What is autophagy
Autophagy is a genetically programmed cell survival pathway involving the degradation of cytoplasmic constituents, and the recycling of ATP and essential building blocks for the maintenance of cellular biosynthesis during nutrient deprivation or metabolic stress 1. This cellular self-consumption process is characterized by sequestration of bulk cytoplasm, long-lived proteins and cellular organelles in double-membrane vesicles, called autophagosomes, which are ultimately delivered to and degraded in the lysosomes 2. Autophagy is a self-catabolic process that maintains intracellular homeostasis and prolongs cell survival under stress via lysosomal degradation of cytoplasmic constituents and recycling of amino acids and energy. Autophagy is the degradation of any type of intracellular components including protein, organelles or any type of particulate structures (e.g. protein aggregates, cellular inclusions, etc.) in lysosomes. Autophagy is used for recycling cellular components, responding to cellular stresses and ridding cells of foreign material. Perturbations in autophagy have been implicated in a number of pathological conditions such as neurodegeneration, cardiac disease and cancer 3. Autophagy is intricately implicated in both health and disease 4. Autophagy is a ubiquitous cellular process used as a mechanism to maintain cellular homeostasis and to respond to cellular stress 5. During autophagy, cellular components are degraded and, in most cases, recycled for later use. Autophagy can be accomplished by several distinct mechanisms and can have distinct degradation targets 6. Autophagy can be nonselective or selective. Nonselective autophagy is used for the turnover of bulk cytoplasm under starvation conditions 7. Selective autophagy targets damaged or superfluous mitochondria (mitophagy) 8, peroxisomes (pexophagy) 9, lipid droplets (lipophagy) 10 and microbes (xenophagy) 11. Autophagy is activated in response to adverse environment conditions such as the deprivation of nutrients, hypoxia, pathogen infection, radiation and oxidative stress as a survival mechanism 12. This process plays a role in cellular homeostasis, development and longevity. Under many conditions, autophagy is considered as a physiologic cytoprotective or pro-survival mechanism; however, completely uncontrolled or excessive autophagy has been associated with cell death 13. The characterization of the regulation of autophagy has become relevant because defective autophagy has been linked to aging 14, neurodegenerative disorders 15, cancer 16, fatty liver and diabetes 17.
Given its importance in normal physiological processes, disruptions in autophagy or its regulation have been associated with a variety of pathological conditions 18. For example, in the nervous system, disruptions in mitophagy, a type of macroautophagy that degrades mitochondria, have been associated with Parkinson disease 19. Autophagic clearance of protein aggregates has been presented as a possible contributor to the pathology of Alzheimer disease 20, Huntington disease 21, amyotrophic lateral sclerosis 22 and Parkinson disease 23. Beyond the nervous system, autophagy has also been implicated in cardiovascular disease 24, cancer 25, immune system function 26 and recently in Niemann-Pick disease 27.
Basal autophagy plays a critical role in cellular homeostasis by eliminating excessive, damaged and/or long-lived proteins and organelles, thus preserving the quality of essential cellular components 28. Under stress, autophagy is commonly induced as a temporary cell survival mechanism. Autophagy defects play a role in the pathogenesis of diverse diseases, including myopathy 29, neuronal degeneration 30, microbial infection 31, inflammatory bowel disease 32, aging 33 and cancer 34.
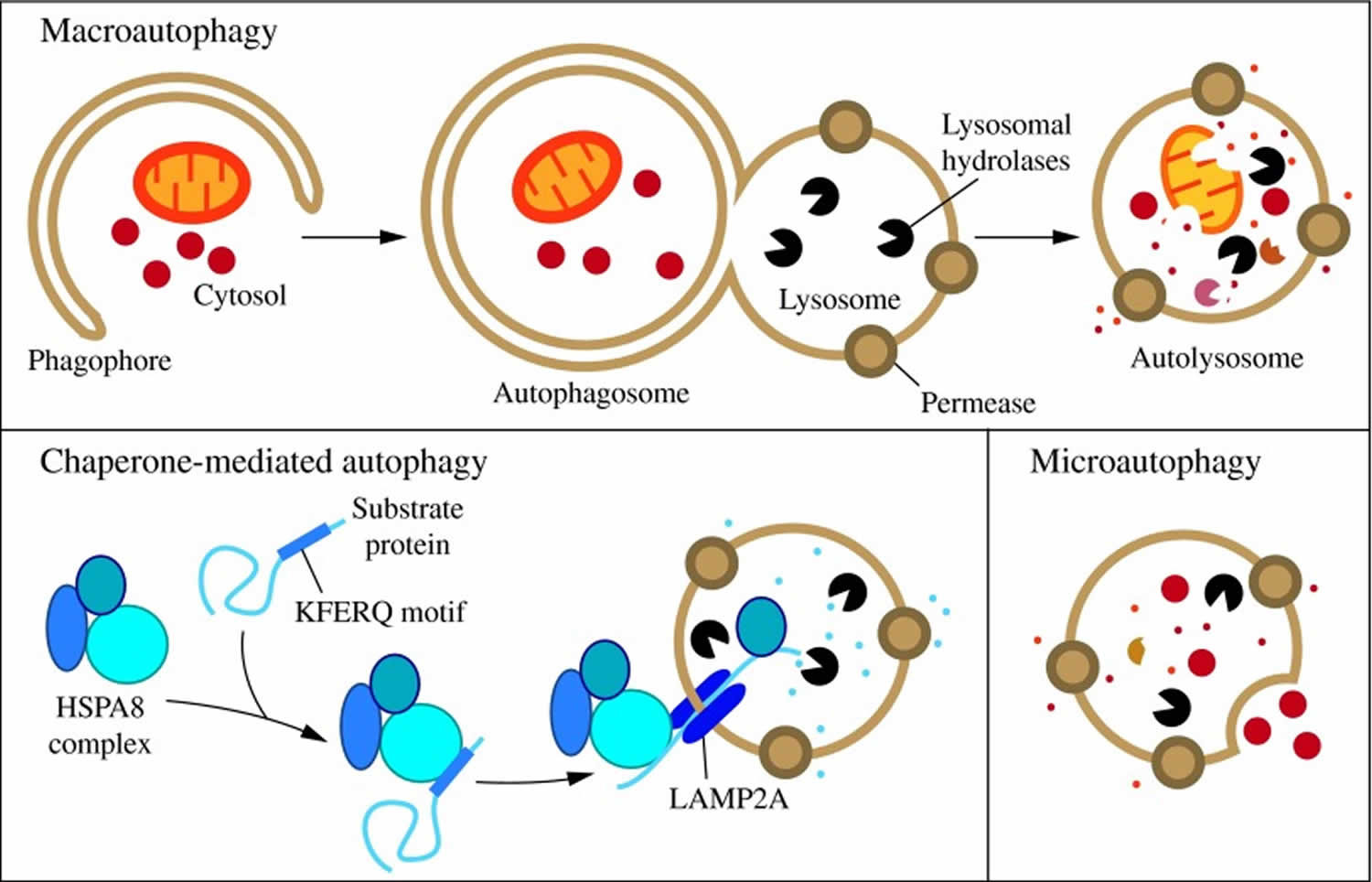
In mammalian cells, there are three primary types of autophagy: microautophagy, macroautophagy, and chaperone-mediated autophagy 35.
- Microautophagy is a type of autophagy in which the lysosomal membrane invaginates or protrudes to internalize cytosolic components in single membrane vesicles that are then rapidly degraded in the lysosomal lumen.
- Macroautophagy is a type of autophagy by which entire regions of the cytosol sequestered inside a double membrane structure (autophagosome) are delivered as a whole into lysosomes through vesicular fusion to enable cargo degradation.
- Chaperone-mediated autophagy is an autophagic pathway responsible for the selective degradation of a subset of cytosolic proteins in lysosomes upon unfolding and translocation across the membrane.
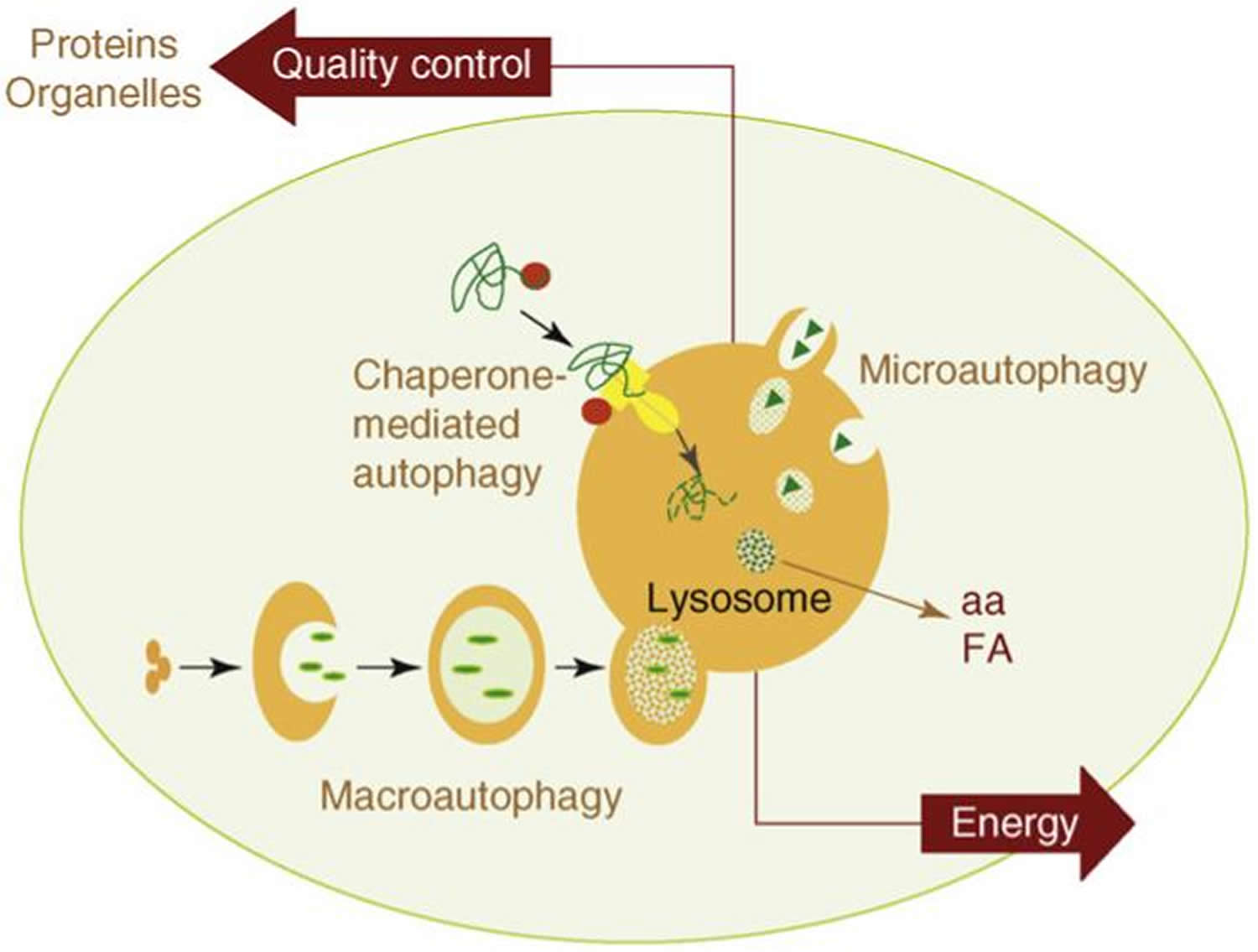
The two most important functions of autophagy are also depicted in this model: (i) as an alternative source of energy, by providing essential components – such as amino acids (aa) or free fatty acids (FA) – that can be used for cellular fueling; and (ii) as an essential component of the cellular mechanisms for quality control, by guaranteeing removal of altered proteins and organelles.
While each is morphologically distinct, all three culminate in the delivery of cargo to the lysosome for degradation and recycling (Figure 1) 36. During microautophagy, invaginations or protrusions of the lysosomal membrane are used to capture cargo 37. Uptake occurs directly at the limiting membrane of the lysosome, and can include intact organelles. Chaperone-mediated autophagy differs from microautophagy in that it does not use membranous structures to sequester cargo, but instead uses chaperones to identify cargo proteins that contain a particular pentapeptide motif; these substrates are then unfolded and translocated individually directly across the lysosomal membrane 38. In contrast to microautophagy and chaperone-mediated autophagy, macroautophagy involves sequestration of the cargo away from the lysosome. In this case, de novo synthesis of double-membrane vesicles—autophagosomes—is used to sequester cargo and subsequently transport it to the lysosome 39.
Figure 1. Three types of autophagy in mammalian cells
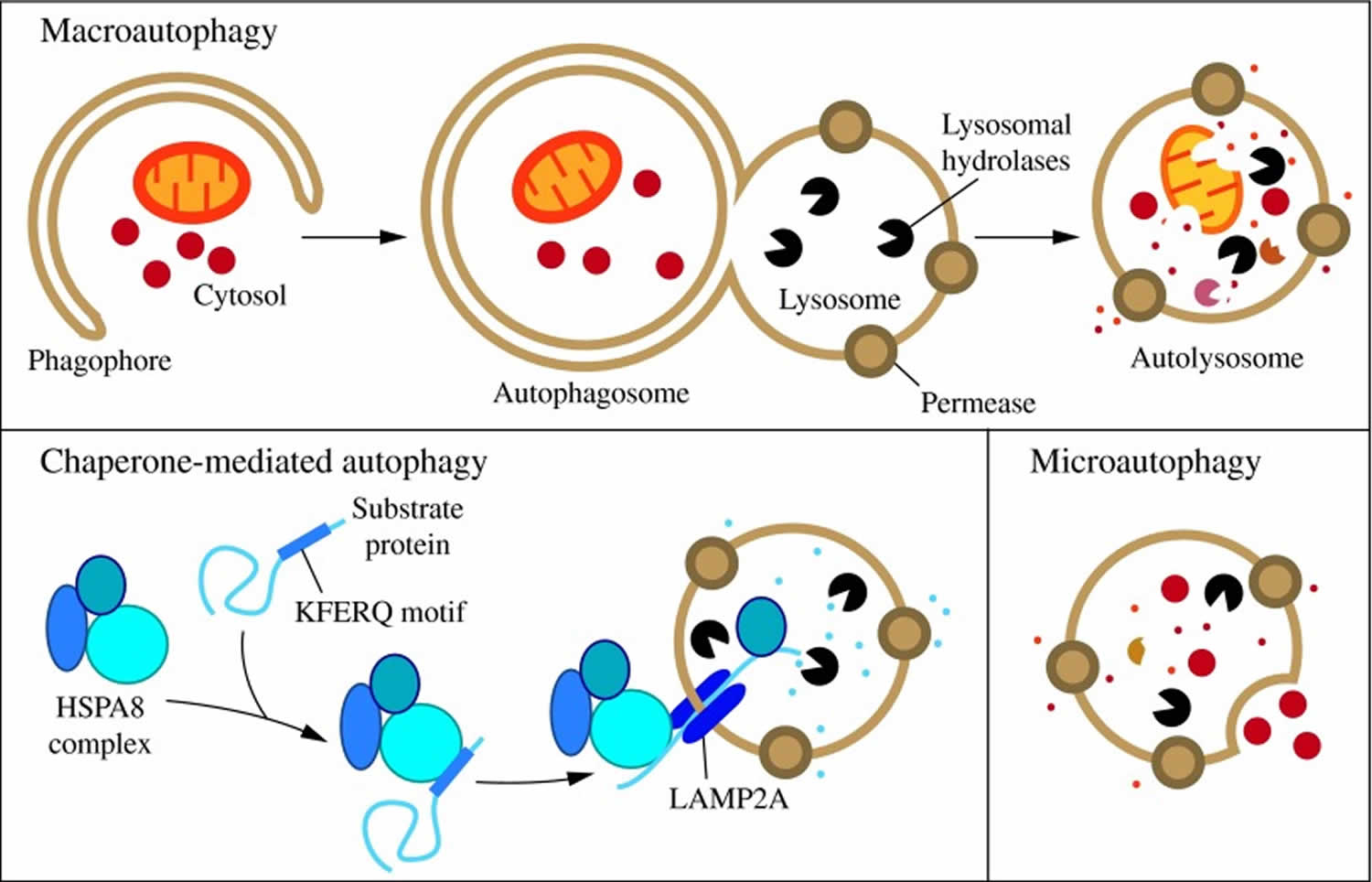
Footnotes: Three types of autophagy in mammalian cells. Macroautophagy relies on de novo formation of cytosolic double-membrane vesicles, autophagosomes, to sequester and transport cargo to the lysosome. Chaperone-mediated autophagy transports individual unfolded proteins directly across the lysosomal membrane. Microautophagy involves the direct uptake of cargo through invagination of the lysosomal membrane. All three types of autophagy lead to degradation of cargo and release of the breakdown products back into the cytosol for reuse by the cell.
[Source 35]Of the three types of autophagy, macroautophagy is the best studied. Macroautophagy occurs at a low level constitutively and can be further induced under stress conditions, such as nutrient or energy starvation, to degrade cytoplasmic material into metabolites that can be used in biosynthetic processes or energy production, allowing for cell survival 39. Under normal growing conditions, macroautophagy aids in cellular maintenance by specifically degrading damaged or superfluous organelles 36. Thus, macroautophagy is primarily a cytoprotective mechanism; however, excessive self-degradation can be deleterious. Accordingly, autophagic dysfunction is associated with a variety of human pathologies, including lung, liver, and heart disease, neurodegeneration, myopathies, cancer, ageing, and metabolic diseases, such as diabetes 40.
Microautophagy
Microautophagy refers to a process by which cytoplasmic contents enter the lysosome through an invagination or deformation of the lysosomal membrane 41. In one early study, isolated rat liver lysosomes were shown by electron microscopy to engulf Percoll particles in vitro by way of protrusions or cup-like invaginations of the lysosomal membrane, forming vesicles within the lysosome. Some of these particles were seen free-floating within the lysosomal lumen, presumably through rupture/lysis of the vesicles 38. A very recent study presented evidence that a microautophagy-like process called endosomal microautophagy transports soluble cytosolic proteins to the vesicles of late endosomal multivesicular bodies 42. Due to the limited number of tools available for the study of microautophagy, we know relatively little about this process, including its regulation and possible roles in human health and disease 37.
Chaperone-Mediated Autophagy
A second type of autophagy, which has so far only been described in mammalian cells, is chaperone-mediated autophagy. Unlike microautophagy and macroautophagy, which can both nonspecifically engulf bulk cytoplasm, chaperone-mediated autophagy is highly specific; common to all chaperone-mediated autophagy substrates is a pentapeptide targeting motif biochemically related to KFERQ 43. Based on sequence analysis and immunoprecipitation experiments, it is estimated that ∼30% of cytosolic proteins contain such a sequence 44. Target proteins containing the KFERQ consensus motif are unfolded through the action of cytosolic chaperones and translocated directly across the lysosomal membrane where they are degraded in the lumen 45. Chaperone-mediated autophagy degrades a wide range of substrate proteins, including certain glycolytic enzymes, transcription factors and their inhibitors, calcium and lipid binding proteins, proteasome subunits, and proteins involved in vesicular trafficking 46.
Macroautophagy
Macroautophagy is distinct from microautophagy and chaperone-mediated autophagy in part because the initial site of sequestration occurs away from the limiting membrane of the lysosome, and involves the formation of cytosolic vesicles that transport the cargo to this organelle. The morphological feature that makes macroautophagy unique from other intracellular vesicle-mediated trafficking processes is that the sequestering vesicles, termed autophagosomes, form de novo rather than through membrane budding; that is, the autophagosome forms by expansion, and does not bud from a preexisting organelle, already containing cargo. Upon induction of macroautophagy in yeast, formation of autophagosomes begins at a single perivacuolar site called the phagophore assembly site 47. In mammalian systems, autophagosome generation is initiated at multiple sites throughout the cytoplasm rather than at a single phagophore assembly site 47. Several studies suggest that endoplasmic reticulum-associated structures called omegasomes may serve as initiation sites in mammals 48.
Following initiation, the membrane begins to expand. At this stage, it is called a phagophore, which is the primary double-membrane sequestering compartment (Figure 2) 49. The source of membrane that makes up the phagophore is highly debated, but various studies have implicated the plasma membrane, endoplasmic reticulum, Golgi complex and mitochondria as possible sources 50. As the phagophore expands, the membrane bends to ultimately generate a spherical autophagosome. The factors that drive curvature of the membrane during nonspecific macroautophagy are not known. In the case of selective macroautophagy, the membrane appears to essentially wrap around the cargo; thus, adjusting to fit the specific target. Upon completion, the phagophore fully surrounds its cargo and fuses to form the double-membrane autophagosome. The size of the autophagosome varies based on organism and cargo type. For example, the diameter of autophagosomes ranges from ∼0.4 to 0.9 μm in yeast, and 0.5 to 1.5 μm in mammals 51.
Figure 2. Morphology of macroautophagy
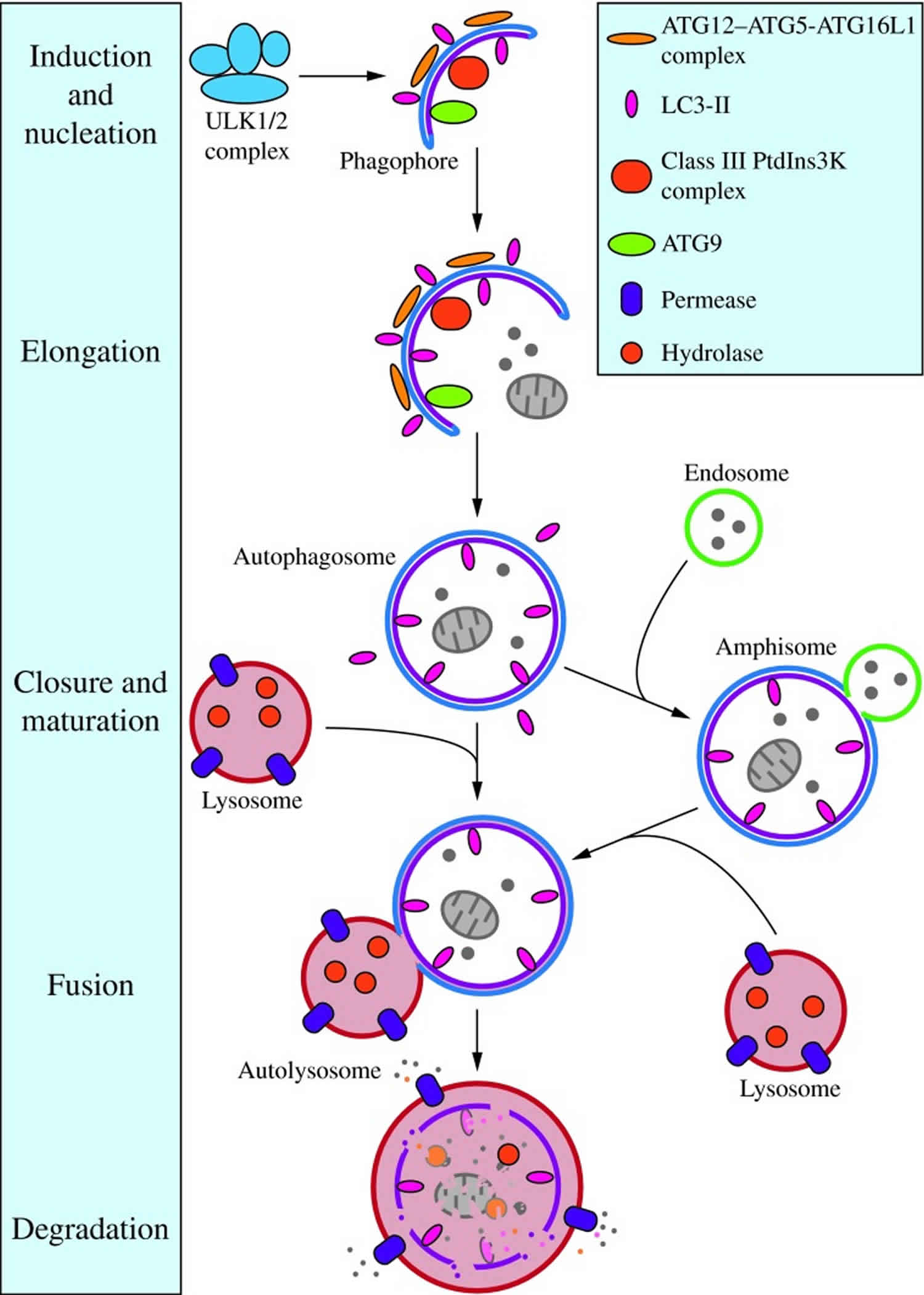
Footnotes: Nucleation of the phagophore occurs following induction by the ULK1/2 complex. Elongation of the phagophore is aided by the ATG12–ATG5-ATG16L1 complex, the class III PtdIns3K complex, LC3-II, and ATG9. Eventually, the expanding membrane closes around its cargo to form an autophagosome and LC3-II is cleaved from the outer membrane of this structure. The outer membrane of the autophagosome will then fuse with the lysosomal membrane to form an autolysosome. In some instances, the autophagosome may fuse with an endosome, forming an amphisome, before fusing with the lysosome. The contents of the autolysosome are then degraded and exported back into the cytoplasm for reuse by the cell. See the text for details. This figure was modified from Figure 1 in Yang and Klionsky (153). ATG, autophagy-related; PtdIns3K, phosphatidylinositol 3-kinase; ULK, unc-51-like kinase (C. elegans).
[Source 35]Autophagy process and regulation
In mammalian cells, autophagy is controlled by the orchestration of more than 30 autophagy related genes (Atg) 52. Autophagy begins with the formation of the “isolation membrane” also known as phagophore. Phagophore membranes seem to originate from endoplasmic reticulum 53, plasma membrane 54, trans-Golgi 55 and mitochondria 56. However, since autophagosomal membranes have a relative lack of transmembrane proteins, phagophore membranes could also originate from de novo membrane formation from cytosolic lipids 57. Phagophores expand and sequester damaged mitochondria and organelles, lipid droplets and portions of the cytoplasm, eventually closing to become autophagosomes 58. Subsequently, autophagosomes mature by fusing with late endosomes and lysosomes, thereby forming autolysosomes. In autolysosomes, the cargo is exposed to lysosomal enzymes to allow its breakdown and resulting nutrients are released into the cytosol 58. Autophagic flux is used to denote the process of autophagosome synthesis, delivery of autophagic substrates to the lysosome, and degradation of autophagic substrates inside the lysosome 59.
Autophagy is stimulated by a variety of stimuli, such as nutrient starvation, pathogen associated molecular patterns (PAMPs), danger associated molecular patterns (DAMPs), oxidative stress, endoplasmic reticulum stress (ER stress) and mitochondria damage 60.
Autophagy and fasting
Nutrient depletion is the most potent known physiological inducer of autophagy 61. In the majority of cultured mammalian cells, nutrient depletion induces autophagy within minutes, with maximal levels observed when cells are cultured in the simultaneous absence of nutrients (such as amino acids and glucose) and growth factors (such as those contained in serum) 62. In mice, following starvation for 24-48 hours, most cells in most tissues display increased numbers of autophagosomes 63. Several critical molecules regulate starvation-induced autophagy (Figure 3A); of these, mTOR and AMPK have been best characterized, and recent studies also suggest a crucial role for sirtuins.
Figure 3. Autophagy and fasting
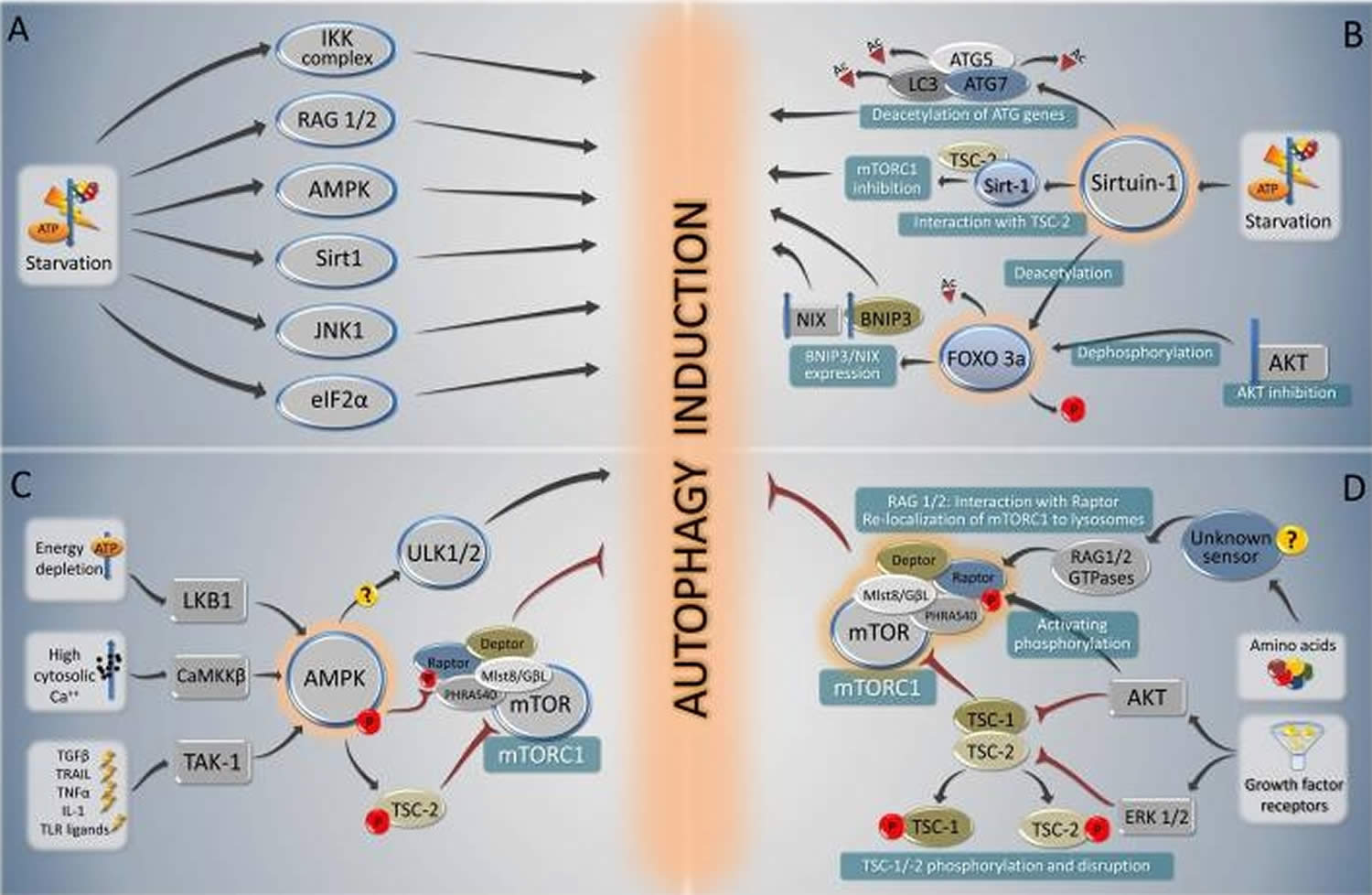
Footnotes: Overview of the major signal transduction pathways that regulate autophagy in response to starvation. A summary of starvation-induced pro-autophagic signaling (A) is followed by a schematic overview of the signaling cascades involving sirtuin-1 and Foxo 3a (B), AMPK (C) and mTORC1 (D).
[Source 61]Autophagy cancer
Recent studies have shed light on the functional role of autophagy in different cellular processes and the potential of autophagy modulation as a novel therapeutic strategy for different pathologic conditions, including cancer 64. Anticancer therapies, such as hormonal agents, chemotherapy and irradiation, frequently induce autophagy, in most cases as a prosurvival response potentially contributing to treatment resistance 65; however, autophagy activation in particular genetic backgrounds and/or completion of the autophagic process beyond reversibility of cell viability can also lead to cell death, thus enhancing treatment efficacy 66. The complex role of autophagy in tumorigenesis and treatment responsiveness makes it hard to decipher how to universally modulate autophagy for maximum therapeutic benefit, indicating that context- and cell type-specific approaches may be required.
Regulation of Autophagy in Cancer
In normal cells, autophagy is regulated by a cellular network that involves upstream signaling pathways integrated by the mammalian target of rapamycin (mTOR) kinase, a master regulator 67. Nutrient and/or growth factor availability activates the PI3K/AKT/mTOR axis, which inhibits autophagy thus stimulating cell growth and proliferation; whereas nutrient and/or growth factor limitation, hypoxia and other stressors deactivate this axis, leading to autophagy induction and suppression of cell growth and proliferation 68. In tumor cells, autophagy regulation is in principal similar, but the regulatory network is even more complex due to frequently abnormal PI3K/AKT/mTOR and other signaling cascade activation and interactions between these pathways 69.
Role of autophagy in cancer
The dual function of autophagy in cancer, as both a tumor suppressor and a protector of cancer cell survival, has been widely recognized and remains a rigorously investigated topic 64. Elucidation of the specific role that autophagy plays at different stages in cancer progression and determination of its cell type and genetic context-dependency will lead to the development of novel, and hopefully more effective, cancer therapeutic and preventative strategies.
Autophagy as a tumor suppressor mechanism
Morphological evidence of autophagosome accumulation in dying cells linked autophagy to cell death and led to the definition of an autophagosome-associated, non-apoptotic form of programmed cell death as autophagic cell death or type II programmed cell death, potentially functioning as a tumor suppressor mechanism similar to apoptosis 70. The tumor suppressive role of autophagy was validated by the discovery and characterization of the autophagy-related genes (Atg) 71, among which Beclin 1 (Atg6) was the first to be reported as a plausible tumor suppressor, as allelic BECN1 deletion is frequently observed in human breast, ovarian and prostate cancers 72, and aging Becn1+/− mutant mice develop tumors (lymphomas, lung and liver cancers), as well as mammary hyperplasia and hepatitis B virus-induced premalignant lesions 73.
Genetic alteration of other autophagy-related genes has also been causally associated with tumorigenesis. Atg4C knockout mice exhibit increased susceptibility to chemical carcinogen -induced fibrosarcomas 74, whereas deletion of the essential autophagy regulator Atg5 results in natural killer (NK) cell malignancies 75. Furthermore, loss of Bax-interacting factor-1 (BIF-1), which is a positive apoptosis and autophagy regulator, suppresses programmed cell death and promotes colon adenocarcinomas 76, and nonsense mutations in the BECN1-binding protein UVRAG (ultraviolet radiation resistance-associated gene), which also positively regulates autophagy, are found in colon and gastric cancers 77.
Both cell autonomous and non-cell autonomous mechanisms have been implicated in autophagy-mediated tumor suppression 78, namely preservation of genome stability and cellular homeostasis and limitation of inflammation, respectively 79.
Autophagy as a protector of cancer cell survival
Counterintuitive to its tumor suppressive role, autophagy has also been extensively documented to support cancer cell survival under stress 80. For example, autophagy induction in immortalized, apoptosis-defective, IL-3-dependent bone marrow cells in response to growth factor withdrawal prolongs cell survival, as supported by the observed cell death acceleration upon autophagy inhibition 81. Defective autophagy due to allelic Becn1 deletion or constitutive AKT activation enhances the susceptibility of apoptosis-incompetent immortalized baby mouse kidney (iBMK) cells to metabolic stress 82. Similarly, partially autophagy-defective Becn1+/− immortalized mouse mammary epithelial cells (iMMECs) are more sensitive to metabolic stress and show accelerated acinar lumen formation in 3D-culture 83.
Autophagy-mediated support of tumor cell survival may play a critical role in cancer progression at later stages, such as dissemination and metastasis, which account for most cancer-associated deaths. In favor of this hypothesis, starvation-induced autophagy is accompanied by suppression of protein synthesis, cell division and motility in an energy conservation effort that sustains cells in a dormant state with the capacity to resume cell growth and proliferation upon regular growth condition restoration 84. For example, autophagy induction in mammary epithelial cells upon their detachment from extraceullular matrix (ECM) sustains cell viability in an anoikis-resistant manner, whereas autophagy upregulation in ovarian cancer cells by the tumor suppressor ARHI (aplasia Ras homolog member I) promotes dormant cell survival in vivo 85. Finally, many studies have clearly documented that in cancer cells, autophagy is upregulated in response to metabolic and genotoxic stress induced by hormonal deprivation, chemotherapy and radiation as a cell survival mechanism, likely contributing to treatment resistance, but also providing a novel therapeutic target in cancer 86.
Autophagy-mediated cancer cell survival is not unexpected given the functions of autophagy in normal cells: basal autophagy maintains cellular homeostasis by removing protein aggregates and damaged organelles, whereas starvation-induced autophagy prolongs cell survival by recycling amino acid and energy, both important functions for cellular fitness and viability preservation. Cancer cells are often under higher metabolic stress than normal cells, which in turn increases tumor dependence on autophagy for survival (autophagy addiction), implying a therapeutic window for preferential cancer cell targeting by pharmacologic autophagy modulation. Metabolic stressors uniquely encountered by tumor cells include: (1) oncogene-induced accelerated cell growth and/or proliferation, which increase metabolic demands even in the presence of ample external nutrients 87, (2) recurrent nutrient and oxygen deprivation during rapid tumor growth or anticancer treatment 88, and (3) inefficient glucose utilization for energy production due to anaerobic glycolysis (Warburg effect) 89. Whether cancer cells are addicted to autophagy in an oncogene- and/or tumor type-dependent manner is currently under investigation.
Autophagy modulation for cancer treatment
The goal of anticancer therapy is to effectively compromise tumor cell growth and survival, so as to cause cancer regression and prevent (or at least delay) cancer recurrence, thus improving patient quality of life and survival. Apoptosis is commonly inactivated in cancer, often in association with disease progression, and renders tumors resistant to chemotherapy- and radiation-induced cell death, undisputedly contributing to treatment resistance and earlier patient demise. During the last decade, our understanding of the proteins and molecular mechanisms responsible for apoptosis regulation, initiation and execution has expanded tremendously 90, leading to rationally designed approaches for apoptosis reactivation, and thus restoration of cell death potential, in cancer cells 91.
Despite recent advances in cancer treatment, many tumors still exhibit unsatisfactory responsiveness to biological agents, chemotherapy and/or radiation, either recurring or continuing to grow during or after treatment 92. Autophagy upregulation is a common occurrence in response to cancer therapies, expected to occur in both tumor and normal cells, but likely playing a more critical role in the survival of the already metabolically stressed cancer cells, as explained above, and thus contributing to treatment resistance. At the same time, however, this prosurvival function of autophagy provides a novel therapeutic opportunity, as concurrent autophagy inhibition may preferentially sensitize tumor cells to anticancer agents by depriving them of an essential survival mechanism that may be dispensable for normal cell viability under similar conditions 93. Drug-induced autophagy may also be therapeutically beneficial by itself, limiting tumor cell proliferation and resulting in autophagic cell death in a cell typeand genetic background-specific manner. Thus, contextspecific pharmacologic autophagy modulation holds great promise as a novel therapeutic approach adding another weapon to the currently available armamentarium against cancer 94.
Autophagy inhibition as a therapeutic strategy in cancer
In cancer cells, autophagy is generally, and often preferentially as compared to normal cells, induced as a prosurvival function in response to treatment-associated genotoxic and metabolic stress 95. Thus, concurrent autophagy inhibition is expected to mostly have a synergistic effect with chemotherapy and/or radiation on cancer cell elimination. The impact of autophagy inhibition on anticancer therapy has been evaluated in multiple tumor models, including glioma 96, myeloma 97, breast 98, colon 99 and prostate cancers 100. For example, preclinical studies supporting autophagy inhibition as an anticancer strategy, treatment-induced autophagy mediated resistance to the HER2 monoclonal antibody trastuzumab, whereas LC3 knockdown via shRNA resulted in resistant cells being re-sensitized to treatment 101; autophagy inhibition by either pharmacological agents or RNAi targeting essential autophagy regulators potentiated imatinib mesylate-induced cell death in chronic myelogenous leukemia (CML) cells 102; autophagy suppression also enhanced the therapeutic efficacy of cisplatin and 5-FU in esophageal and colon cancers, respectively 103.
In the absence of drugs specifically targeting autophagy regulators, indirect autophagy inhibition by the lysosomotropic antimalaria drugs chloroquine and hydroxychloroquine, which interfere with lysosomal acidification and thus, block the autophagic process at its final step, are currently under clinical investigation in combination with standard treatment in multiple tumor types. Treatment with chloroquine or hydroxychloroquine is likely not the same as direct autophagy inhibition, given that chloroquine and hydroxychloroquine are known to exert additional functions, such as immunomodulation and possibly DNA damage as alkylating agents at higher doses. Nevertheless, the results of clinical trials are eagerly awaited as an initial proof-of-principle that autophagy inhibition has a role in cancer treatment. In the event that these studies fail to show the trends anticipated, before declaring autophagy inhibition as an unsuccessful therapeutic strategy in cancer, it will be necessary to assess whether tumors with particular oncogenic changes are better candidates than others for such treatment, in which case trials should be redesigned to specifically target these malignancies.
Autophagy induction as an alternate anticancer strategy
Although autophagy inhibitors combined with standard treatment are emerging as promising anticancer agents, certain cancer cell lines and xenograft tumors were sensitized to therapeutic regimens involving autophagy induction rather than inhibition. This was commonly observed in an apoptosis-defective background, where cancer cells were committed to non-apoptotic cell death modes, including necrosis, necroptosis and possibly autophagic cell death 95. Alkylating agents, such as actinomycin D and arsenic trioxide; hormonal therapies, including tamoxifen and vitamin D analogues; natural compounds, such as resveratrol; cytokines, such as IFNγ; gene therapies, including p53 and p27kip1; have all been implicated in the induction of autophagic cell death in various cancer cell lines in vitro, as autophagosomes are commonly observed in dying cells 104. However, it is often unclear whether autophagy plays an active role in the cell death process or it is a mere bystander representing the stressed cell’s futile attempts to preserve viability by upregulating an energy and amino acid recycling program. Thus, the presence of autophagosomes in dying cells is not necessarily synonymous with cell death by autophagy, unless knockdown of essential autophagy regulators prolongs cell survival under the same stress conditions 105. For example, the autophagy inducer STF-62247 killed VHL-deficient renal cancer cells in association with autophagosome accumulation and reduction of autophagy regulator levels compromised sensitivity to this drug 106.
The implication of autophagy as a cell death mechanism in tumors with inactivated apoptosis is not surprising, given that cells with functional apoptosis undergo a rapid and ‘clean’ (not associated with inflammation) apoptotic cell death when severely stressed, making observation, and thus study, of autophagy in an apoptosis-competent background difficult 107. Disabled apoptosis is a frequent occurrence in cancer, thus tumor cells under extreme stress often die by other mechanisms, as already mentioned. However, the conditions-beyond apoptosis inactivation-under which autophagy functions as a primary cell death mechanism remain to be defined; such knowledge will be critical for the rational design and targeted application of therapeutic regimens exploiting autophagy induction for more effective tumor cell killing.
Autophagy modulation for cancer prevention
In the long run, the most effective (and also the least expensive) way to treat cancer is to prevent it from arising in the first place. Cancer prevention strategies must be accessible, easily implemented, well tolerated and safe over time. Accumulation of protein aggregates due to defective autophagy is a well-recognized culprit in neurodegeneration, including Alzheimer’s, Parkinson’s and polyglutamine diseases, and in hepatic dysfunction, but its contribution to cancer development and progression was not so clear 108. Recent studies indicated that protein aggregation in autophagy-defective tumor cells is a likely source of genotoxic stress, mostly due to the resultant endoplasmic reticulum and oxidative stress, in turn contributing to the genomic damage and instability, and thus increased tumorigenicity, associated with autophagy defects 109. Since basal autophagy plays a critical role in cellular homeostasis by degrading aged or malfunctioning organelles and damaged or misfolded proteins, thus maintaining genome integrity, and as such exhibiting a tumor suppressive effect, it is reasonable to target autophagy for cancer prevention. Autophagy stimulators are currently being explored as preventative agents in neurodegeneration 110; their predicted role in cancer prevention is also worthy of investigation and has potentially significant clinical implications, as supported by accumulating scientific evidence.
For example, cancer risk highly correlates with age 111. Caloric restriction involving limited food intake and physical exercise induces autophagy and delays the aging process, thus extending lifespan and possibly inhibiting tumorigenesis. The longevity resulting from caloric restriction was further validated in Tp53−/− mice, rhesus monkeys, yeast, Caenorthabditis elegans and Drosophila. Thus, caloric restriction may be one way to modulate autophagy for cancer prevention. However, this hypothesis is far from clinical application, as it still needs to be extensively investigated and ultimately validated in humans. Diet-associated autophagy upregulation may be another approach to cancer prevention. Autophagy induction by carotenoids, lycopene, lutein, polyphenols, resveratrol, curcumin and epigallocatechin-3-gallate has been implicated in the ability of these dietary compounds to inhibit ROS and reactive nitrogen species accumulation. Pharmacological activation of autophagy by drugs, such as rapamycin analogs, class I PI3K inhibitors, chloroquine and metformin, has also been reported to inhibit malignant transformation, and thus likely prevent cancer, by limiting genomic damage. The possibility of preventing cancer by autophagy modulation is intriguing, but clearly in need of further investigation, as the most effective and safest way to manipulate autophagy for cancer prevention remains to be defined.
Autophagy in cancer summary
The role of autophagy in cancer and treatment responsiveness is undoubtedly complicated. The double-edged sword function of autophagy, as both a tumor suppressor and a protector of cancer cell survival, likely impacts anticancer treatment efficacy in opposing ways. Exploitation of the functional autophagy status in tumors and pharmacologic autophagy modulation for cancer treatment and prevention presents novel opportunities in cancer management. Autophagy defects are associated with susceptibility to metabolic stress, DNA damage accumulation, genomic instability and accelerated tumorigenicity. It is, thus, reasonable to hypothesize that autophagy upregulation may preserve cellular fitness and genome integrity to prevent cancer development and progression; chronically autophagy-deficient tumors may also be particularly sensitive to genotoxic and/or metabolic stressinducing anticancer agents, such as DNA damaging and antiangiogenic drugs. On the other hand, autophagy-competent tumors (even if their autophagy potential is partially compromised due to cancer-associated cellular events, such as constitutive PI3K/AKT/mTOR axis activation) likely utilize, and potentially rely on, this pathway for survival under metabolic stress conditions, such as during rapid tumor growth, metastasis and treatment. In the latter case, concurrent (i.e., acute) autophagy inhibition is expected to increase the efficacy of any anticancer modality, including radiation, chemotherapy, biological agents and combinatorial regimens.
In conclusion, the multifaceted nature of autophagy and its diverse crosstalk with other biological processes, including cell death pathways, must be carefully considered when the autophagic system is targeted for anticancer benefit. Areas of great interest in cancer research and with potentially significant therapeutic implications include autophagy regulation in tumor cells, impact of autophagy functional status in tumors on cancer progression and response to treatment, and elucidation of how to best modulate autophagy for therapeutic benefit and cancer prevention, so as to achieve the ultimate goal of cancer eradication 112.
References- Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12:1509–1518
- Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4:740–743.
- Denny P, Feuermann M, Hill DP, Lovering RC, Plun-Favreau H, Roncaglia P. Exploring autophagy with Gene Ontology. Autophagy. 2018;14(3):419-436. doi:10.1080/15548627.2017.1415189. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5915032/
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995.
- Wen X, Klionsky DJ. An overview of macroautophagy in yeast. J Mol Biol. 2016;428(9 Pt A):1681–1699. doi:10.1016/j.jmb.2016.02.021 PubMed PMID: 26908221; PubMed Central PMCID: PMCPMC4846508
- Wen X, Klionsky DJ. An overview of macroautophagy in yeast. J Mol Biol. 2016;428(9 Pt A):1681–1699. doi:10.1016/j.jmb.2016.02.021
- A role for ubiquitin in selective autophagy. Kirkin V, McEwan DG, Novak I, Dikic I. Mol Cell. 2009 May 15; 34(3):259-69.
- Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Narendra D, Tanaka A, Suen DF, Youle RJ. J Cell Biol. 2008 Dec 1; 183(5):795-803
- Excess peroxisomes are degraded by autophagic machinery in mammals. Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, Chiba T, Tanaka K, Kominami E. J Biol Chem. 2006 Feb 17; 281(7):4035-41.
- Autophagy regulates adipose mass and differentiation in mice. Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. J Clin Invest. 2009 Nov; 119(11):3329-39.
- Bacterial subversion of host innate immune pathways. Baxt LA, Garza-Mayers AC, Goldberg MB. Science. 2013 May 10; 340(6133):697-701.
- Autophagy and the integrated stress response. Kroemer G, Mariño G, Levine B. Mol Cell. 2010 Oct 22; 40(2):280-93. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3127250/
- Life, death and burial: multifaceted impact of autophagy. Galluzzi L, Morselli E, Vicencio JM, Kepp O, Joza N, Tajeddine N, Kroemer G. Biochem Soc Trans. 2008 Oct; 36(Pt 5):786-90.
- The Interplay between Autophagy and Aging. Pyo JO, Yoo SM, Jung YK. Diabetes Metab J. 2013 Oct; 37(5):333-9.
- Dual roles for autophagy: degradation and secretion of Alzheimer’s disease Aβ peptide. Nilsson P, Saido TC. Bioessays. 2014 Jun; 36(6):570-8.
- Autophagy and cancer cell metabolism. Lozy F, Karantza V. Semin Cell Dev Biol. 2012 Jun; 23(4):395-401.
- Role of autophagy in the progression from obesity to diabetes and in the control of energy balance. Quan W, Jung HS, Lee MS. Arch Pharm Res. 2013 Feb; 36(2):223-9. https://www.ncbi.nlm.nih.gov/pubmed/23371805/
- Klionsky DJ, Codogno P. The mechanism and physiological function of macroautophagy. J Innate Immun. 2013;5(5):427–433. doi:10.1159/000351979
- Wang B, Abraham N, Gao G, et al. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl Neurodegener. 2016;5:19. doi:10.1186/s40035-016-0065-1
- Salminen A, Kaarniranta K, Kauppinen A, et al. Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog Neurobiol. 2013;106–107:33–54. doi:10.1016/j.pneurobio.2013.06.002
- Zhao T, Hong Y, Li XJ, et al. Subcellular clearance and accumulation of Huntington disease protein: a mini-review. Front Mol Neurosci. 2016;9:27. doi:10.3389/fnmol.2016.00027
- Monahan Z, Shewmaker F, Pandey UB. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016;1649(Pt B):189–200. doi:10.1016/j.brainres.2016.05.022
- Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93(5):1015–1034. doi:10.1016/j.neuron.2017.01.022
- Ikeda Y, Shirakabe A, Brady C, et al. Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. J Mol Cell Cardiol. 2015;78:116–122. doi:10.1016/j.yjmcc.2014.09.019
- Mowers EE, Sharifi MN, Macleod KF, et al. Autophagy in cancer metastasis. Oncogene. 2017;36(12):1619–1630. doi:10.1038/onc.2016.333
- Lupfer C, Thomas PG, Anand PK, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14(5):480–488. doi:10.1038/ni.2563
- Canonico B, Cesarini E, Salucci S, et al. Defective autophagy, mitochondrial clearance and lipophagy in niemann-pick type B lymphocytes. PLoS One. 2016;11(10):e0165780. doi:10.1371/journal.pone.0165780
- Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–673
- Winslow AR, Rubinsztein DC. Autophagy in neurodegeneration and development. Biochim Biophys Acta. 2008;1782:723–729.
- Orvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 2009;16:57–69.
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263.
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268.
- Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria and aging. Physiology (Bethesda) 2008;23:248–262.
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967
- Parzych KR, Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxidants & Redox Signaling. 2014;20(3):460-473. doi:10.1089/ars.2013.5371. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3894687/
- Yang Z. and Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22: 124–131, 2010
- Mijaljica D, Prescott M, and Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7: 673–682, 2011
- Massey A, Kiffin R, and Cuervo AM. Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol 36: 2420–2434, 2004
- Yorimitsu T. and Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 12: 1542–1552, 2005
- Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, and Vandenabeele P. Autophagy: for better or for worse. Cell Res 22: 43–61, 2012
- Marzella L, Ahlberg J, and Glaumann H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch B Cell Pathol Incl Mol Pathol 36: 219–234, 1981
- Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, and Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20: 131–139, 2011
- Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15: 305–309, 1990
- Chiang H-L. and Dice JF. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J Biol Chem 263: 6797–6805, 1988
- Orenstein SJ. and Cuervo AM. Chaperone-mediated autophagy: molecular mechanisms and physiological relevance. Semin Cell Dev Biol 21: 719–726, 2010
- Arias E. and Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 23: 184–189, 2011
- Chen Y. and Klionsky DJ. The regulation of autophagy—unanswered questions. J Cell Sci 124: 161–170, 2011
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, and Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11: 1433–1437, 2009
- He C. and Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43: 67–93, 2009
- Mizushima N, Yoshimori T, and Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132, 2011
- Mizushima N. and Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 27: 19–40, 2007
- The machinery of macroautophagy. Feng Y, He D, Yao Z, Klionsky DJ. Cell Res. 2014 Jan; 24(1):24-41.
- A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. Nat Cell Biol. 2009 Dec; 11(12):1433-7.
- Plasma membrane contributes to the formation of pre-autophagosomal structures. Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Nat Cell Biol. 2010 Aug; 12(8):747-57.
- The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Ge L, Melville D, Zhang M, Schekman R. Elife. 2013 Aug 6; 2():e00947.
- Mitochondria supply membranes for autophagosome biogenesis during starvation. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Cell. 2010 May 14; 141(4):656-67.
- The autophagosome: origins unknown, biogenesis complex. Lamb CA, Yoshimori T, Tooze SA. Nat Rev Mol Cell Biol. 2013 Dec; 14(12):759-74.
- Autophagy: process and function. Mizushima N. Genes Dev. 2007 Nov 15; 21(22):2861-73.
- Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445-544. doi:10.4161/auto.19496. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3404883/
- Autophagy and the integrated stress response. Kroemer G, Mariño G, Levine B. Mol Cell. 2010 Oct 22; 40(2):280-93.
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Molecular cell. 2010;40(2):280-293. doi:10.1016/j.molcel.2010.09.023. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3127250/
- Inhibition of macroautophagy triggers apoptosis. Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Mol Cell Biol. 2005 Feb; 25(3):1025-40.
- The role of the Atg1/ULK1 complex in autophagy regulation. Mizushima N. Curr Opin Cell Biol. 2010 Apr; 22(2):132-9.
- Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochim Biophys Acta. 2009;1793:1516–1523.
- Al-Ejeh F, Kumar R, Wiegmans A, Lakhani SR, Brown MP, Khanna KK. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene. 2010;29:6085–6098.
- Buytaert E, Callewaert G, Vandenheede JR, Agostinis P. Deficiency in apoptotic effectors Bax and Bak reveals an autophagic cell death pathway initiated by photodamage to the endoplasmic reticulum. Autophagy. 2006;2:238–240
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295.
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293.
- Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–6446
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213.
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822.
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676.
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820.
- Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583.
- Iqbal J, Kucuk C, Deleeuw RJ, Srivastava G, Tam W, Geng H, et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia. 2009;23:1139–1151.
- Coppola D, Khalil F, Eschrich SA, Boulware D, Yeatman T, Wang HG. Downregulation of Bax-interacting factor-1 in colorectal adenocarcinoma. Cancer. 2008;113:2665–2670.
- Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol. 2008;39:1059–1063.
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967.
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075
- White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316.
- Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040.
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381.
- Karantza-Wadsworth V, White E. A mouse mammary epithelial cell model to identify molecular mechanisms regulating breast cancer progression. Methods Enzymol. 2008;446:61–76
- Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007;120:379–383.
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806.
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700.
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548
- Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007;120:379–383
- Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3010857/
- Hoyer-Hansen M, Jaattela M. Autophagy: an emerging target for cancer therapy. Autophagy. 2008;4:574–580.
- Tan TT, White E. Therapeutic targeting of death pathways in cancer: mechanisms for activating cell death in cancer cells. Adv Exp Med Biol. 2008;615:81–104.
- Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim Biophys Acta. 2010;1806:220–229
- Gewirtz DA. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy. 2009;5:1232–1234
- Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res. 2010;70:3431–3434
- Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy. 2006;2:85–90.
- Shingu T, Fujiwara K, Bogler O, Akiyama Y, Moritake K, Shinojima N, et al. Stage-specific effect of inhibition of autophagy on chemotherapy-induced cytotoxicity. Autophagy. 2009;5:537–539
- Shanmugam M, McBrayer SK, Qian J, Raikoff K, Avram MJ, Singhal S, et al. Targeting glucose consumption and autophagy in myeloma with the novel nucleoside analogue 8-aminoadenosine. J Biol Chem. 2009;284:26816–26830
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One. 2009;4:6251.
- Carew JS, Medina EC, Esquivel JA, 2nd, Mahalingam D, Swords R, Kelly K, et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J Cell Mol Med. 2010;14:2448–2459
- Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009;69:700–708
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One. 2009;4:6251
- Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119:1109–1123
- Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol. 2009;16:761–771.
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134.
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010.
- Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14:90–102
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation and tumorigenesis. Cancer Cell. 2006;10:51–64.
- Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–12544.
- Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–116.
- Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4:895–901
- Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140
- Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biology & Therapy. 2011;11(2):157-168. doi:10.4161/cbt.11.2.14622. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3230307/





{kind=link}