergoes the multistage autologous rib graft reconstructive surgery for microtia (usually around 5.5-7 years). This surgery is typically performed after the cartilaginous auricular framework is placed and the ear lobule is created. Surgery is not typically recommended until the child is old enough and mature enough to cooperate in the office for the critical postoperative care, including packing removal and ear canal debridement (usually at 5.5 years). For families choosing the Medpor implant, atresia surgery is recommended prior to placement of the Medpor framework 12.
Timing of surgery
External ear reconstruction is usually delayed until the child is about 5.5-7 years, with atresia repair either preceding the Medpor microtia repair or following the series of operations required for rib graft microtia repair. This delay allows for growth of the rib cage, enabling sufficient costal cartilage to be harvested for sculpting the auricle. A child who is large for his/her age may be operated on earlier. The atresia surgery is typically performed after the cartilaginous auricular framework is placed, the ear lobule created, and the sculpted auricle elevated off the skull with a skin graft.
For the child with Grade 1 or 2 microtia that does not require reconstructive surgery, atresia surgery can be performed at the age of five if the child is cooperative. Waiting until the child is 6 or 7 allows the child to achieve a level of maturity and cooperation absolutely critical for the postoperative dressing changes, packing removal, and ear canal cleaning to ensure a good result. A potentially poor result can be salvaged postoperatively in the office but requires the cooperation of the patient.
Although reconstructive surgeons using the Medpor implant have implanted children as young as 3 years, the authors do not advocate atresia repair at that age because, in this author’s experience, the child is simply too young to cooperate with the necessary postoperative packing removal and ear canal debridement. In addition, younger children may have a higher rate of meatal or canal stenosis necessitating revision surgery. Finally, younger children have a higher incidence of Eustachian tube dysfunction and middle ear fluid. Waiting until the child is older allows the Eustachian tube to mature and aerate or ventilate the middle ear space more effectively so that the middle ear stays clear of fluid, inflammation, and infection. The younger child is certainly not bothered psychologically by the ear deformity and is not yet in school where other children could tease him or her.
Corrective surgery may also be delayed until adulthood, when the patient can make an informed decision. The benefits of binaural hearing and improved sound localization must be weighed against the potential complications of surgery, including canal stenosis and new bone growth seen in younger patients. Research is ongoing regarding the optimal timing of atresia surgery and whether the child or adult can “use” their ne ear in tests of binaural (use of both ears in a coordinated fashion) hearing – hearing in background noise and sound localization.
Surgery in patients with bilateral congenital aural atresia is performed with the goal to restore useful hearing, unaided or aided. Surgery can take place just before the child enters school, usually when aged 5-6 years. Unlike classical otologic surgery, the better ear (anatomically) is chosen for surgery to repair congenital aural atresia.
Aural atresia repair complications
Injury to the facial nerve is one of the most feared complications of surgery for correction of congenital aural atresia (congenital aural atresia). This complication has historically deterred surgeons from correcting this condition. However, in experienced hands and with improved imaging techniques, the complication rate has been below 0.1%.
The facial nerve is estimated to be aberrant in 25-30% of patients. In a review of facial nerve injuries in more than 1000 patients with congenital aural atresia, Jahrsdoerfer and Lambert (1998) reported injury in 10 patients. Patients with low-set ears, canal stenosis, and cholesteatoma had facial nerves vulnerable to injury during surgery 22.
Complications of atresia surgery can be minimized with proper selection of patients and with careful attention to surgical detail. Most common complications include meatal or canal stenosis in 15-20%, usually requiring revision surgery, and chronic drainage/infection in 10%, usually due to sloughing of the skin graft with “mucosalization” of the canal, which requires revision with a new skin graft or possible temporizing measures such as cauterization or Gentian violet if the area is small.
Less commonly, sensorineural hearing loss (5%) is most likely related to the high energy of the drill on the ossicular chain being conducted to the cochlea. About 15-20% of patients also lose the initial gains in hearing, due either to lateralization of the tympanic membrane or refixation of the ossicular chain. Approximately 15-20% of patients require a revision procedure at some point in the future, usually due to stenosis of the canal, loss of early hearing gains, or sloughing of the skin graft with chronic moisture in the canal.
In a large review of revision atresia surgery operations, meatal stenosis (narrowing of the ear canal) was the most common complication (58% of 107 revision operations), followed by conductive hearing loss and chronic moisture/infection (19-20%). Improvement in hearing, reopening of the canal, and resolution of the drainage were possible in these patients 23.
Follow-up
Surgical success is based on restoration and stabilization of hearing and maintenance of a patent ear canal. The first audiometric examination is performed after debriding the ear canal at the one month postoperative visit.
As mentioned, the ear canal will need routine cleaning and debridement once or twice a year for the rest of the patient’s life. The debridement simply involves elevating the dead skin that accumulates on the skin graft and removing it. Often, the dead skin reduces the efficiency of the vibration of the new eardrum, and patients can tell that their hearing is a bit muffled. Removal of this dead skin layer restores the clarity of the hearing in the ear as the dead skin peels off the tympanic membrane.
Over-the-counter alcohol-based eardrops (eg, SwimEar drops) are used weekly and after swimming to dry the canal and prevent moisture buildup.
Treatment of microtia
The reconstructive options for microtia are broadly divided into:
- No treatment;
- A subcutaneously placed autologous costal cartilage framework;
- Implanted artificial material including a porous polyethylene implant placed either subcutaneously or under a vascularized fascial flap and skin graft;
- A prosthetic ear affixed to the skin with a medical grade adhesive or by osseointegrated implants.
Patients should be observed by a multidisciplinary team where options for both hearing and pinna reconstruction must be considered and coordinated. This surgery should be undertaken by experienced teams as there is a steep learning curve.
Surgeons performing the autologous costal cartilage method must consider the development of costal cartilage, which occurs at least from 6 years old; ideally performing the method in 9 years old or higher would meet the requirements of framework carving. Different surgical techniques have different age requirements 24. Puberty, usually occurring at 12–15 years, is not the suitable age for inexperienced surgeons, because the costal cartilage is prone to hollow during the rapid pubertal growth and care must be taken to maintain consistency of the cartilage framework.
In adulthood, costal cartilage becomes calcified with gradually worsening elasticity, which is not optimal for surgery. Costal cartilage B-ultrasound can be used to determine whether the costal cartilage is hollow or calcified. If the patient is a candidate for aural atresia repair and wishes to pursue a canal reconstruction surgery, the canal surgery must be combined with or follow the autologous rib cartilage microtia repair in order to prevent compromise to the often tenuous vascularity of the skin flap covering the cartilage graft reconstruction 25.
For porous polyethylene microtia repair, surgical reconstruction can be performed in as young as 3 years old, but International Microtia and Atresia Workgroup recommends this surgery after 5 years of age 16. If the patient is a candidate for aural atresia repair and wishes to pursue the canal reconstruction surgery, the canal surgery must be performed before the porous polyethylene microtia repair. Canal surgery following previous polyethylene repair risks infection and/or extrusion of the polyethylene graft 26.
The quality of the tissues and post-auricular skin must be assessed as this will affect how best to approach the surgical procedure and the results. Loose skin with good laxity is optimal for autologous cartilage microtia reconstruction. The laxity of the skin and presence of previous incisions or trauma will determine if further measures are required such as temporoparietal fascial flaps with skin grafts or skin expansion may be considered to help cover the framework.
Aural atresia prognosis
The goal of congenital aural atresia (congenital aural atresia) surgery is to restore hearing to a speech-reception threshold of less than or equal to 30 dB HL without the need for amplification and to give the patient a clean, dry, skin-lined ear canal and eardrum 27.
Hearing outcomes after atresia surgery can vary widely. A literature review by Nadaraja et al indicated that atresia repair will lead to normal or near-normal hearing in 50-80% of patients 28.
The two most important factors in achieving consistently good hearing outcomes in atresia surgery are careful preoperative selection of patients and meticulous surgical technique at each step of the operation. Surgery is not recommended for patients with unilateral atresia and Jahrsdoerfer scores of 6 or below.
Surgery is attempted on the patient with bilateral atresia and a score of 5 or 6. Although excellent hearing outcomes are difficult, the patient may be able to wear a conventional hearing aid in the new canal to bring the hearing thresholds into the normal range. Surgery is not recommended for patients with 4 or below. The most important anatomic feature for successful surgery is middle ear aeration; without an aerated middle ear space, the patient is not considered a candidate for surgery 29.
A retrospective study by Ahn et al 30 of patients with congenital aural atresia who underwent canaloplasty with implantation of a partial ossicular replacement prosthesis (PORP) indicated that the length of the prosthesis is significantly associated with the procedure’s long-term effectiveness. At 2-year follow-up, it was found that the mean length of the prosthesis in patients with a successful surgical outcome was 2.30 mm, compared with 2.77 mm in patients with an unsuccessful outcome 30.
A study by Shonka et al 14 examined the predictive ability of the Jahrsdoerfer scale for hearing outcomes. In this series of 116 patients, patients with a score greater than 7 had an 85-90% chance of achieving normal or near-normal hearing (as defined by an SRT ≤ 30 dB HL) in the short-term; patients with lower Jahrsdoerfer scores had a 45-50% chance of achieving this result. In this same study, aeration of the middle ear was the single best predictor of hearing success in atresia surgery; without an aerated middle ear space, or with a poorly aerated middle ear space, hearing outcomes were not good 14. The importance of middle ear aeration has been supported by other studies 31.
A retrospective study by Sakamato et al 32 indicated that in patients with congenital aural atresia undergoing canal tympanoplasty, an external auditory canal area of over 72.3 mm2 is the most important predictor of a long-term favorable outcome. The study, which involved 51 ears, also found that a mesotympanic depth of over 5.5 mm, a mesotympanic height of over 4.6 mm, and an external auditory canal diameter of over 9.5 mm are also significant predictive factors for such outcomes.
Some patients with atresia may require reconstruction of the ear bones with an artificial ear bone, or prosthesis. This type of reconstruction tends not to be as reliable as using the patient’s own intact native ossicular chain. Anatomy of the middle ear and ossicles occasionally dictates (approximately 8-10% of the time) that a prosthesis must be used. This same study argued that a partial replacement prosthesis is superior to a total ossicular replacement prosthesis 33. Interestingly, other atresia surgeons have not found a difference between reconstruction using the patient’s native ossicular chain and an ossicular prosthesis 34.
One criticism of atresia surgery case series has been lack of long-term follow-up. Lambert 35 examined the stability of hearing results after atresia surgery and found that almost two thirds of patients maintained an speech-reception threshold less than or equal to 30 dB HL for the longer follow-up (>1 year; mean, 2.8 years); about one third required a revision procedure. Similarly, De la Cruz 36 reported a long-term (≥ 6 months) air-bone gap (ABG) of 30 dB or less in 51% of primary cases and 39% of revisions. Revision surgery may not hold up as well long term 37.
A retrospective study by Imbery et al 2 found that in children and adults who underwent primary congenital aural atresia repair, hearing, as measured by a three-tone (500, 1000, 2000 Hz) air conduction pure-tone average, improved by an average of 30.5 dB, with this gain then declining by 8.2 dB over a mean 4.4-year follow-up. The investigators noted that in 64% of the patients, hearing loss was less than 10 dB, while 36% experienced a loss of 10 dB or more 2.
Until several years ago, surgeons counseled patients and families against repair of unilateral atresia, given the mediocre hearing gains and risk of major complications. Since the introduction of high-resolution CT scanning with improved preoperative evaluation, the repair of unilateral atresia has become more widely accepted. Clearly, a learning curve exists for this challenging operation, and one report suggests a minimum of 32 ears to achieve proficiency in achieving good short-term hearing results, and 48 ears for good long-term hearing outcomes. The anterior surgical approach as first proposed by Jahrsdoerfer has withstood the test of time and scrutiny. As the seemingly subtle deficits of unilateral hearing loss in children are further elucidated 3, some habilitative intervention for children with atresia will be more often recommended. Atresia surgery is one of the many options families face when setting their children up for success in the home and in the classroom.
References- Kesser BW, Krook K, Gray LC. Impact of unilateral conductive hearing loss due to aural atresia on academic performance in children. Laryngoscope. 2013 Sep. 123(9):2270-5.
- Imbery TE, Gray L, Champaloux E, Kesser BW. Long-term Audiometric Outcomes After Atresiaplasty for Congenital Aural Atresia. Otol Neurotol. 2020 Mar. 41 (3):371-8.
- Gray L, Kesser B, Cole E. Understanding speech in noise after correction of congenital unilateral aural atresia: effects of age in the emergence of binaural squelch but not in use of head-shadow. Int J Pediatr Otorhinolaryngol. 2009 Sep. 73(9):1281-7.
- Li CL, Chen Y, Chen YZ, Fu YY, Zhang TY. Congenital Aural Stenosis: Clinical Features and Long-term Outcomes. Sci Rep. 2016;6:27063. doi: 10.1038/srep27063
- Dostal A, Nemeckova J, Gaillyova R, Vranova V, Zezulkova D, Lejska M. Identification of 2.3-Mb gene locus for congenital aural atresia in 18q22.3 deletion: a case report analyzed by comparative genomic hybridization. Otol Neurotol. 2006 Apr. 27(3):427-32.
- Kountakis SE, Helidonis E, Jahrsdoerfer RA. Microtia grade as an indicator of middle ear development in aural atresia. Arch Otolaryngol Head Neck Surg. 1995 Aug. 121(8):885-6.
- Montino S, Agostinelli A, Trevisi P, Martini A, Ghiselli S. Check-list for the assessment of functional impairment in children with congenital aural atresia. Int J Pediatr Otorhinolaryngol. 2017 Nov. 102:174-9.
- Cole RR, Jahrsdoerfer RA. The risk of cholesteatoma in congenital aural stenosis. Laryngoscope. 1990 Jun. 100(6):576-8.
- Kiese-Himmel C, Kruse E. [Unilateral hearing loss in childhood. An empirical analysis comparing bilateral hearing loss]. Laryngorhinootologie. 2001 Jan. 80(1):18-22.
- Jahrsdoerfer RA, Kesser BW. Issues on aural atresia for the facial plastic surgeon. Facial Plast Surg. 1995 Oct. 11(4):274-7.
- Reinisch JF, Lewin S. Ear reconstruction using a porous polyethylene framework and temporoparietal fascia flap. Facial Plast Surg. 2009 Aug. 25(3):181-9.
- Roberson JB Jr, Reinisch J, Colen TY, Lewin S. Atresia repair before microtia reconstruction: comparison of early with standard surgical timing. Otol Neurotol. 2009 Sep. 30(6):771-6.
- Jahrsdoerfer RA, Yeakley JW, Aguilar EA, et al. Grading system for the selection of patients with congenital aural atresia. American Journal of Otology. 1992. 13:6-12.
- Shonka DC, Jahrsdoerfer RA, Kesser BW. The Jahrsdoerfer Grading Scale in Surgery for Congenital Aural Atresia. Arch Otolaryngol Head Neck Surg. Aug. 2008. 134:873-7.
- Dostal A, Nemeckova J, Gaillyova R, et al. Identification of 2.3-Mb gene locus for congenital aural atresia in 18q22.3 deletion: a case report analyzed by comparative genomic hybridization. Otol Neurotol. 2006 Apr. 27(3):427-32.
- Zhang TY, Bulstrode N, Chang KW, et al. International Consensus Recommendations on Microtia, Aural Atresia and Functional Ear Reconstruction. J Int Adv Otol. 2019;15(2):204-208. doi:10.5152/iao.2019.7383 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6750779
- Lo JF, Tsang WS, Yu JY, Ho OY, Ku PK, Tong MC. Contemporary hearing rehabilitation options in patients with aural atresia. Biomed Res Int. 2014. 2014:761579.
- Wazen JJ, Caruso M, Tjellstrom A. Long-term results with the titanium bone-anchored hearing aid: the U.S. experience. Am J Otol. 1998 Nov. 19(6):737-41.
- Farnoosh S, Mitsinikos FT, Maceri D, Don DM. Bone-Anchored Hearing Aid vs. Reconstruction of the External Auditory Canal in Children and Adolescents with Congenital Aural Atresia: A Comparison Study of Outcomes. Front Pediatr. 2014. 2:5.
- van der Pouw KT, Snik AF, Cremers CW. Audiometric results of bilateral bone-anchored hearing aid application in patients with bilateral congenital aural atresia. Laryngoscope. 1998 Apr. 108(4 Pt 1):548-53.
- Attaway J, Stone CL, Sendor C, Rosario ER. Effect of Amplification on Speech and Language in Children With Aural Atresia. Am J Audiol. 2015 Sep 1. 24 (3):354-359.
- Jahrsdoerfer RA, Lambert PR. Facial nerve injury in congenital aural atresia surgery. Am J Otol. 1998 May. 19(3):283-7.
- Oliver ER, Hughley BB, Shonka DC, Kesser BW. Revision aural atresia surgery: indications and outcomes. Otol Neurotol. 2011 Feb. 32(2):252-8.
- Wilkes GH, Wong J, Guilfoyle R. Microtia reconstruction. Plast Reconstr Surg. 2014;134:464e–479e. doi: 10.1097/PRS.0000000000000526
- Siegert R. Combined reconstruction of congenital auricular atresia and severe microtia. Adv Otorhinolaryngol. 2010;68:95–107. doi: 10.1159/000314565
- Tahiri Y, Reinisch J. Porous Polyethylene Ear Reconstruction. Clin Plast Surg. 2019;46:223–30. doi: 10.1016/j.cps.2018.11.006
- Kesser BW, Cole ED, Gray LC. Emergence of Binaural Summation After Surgical Correction of Unilateral Congenital Aural Atresia. Otol Neurotol. 2016 Jun. 37 (5):499-503.
- Nadaraja GS, Gurgel RK, Kim J, Chang KW. Hearing outcomes of atresia surgery versus osseointegrated bone conduction device in patients with congenital aural atresia: a systematic review. Otol Neurotol. 2013 Oct. 34 (8):1394-9.
- Byun H, Moon IJ, Woo SY, et al. Objective and Subjective Improvement of Hearing in Noise After Surgical Correction of Unilateral Congenital Aural Atresia in Pediatric Patients: A Prospective Study Using the Hearing in Noise Test, the Sound-Spatial-Quality Questionnaire, and the Glasgow Benefit Inventory. Ear Hear. 2015 Jul-Aug. 36 (4):e183-9.
- Ahn J, Ryu G, Kang M, Cho YS. Long-term Hearing Outcome of Canaloplasty With Partial Ossicular Replacement in Congenital Aural Atresia. Otol Neurotol. 2018 Jun. 39 (5):602-8.
- Oliver ER, Lambert PR, Rumboldt Z, Lee FS, Agarwal A. Middle ear dimensions in congenital aural atresia and hearing outcomes after atresiaplasty. Otol Neurotol. 2010 Aug. 31(6):946-53.
- Sakamoto T, Kikuta S, Kikkawa YS, et al. Prognostic factors for long-term hearing preservation after canal-tympanoplasty for congenital aural atresia. Eur Arch Otorhinolaryngol. 2015 Nov. 272 (11):3151-6.
- Dobratz E, Rastogi A, Jahrsdoerfer RA, and Kesser BW. To POP or not: Ossiculoplasty in congenital aural atresia surgery. Laryngoscope. Aug. 2008. 118:1452-7.
- Chang H, Song JJ, Choi BY, Lee JH, Oh SH, Chang SO. Partial ossicular replacement versus type II tympanoplasty in congenital aural atresia surgery: a matched group study. Otol Neurotol. 2009 Aug. 30(5):609-13.
- Lambert PR. Congenital aural atresia: stability of surgical results. Laryngoscope. 1998 Dec. 108(12):1801-5.
- De la Cruz A, Teufert KB. Congenital aural atresia surgery: long-term results. Otolaryngol Head Neck Surg. 2003 Jul. 129(1):121-7.
- Chang SO, Choi BY, Hur DG. Analysis of the long-term hearing results after the surgical repair of aural atresia. Laryngoscope. 2006 Oct. 116(10):1835-41.





{kind=link}