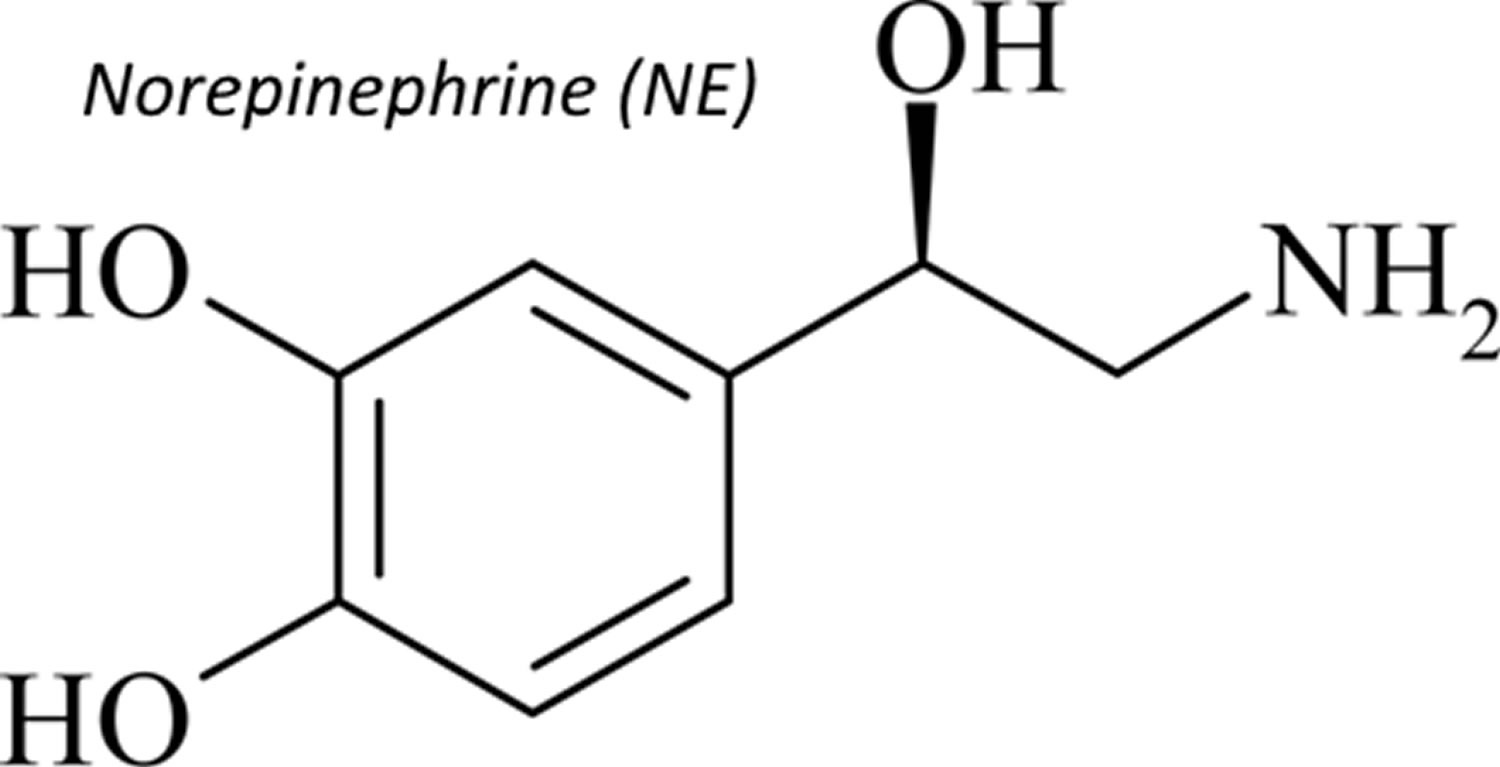
What is norepinephrine
Norepinephrine also called noradrenaline (noradrenalin), is a naturally occurring catecholamine hormone that functions as a neurotransmitter in the sympathetic nervous system. Norepinephrine is a precursor of epinephrine that is also secreted by the adrenal glands medulla and is a widespread central nervous system and autonomic neurotransmitter. However, only a small amount of plasma norepinephrine comes from the adrenal gland under resting conditions, but during some stress responses, such as acute glucoprivation (fall in blood glucose level), the adrenal contribution to plasma norepinephrine increases. Norepinephrine directly stimulates adrenergic receptors. Stimulation of alpha-adrenergic receptors causes vasoconstriction (constriction of blood vessels to increase blood pressure) of the radial smooth muscle of the iris, arteries, arterioles, veins, urinary bladder, and the sphincter of the gastrointestinal tract. Stimulation of beta-1 adrenergic receptors causes an increase in myocardial contractility, heart rate, automaticity, and atrioventricular (AV) conduction while stimulation of beta-2 adrenergic receptors causes bronchiolar and vascular smooth muscle dilatation.
Norepinephrine is the principal transmitter of most postganglionic sympathetic fibers and of the diffuse projection system in the brain arising from the locus ceruleus. Norepinephrine is also found in plants and is used pharmacologically as a sympathomimetic.
Norepinephrine in the bloodstream emanates mainly from networks of sympathetic nerves that enmesh blood vessels— especially arterioles— throughout the body and pervade organs such as the heart and kidneys. The caliber of the arterioles determines total peripheral resistance to blood flow. The sympathetic innervation of the smooth muscle cells in arteriolar walls therefore represents a focal point in neural regulation of blood pressure. In the heart, sympathetic nerves form lattice-like networks around myocardial cells and also supply coronary arterial vessels. Because of the close architectural association between the sympathetic nerves and myocardial and arteriolar smooth muscle cells, one might predict an important role of sympathetic nerves in regulation of cardiovascular performance.
Only a very small proportion of norepinephrine released from sympathetic nerves reaches the bloodstream unchanged. The main route of inactivation of norepinephrine is by reuptake into the nerve terminals. Under resting conditions, however, most of the norepinephrine produced in sympathetic nerves is metabolized before entry of the transmitter into the interstitial fluid or plasma (Figure 1).
Norepinephrine (NE) is the primary neurotransmitter for postganglionic sympathetic adrenergic nerves. It is synthesized inside the nerve axon, stored within vesicles, then released by the nerve when an action potential travels down the nerve. Below are the details for release and synthesis of norepinephrine (NE):
- The amino acid tyrosine is transported into the sympathetic nerve axon.
- Tyrosine (Tyr) is converted to DOPA (levodopa or L-3,4-dihydroxyphenylalanine) by tyrosine hydroxylase (rate-limiting step for norepinephrine synthesis).
- DOPA (levodopa or L-3,4-dihydroxyphenylalanine) is converted to dopamine (DA) by DOPA decarboxylase.
- Dopamine is transported into vesicles then converted to norepinephrine (NE) by dopamine β-hydroxylase (DBH); transport into the vesicle can by blocked by the drug reserpine.
- An action potential traveling down the axon depolarizes the membrane and causes calcium to enter the axon.
- Increased intracellular calcium causes the vesicles to migrate to the axonal membrane and fuse with the membrane, which permits the norepinephrine (NE) to diffuse out of the vesicle into the extracellular (junctional) space. Dopamine β-hydroxylase (DBH) and depending on the nerve other secondary neurotransmitters (e.g., ATP), is released along with the norepinephrine (NE).
- The norepinephrine (NE) binds to the postjunctional receptor and stimulates the effector organ response.
Figure 1. Norepinephrine synthesis and release
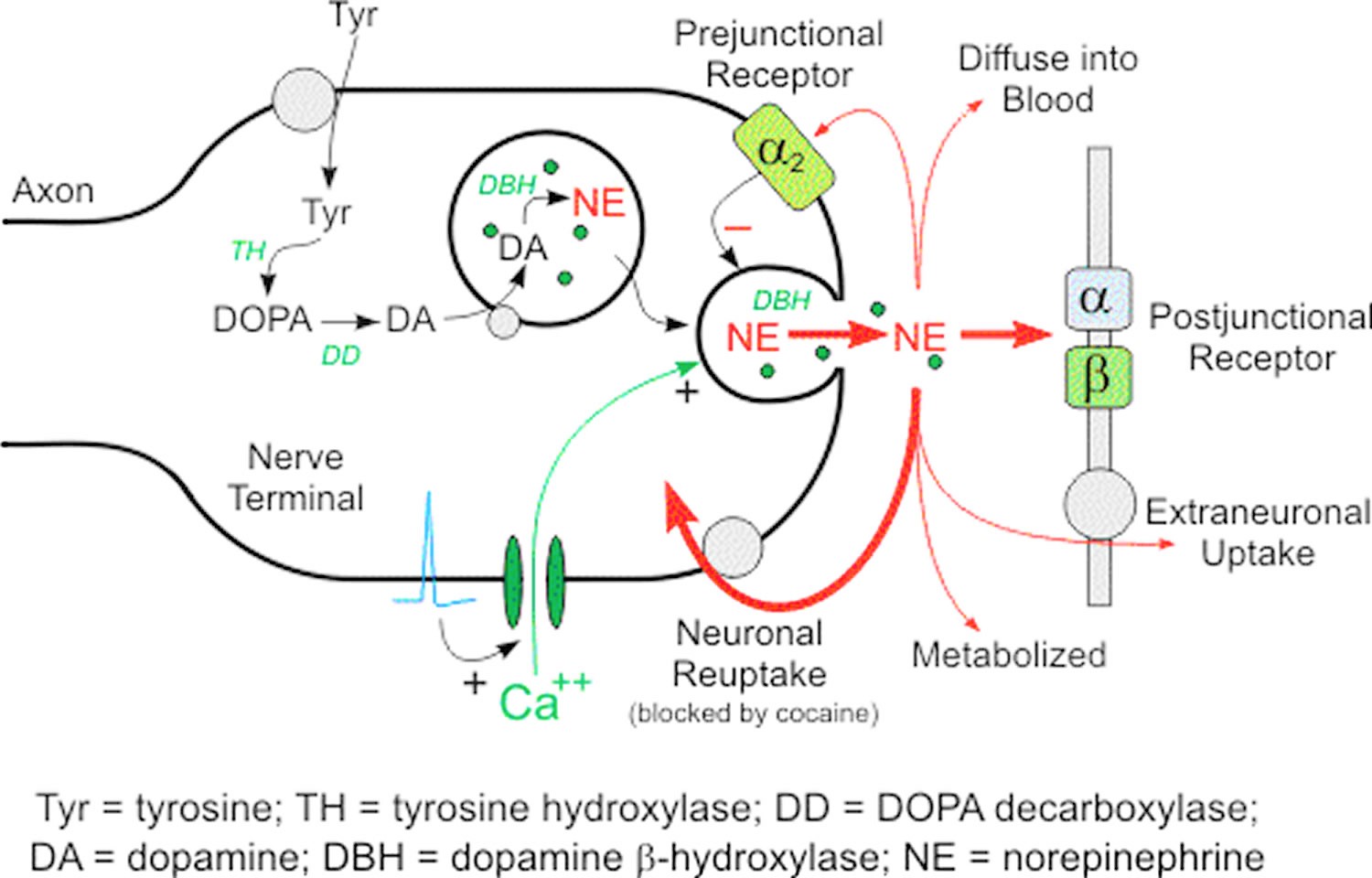
Footnotes: Norepinephrine in the cytoplasm of sympathetic nerves has two sources; most come from continuous vesicular leakage, and a small, variable amount comes from uptake of norepinephrine released in response to sympathetic nerve traffic. Determinants of plasma norepinephrine (NE). Plasma norepinephrine (NE) levels are determined mainly by release from vesicles by exocytosis and reuptake via the cell membrane norepinephrine transporter via the uptake-1 process.
Since plasma norepinephrine (NE) is derived from sympathetic nerves, plasma norepinephrine (NE) levels have been widely used to indicate sympathetic nervous system activity. The relationship between plasma norepinephrine levels and sympathetic nerve traffic is not simple. The plasma concentration depends on both the rate of release of norepinephrine into the plasma and the rate of removal from the plasma. Thus, a high plasma norepinephrine level does not necessarily indicate a high rate of sympathetic nerve traffic; a decrease in removal from the plasma also can increase plasma norepinephrine levels, without a change in the rate of sympathetic nerve traffic.
Secondly, the sympathetic nervous system consists of myriad nerves distributed throughout the body, and stressors can activate sympathetic nerve traffic heterogeneously to different organs. For blood sampling from humans, most researchers use the antecubital vein. Since sympathetic nervous activity in the forearm and hand arm influences levels of norepinephrine in antecubital venous plasma, those levels may not accurately reflect changes in sympathetic nervous activity elsewhere in the body during stress.
Thirdly, since only a small proportion of norepinephrine released from sympathetic nerve endings actually reaches the circulation unchanged, small variations in efficiency of the cell membrane norepinephrine transporter can markedly alter the amount of norepinephrine reaching the plasma.
Fourthly, any of several endogenous biochemicals—including norepinephrine itself, by activating presynaptic α2-adrenoceptors (alpha 2-adrenergic receptors) — have the potential to modulate release of norepinephrine from the nerve terminals. In clinical studies, α2-adrenoceptor (alpha 2-adrenergic receptors) stimulation has been shown to inhibit norepinephrine release into the bloodstream in the heart and forearm.
Fifthly, in some pathological states and in response to a variety of sympathomimetic amines, norepinephrine is released from sympathetic nerve terminals by a nonexocytotic mechanism differing from the calcium-dependent, exocytotic mechanism of release in response to sympathetic nerve traffic. Cardiac ischemic anoxia (low heart oxygen state) is an example of such a pathologic state. Increased net leakage of norepinephrine from vesicular storage sites builds up norepinephrine concentrations in the axoplasm, and exit of norepinephrine via the cell membrane norepinephrine transporter can then lead to norepinephrine entry into the interstitial fluid. Sympathomimetic amines such as tyramine and amphetamine increase plasma norepinephrine levels by this nonexocytotic mechanism.
While these considerations do not invalidate plasma norepinephrine levels in arm venous blood in diagnosis, assessment of drug effects, or prognosis, it is evident that plasma norepinephrine levels must be interpreted with care, keeping in mind the purpose of the test, the characteristics of the patient, the possible interacting effects of medications, and other factors that can influence the obtained results. Without other neurochemical information, one cannot distinguish norepinephrine release from neuronal reuptake as determinants of norepinephrine spillover, in the whole body or in specific organs. Many studies have noted that both plasma norepinephrine concentrations and directly recorded skeletal muscle sympathetic activity increase with subject age; the increased plasma norepinephrine concentrations appear to reflect both increased spillover and decreased clearance 1.
Clonidine is an α2-adrenoceptor agonist (α2-adrenoceptor activator) that acts in the central nervous system to decrease sympathetic nervous system outflows and also in the periphery at presynaptic receptors to decrease norepinephrine release from sympathetic nerve terminals. By both effects, clonidine decreases plasma norepinephrine levels. In patients with pheochromocytoma, a tumor that produces catecholamines, plasma norepinephrine levels can be increased because of release of norepinephrine into the bloodstream independently of the sympathetic nervous system. In such patients, failure of clonidine to reduce plasma norepinephrine constitutes a positive diagnostic test result 2. Conversely, the combination of a high plasma norepinephrine level and a large fall in blood pressure in response to clonidine may identify patients with “hypernoradrenergic hypertension” 3.
Yohimbine exerts effects opposite to those of clonidine. Intravenous infusion of yohimbine increases sympathetic neural outflows and blocks α2-adrenoceptors on sympathetic nerve terminals, thereby increasing plasma norepinephrine levels. Yohimbine challenge testing can assess whether a patient with neurogenic orthostatic hypotension has releasable norepinephrine stores 4, which can be a target for treatment. Yohimbine challenge testing can also reveal excessive norepinephrine release in patients with anxiety or panic disorder 5.
Indirectly acting sympathomimetic amines, such as dextroamphetamine and tyramine, release norepinephrine from sympathetic nerve endings. These drugs are substrates for both the cell membrane norepinephrine and vesicular monoamine transporters. By intravesicular alkalinization, they enhance norepinephrine leakage from storage vesicles into the axoplasm. They also interfere with the efficiency of the cell membrane norepinephrine transporter, resulting in transport of the axoplasmic norepinephrine into the extracellular fluid. In humans, infusion of tyramine or dextroamphetamine therefore increases plasma norepinephrine levels. During tyramine infusion, plasma dihydroxyphenylethylene glycol (DHPG) levels increase to a proportionately larger extent than do plasma norepinephrine levels 6, probably because of the greater buildup of norepinephrine in the axoplasm than in the extracellular fluid.
Foodstuffs such as hard cheeses and red wines contain large amounts of tyramine. Normally, dietary tyramine is metabolized in the gastrointestinal tract and liver before the amine can enter the systemic circulation. In patients taking an MAO inhibitor (monoamine oxidase inhibitor), tyramine is able to reach the sympathetic nerve terminals, and after neuronal and vesicular uptake of tyramine, paroxysmal hypertension can result from release of vesicular norepinephrine—a phenomenon termed the “cheese effect”. Because of the susceptibility to severe hypertension due to the cheese effect, MAO inhibitors (monoamine oxidase inhibitors) have not had wide usage as antidepressants, despite their clinical efficacy.
α-Methyl-para-tyrosine blocks tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis. Repeated administration of α-methyl-para-tyrosine decreases plasma levels of DOPA (levodopa), dihydroxyphenylacetic acid, norepinephrine, and dihydroxyphenylethylene glycol. The drug is used clinically before surgery to remove a pheochromocytoma 7.
Norepinephrine biosynthesis
Norepinephrine is synthesized from the amino acid tyrosine by a series of enzymatic steps in the adrenal medulla and postganglionic neurons of the sympathetic nervous system. While the conversion of tyrosine to dopamine occurs predominantly in the cytoplasm, the conversion of dopamine to norepinephrine by dopamine β-monooxygenase occurs predominantly inside neurotransmitter vesicles 8. The metabolic pathway is:
Phenylalanine → Tyrosine → L-DOPA (levodopa) → Dopamine → Norepinephrine
Thus the direct precursor of norepinephrine is dopamine, which is synthesized indirectly from the essential amino acid phenylalanine or the non-essential amino acid tyrosine 8. These amino acids are found in nearly every protein and, as such, are provided by ingestion of protein-containing food, with tyrosine being the most common.
Figure 2. Norepinephrine biosynthesis
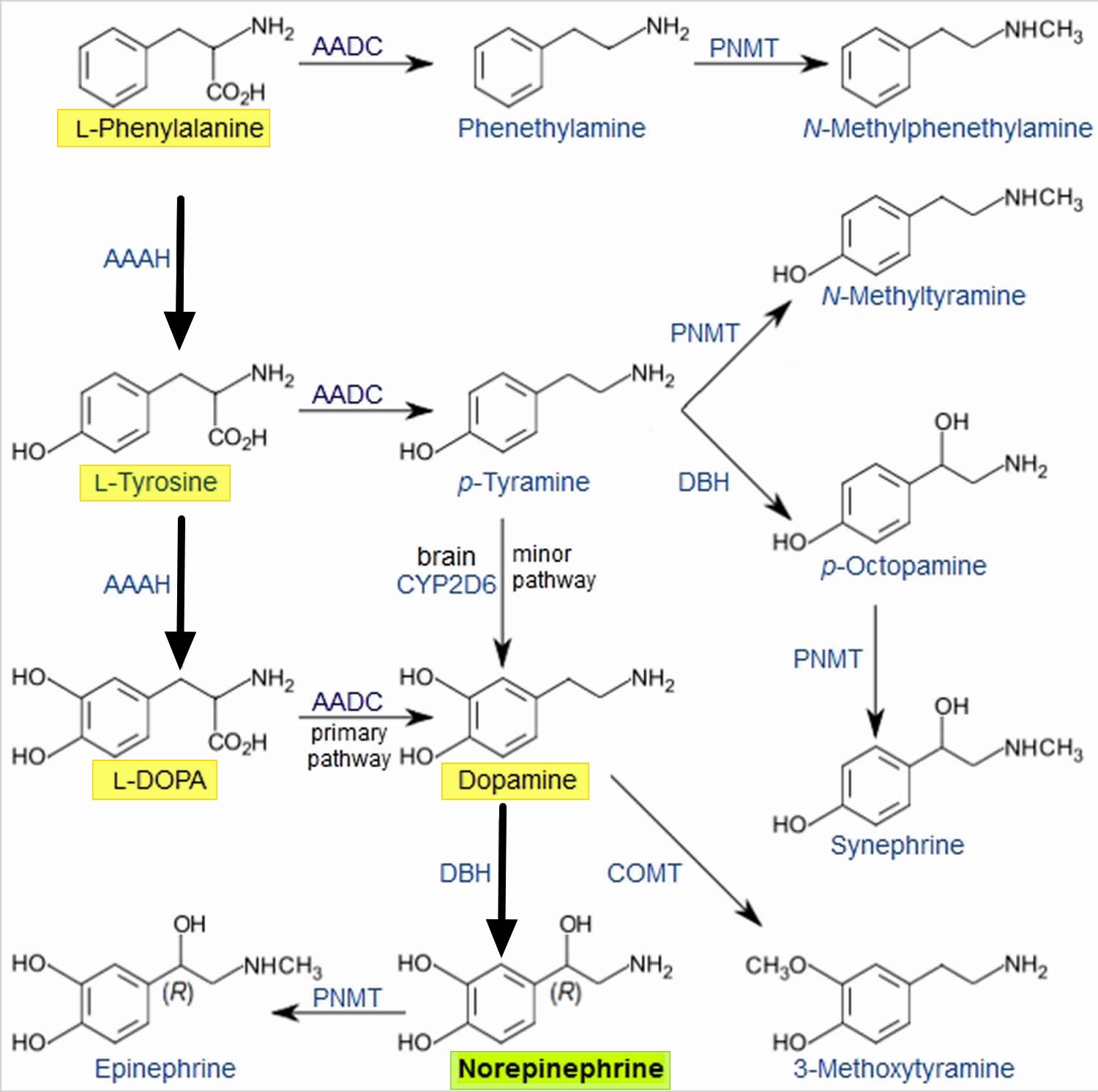
Phenylalanine is converted into tyrosine by the enzyme phenylalanine hydroxylase, with molecular oxygen (O2) and tetrahydrobiopterin as cofactors. Tyrosine is converted into L-DOPA by the enzyme tyrosine hydroxylase, with tetrahydrobiopterin, O2, and probably ferrous iron (Fe2+) as cofactors. L-DOPA is converted into dopamine by the enzyme aromatic L-amino acid decarboxylase (also known as DOPA decarboxylase), with pyridoxal phosphate as a cofactor. Dopamine is then converted into norepinephrine by the enzyme dopamine β-monooxygenase (formerly known as dopamine β-hydroxylase), with O2 and ascorbic acid as cofactors 8.
Norepinephrine itself can further be converted into epinephrine by the enzyme phenylethanolamine N-methyltransferase with S-adenosyl-L-methionine as cofactor 8.
Norepinephrine function
Sympathetic nervous system
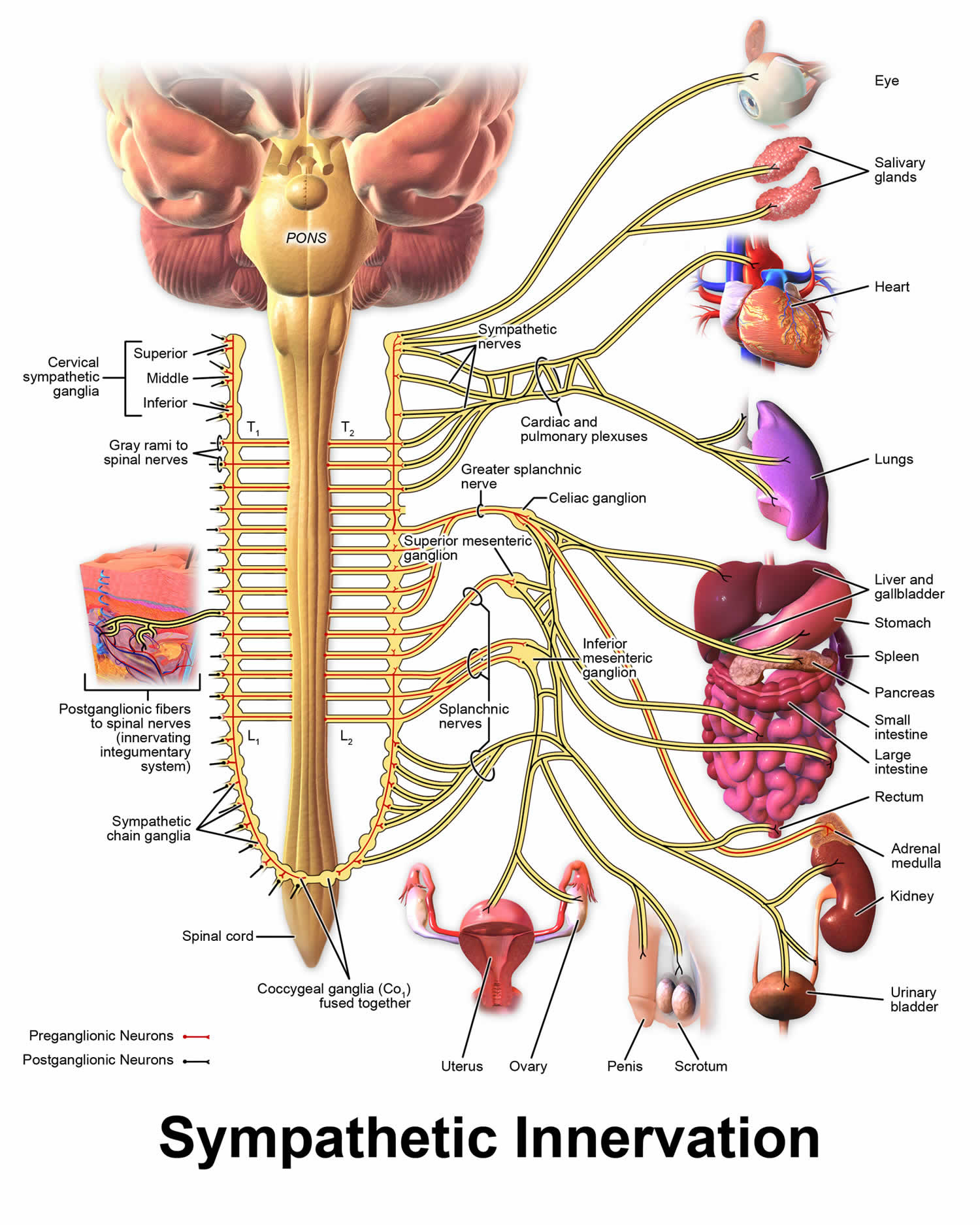
Norepinephrine is the main neurotransmitter used by the sympathetic nervous system, which consists of about two dozen sympathetic chain ganglia located next to the spinal cord, plus a set of prevertebral ganglia located in the chest and abdomen 9. These sympathetic ganglia are connected to numerous organs, including the eyes, salivary glands, heart, lungs, liver, gallbladder, stomach, intestines, kidneys, urinary bladder, reproductive organs, muscles, skin, and adrenal glands 9. Sympathetic activation of the adrenal glands causes the part called the adrenal medulla to release norepinephrine (as well as epinephrine) into the bloodstream, from which, functioning as a hormone, it gains further access to a wide variety of tissues 9.
Broadly speaking, the effect of norepinephrine on each target organ is to modify its state in a way that makes it more conducive to active body movement, often at a cost of increased energy use and increased wear and tear 10. This can be contrasted with the acetylcholine-mediated effects of the parasympathetic nervous system, which modifies most of the same organs into a state more conducive to rest, recovery, and digestion of food, and usually less costly in terms of energy expenditure 10.
The sympathetic effects of norepinephrine include:
- In the eyes, an increase in production of tears, making the eyes more moist, and pupil dilation through contraction of the iris dilator 11.
- In the heart, an increase in the amount of blood pumped 12.
- In brown adipose tissue, an increase in calories burned to generate body heat.
- Multiple effects on the immune system. The sympathetic nervous system is the primary path of interaction between the immune system and the brain, and several components receive sympathetic inputs, including the thymus, spleen, and lymph nodes. However the effects are complex, with some immune processes activated while others are inhibited.
- In the arteries, constriction of blood vessels, causing an increase in blood pressure.
- In the kidneys, release of renin and retention of sodium in the bloodstream.
- In the liver, an increase in production of glucose, either by glycogenolysis after a meal or by gluconeogenesis when food has not recently been consumed. Glucose is the body’s main energy source in most conditions.
- In the pancreas, increased release of glucagon, a hormone whose main effect is to increase the production of glucose by the liver.
- In skeletal muscles, an increase in glucose uptake.
- In adipose tissue (i.e., fat cells), an increase in lipolysis, that is, conversion of fat to substances that can be used directly as energy sources by muscles and other tissues.
- In the stomach and intestines, a reduction in digestive activity. This results from a generally inhibitory effect of norepinephrine on the enteric nervous system, causing decreases in gastrointestinal mobility, blood flow, and secretion of digestive substances.
Figure 3. Sympathetic nervous system
Central nervous system
Brain areas containing noradrenergic neurons.
The noradrenergic neurons in the brain form a neurotransmitter system, that, when activated, exerts effects on large areas of the brain. The effects are manifested in alertness, arousal, and readiness for action.
Noradrenergic neurons (i.e., neurons whose primary neurotransmitter is norepinephrine) are comparatively few in number, and their cell bodies are confined to a few relatively small brain areas, but they send projections to many other brain areas and exert powerful effects on their targets. These noradrenergic cell groups were first mapped in 1964 by Annica Dahlström and Kjell Fuxe, who assigned them labels starting with the letter “A” (for “aminergic”) 13. In their scheme, areas A1 through A7 contain the neurotransmitter norepinephrine (A8 through A14 contain dopamine). Noradrenergic cell group A1 is located in the caudal ventrolateral part of the medulla, and plays a role in the control of body fluid metabolism 14. Noradrenergic cell group A2 is located in a brainstem area called the solitary nucleus; these cells have been implicated in a variety of responses, including control of food intake and responses to stress 15. Cell groups A5 and A7 project mainly to the spinal cord 16.
Figure 4. Noradrenergic neurons in the brain
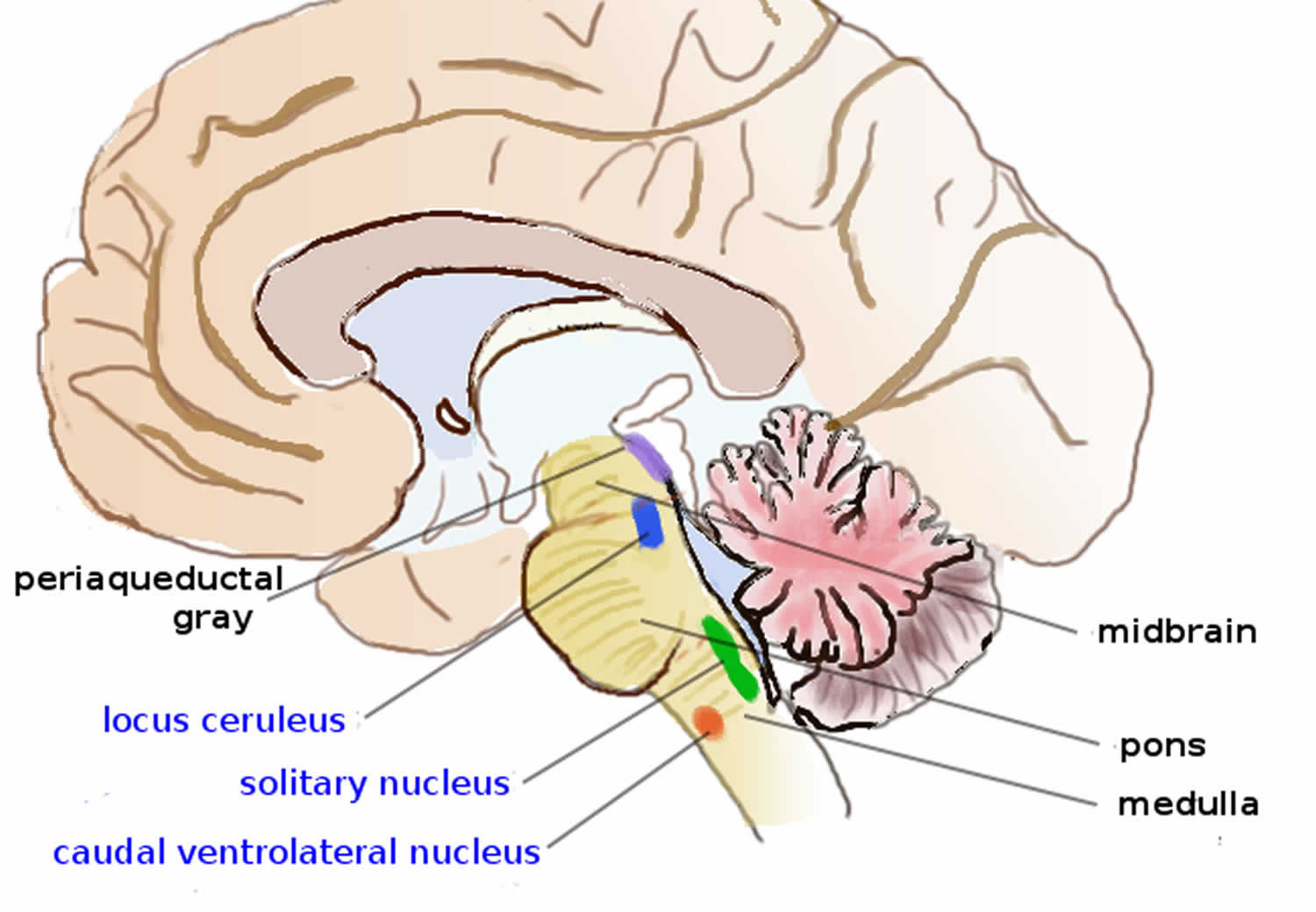
The most important source of norepinephrine in the brain is the locus coeruleus, which contains noradrenergic cell group A6 and adjoins cell group A4. The locus coeruleus is quite small in absolute terms—in primates it is estimated to contain around 15,000 neurons, less than one millionth of the neurons in the brain—but it sends projections to every major part of the brain and also to the spinal cord 17.
The level of activity in the locus coeruleus correlates broadly with vigilance and speed of reaction. Locus coeruleus activity is low during sleep and drops to virtually nothing during the REM (dreaming) state 18. It runs at a baseline level during wakefulness, but increases temporarily when a person is presented with any sort of stimulus that draws attention. Unpleasant stimuli such as pain, difficulty breathing, bladder distension, heat or cold generate larger increases. Extremely unpleasant states such as intense fear or intense pain are associated with very high levels of locus coeruleus activity 17.
Norepinephrine released by the locus coeruleus affects brain function in a number of ways. It enhances processing of sensory inputs, enhances attention, enhances formation and retrieval of both long term and working memory, and enhances the ability of the brain to respond to inputs by changing the activity pattern in the prefrontal cortex and other areas 19. The control of arousal level is strong enough that drug-induced suppression of the locus coeruleus has a powerful sedating effect 18.
There is great similarity between situations that activate the locus coeruleus in the brain and situations that activate the sympathetic nervous system in the periphery: thelocus coeruleus essentially mobilizes the brain for action while the sympathetic system mobilizes the body. It has been argued that this similarity arises because both are to a large degree controlled by the same brain structures, particularly a part of the brainstem called the nucleus gigantocellularis 17.
Norepinephrine drug uses
Norepinephrine is similar to norepinephrine (adrenaline). Norepinephrine works by constricting (narrowing) the blood vessels and increasing blood pressure and blood glucose (sugar) levels.
- For blood pressure control in certain acute hypotensive states (e.g., pheochromocytomectomy, sympathectomy, poliomyelitis, spinal anesthesia, myocardial infarction, septicemia, blood transfusion, and drug reactions).
- As an adjunct in the treatment of cardiac arrest and profound hypotension.
Norepinephrine is used to treat life-threatening low blood pressure (hypotension) that can occur with certain medical conditions or surgical procedures. This medication is often used during CPR (cardio-pulmonary resuscitation).
How is norepinephrine given?
Norepinephrine is injected into a vein through an IV. You will receive this injection in a hospital or emergency setting.
Norepinephrine is usually given for as long as needed until your body responds to the medication. Some people must receive norepinephrine for several days.
Your blood pressure, breathing, and other vital signs will be watched closely while you are receiving norepinephrine.
Tell your caregivers if you feel any pain, irritation, cold feeling, or other discomfort of your skin or veins where the medicine is injected. Norepinephrine can damage the skin or tissues around the injection site if the medication accidentally leaks out of the vein.
What other drugs will affect norepinephrine?
If possible before you receive norepinephrine, tell your doctor about all other medicines you use, especially:
- blood pressure medications;
- an MAO inhibitor (monoamine oxidase inhibitor) –isocarboxazid, linezolid, methylene blue injection, phenelzine, rasagiline, selegiline, tranylcypromine, and others; or
- an antidepressant–amitriptyline, amoxapine, clomipramine, desipramine, doxepin, imipramine, nortriptyline, protriptyline, trimipramine.
This list is not complete. Other drugs may interact with norepinephrine, including prescription and over-the-counter medicines, vitamins, and herbal products. Not all possible interactions are listed in this medication guide.
Norepinephrine dose
Dose Adjustments
- Elderly: Initiate at the low end of the dosing range due to increased likelihood for decreased hepatic, renal, or cardiac function or presence of concomitant diseases or other drug therapy. Avoid administration into veins of the leg.
- High dosage: Vast individual variation occurs in the dose required to attain and maintain an adequate blood pressure. In all cases, doses should be titrated based on individual patient response. Occasionally, much larger doses (as high as 68 mg base) may be needed if the patient remains hypotensive, however, blood volume depletion should always be suspected and corrected when present; monitoring of central venous pressure may be helpful in detecting and treating this situation.
Adult Dose for Hypotension
Uses: For blood pressure control in certain acute hypotensive cases (e.g., pheochromocytomectomy, sympathectomy, poliomyelitis, spinal anesthesia, myocardial infarction, septicemia, blood transfusion, and drug reactions); and as an adjunct in the treatment of profound hypotension
- Initial dose: 8 to 12 mcg/min continuous IV infusion
- Maintenance dose: 2 to 4 mcg/min continuous IV infusion
- Duration of therapy: Continue infusion until adequate blood pressure and tissue perfusion are maintained without therapy.
Comments:
- Doses given in terms of norepinephrine base.
- After observing response to initial dose, adjust the rate of flow to establish and maintain a low normal blood pressure (usually 80 to 100 mmHg systolic) sufficient to maintain the circulation to vital organs.
- In previously hypertensive patients, blood pressure should be raised no higher than 40 mmHg below the preexisting systolic pressure.
- Doses should be titrated based on individual patient response.
- Infusions should be reduced gradually, avoiding abrupt withdrawal.
Adult Dose for Sepsis
Society of Critical Care Medicine recommendations: 0.01 to 3 mcg/kg/min
Use: For use in patients during sepsis or septic shock to increase blood pressure
Adult Dose for Cardiac Arrest
Use: As an adjunct in the treatment of cardiac arrest
- Initial dose: 8 to 12 mcg/min continuous IV infusion
- Maintenance dose: 2 to 4 mcg/min continuous IV infusion
- Duration of therapy: Continue infusion until adequate blood pressure and tissue perfusion are maintained without therapy.
Comments:
- Doses given in terms of norepinephrine base.
- After observing response to initial dose, adjust the rate of flow to establish and maintain a low normal blood pressure (usually 80 to 100 mmHg systolic) sufficient to maintain the circulation to vital organs.
- In previously hypertensive patients, blood pressure should be raised no higher than 40 mmHg below the preexisting systolic pressure.
- Doses should be titrated based on individual patient response.
- Infusions should be reduced gradually, avoiding abrupt withdrawal.
American Heart Association recommendations: 0.1 to 0.5 mcg/kg/min IV infusion; titrate to effect
Use: For use in the treatment of post cardiac arrest care for severe hypotension (e.g., systolic blood pressure less than 70 mmHg) and a low total peripheral resistance
Comments:
- A 70 kg adult patient would receive a dose from 7 to 35 mcg/min.
Renal Dose Adjustments
Data not available
Liver Dose Adjustments
Data not available
Norepinephrine side effects
Get emergency medical help if you have any of these signs of an allergic reaction: hives; difficult breathing; swelling of your face, lips, tongue, or throat.
Tell your caregivers at once if you have a serious side effect such as:
- pain, burning, irritation, discoloration, or skin changes where the injection is given;
- sudden numbness, weakness, or cold feeling anywhere in your body;
- slow or uneven heart rate;
- blue lips or fingernails, mottled skin;
- little or no urinating;
- trouble breathing;
- problems with vision, speech, or balance; or
- dangerously high blood pressure-severe headache, blurred vision, buzzing in your ears, anxiety, confusion, chest pain, shortness of breath, uneven heartbeats, seizure.
This is not a complete list of side effects and others may occur.
Norepinephrine vs Epinephrine
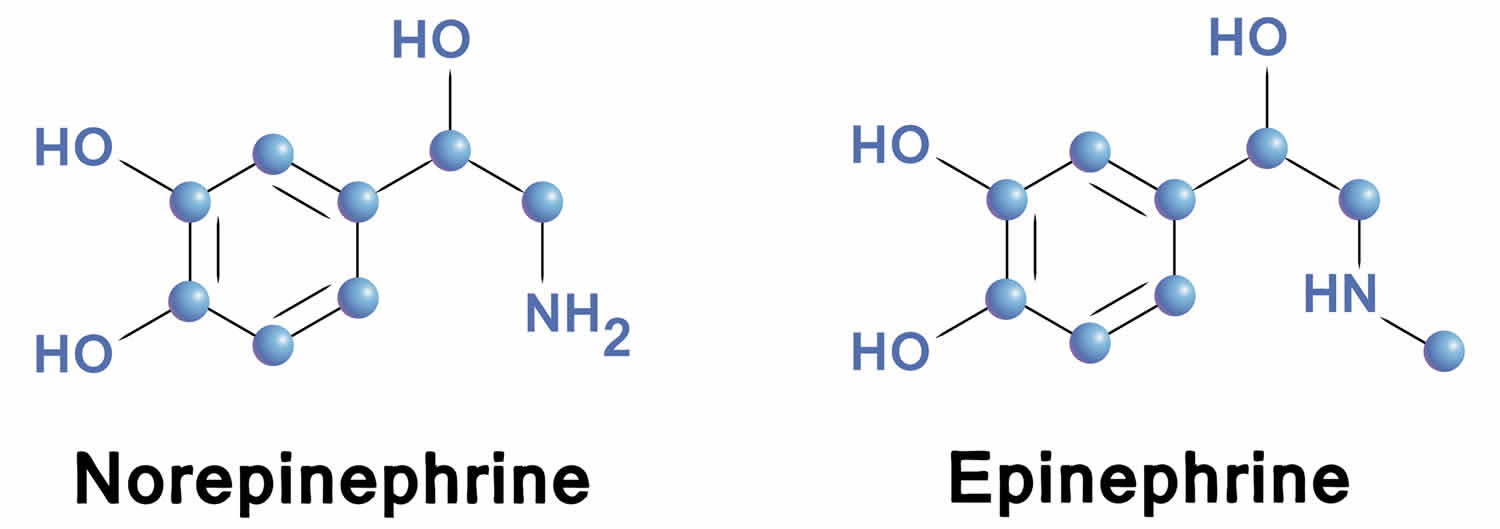
Norepinephrine differs from epinephrine by the absence of a methyl group on the nitrogen atom. Epinephrine is synthesized from norepinephrine within the adrenal glands medulla, which are small glands located above your kidneys. Preganglionic fibers of the sympathetic nervous system synapse within the adrenal glands. Activation of these preganglionic fibers releases acetylcholine, which binds to postjunctional nicotinic receptors in the tissue. This leads to stimulation of norepinephrine synthesis within adenomedullary cells, but unlike sympathetic neurons, there is an additional enzyme (phenylethanolamine-N-methyltransferase) that adds a methyl group to the norepinephrine molecule to form epinephrine. The epinephrine is released into the blood perfusing the glands and carried throughout the body.
In response to stressors posing global metabolic threats, such as acute glucoprivation (low blood glucose), emotional distress, and hemorrhagic hypotension, increments in plasma epinephrine levels exceed those of norepinephrine levels 20.
Drugs that stimulate nicotinic, angiotensin II, or glucagon receptors increase plasma epinephrine levels 7. Epinephrine stimulates β2-adrenoceptors more potently than does norepinephrine. Physiological increases in circulating epinephrine concentrations decrease the serum K+ concentration by increasing active Na+-K+ transport across cell membranes, especially in skeletal muscle 21. β2-Adrenoceptor agonists can be used clinically to treat hyperkalemia (high blood potassium); conversely, exercise can induce hyperkalemia in patients taking β-adrenoceptor blockers.
β-Adrenoceptor agonists also increase norepinephrine release into plasma by stimulating sympathetic outflows reflexively in response to decreased vascular resistance and by occupying β-adrenoceptors on sympathetic nerves. Concurrently, plasma epinephrine levels fall 22. Occupation of cardiac β-adrenoceptors increases cardiac norepinephrine spillover 23.
Because of the trophic effect of adrenocortical steroids on activity of phenylethanolamine-N-methyltransferase in adrenomedullary chromaffin cells, manipulations of hypothalamo-pituitary-adrenocortical activity affect plasma epinephrine levels more than they do plasma norepinephrine levels 7. Indeed, plasma epinephrine levels in many situations vary more closely with those of corticotropin than those of norepinephrine 7.
Secondary adrenocortical insufficiency may result from exogenous glucocorticoid administration. The mechanism involves suppression of intra-adrenal cortisol production through negative feedback of the hypothalamo-pituitary-adrenocortical axis. Low epinephrine levels in severe asthma patients treated with glucocorticoids may be explained by iatrogenic adrenocortical insufficiency 24. Similar impairment of adrenal medullary function might be expected in other patients on glucocorticoid treatment regimens.
Since cells of the adrenal medulla secrete their contents directly into the bloodstream, plasma epinephrine levels generally reflect neural outflow to the adrenal glands medulla. Thus, increments in adrenomedullary secretion of catecholamines resulting from manipulations of circulatory reflexes or from administration of drugs into the brain correlate with increments in directly recorded adrenal nerve activity 25.
Plasma levels of epinephrine are very low in antecubital venous plasma of healthy volunteers at rest—as little as 30 pmol/liter—lower than plasma levels of norepinephrine, which normally average about 1 nM. In contrast with plasma levels of norepinephrine, which generally increase with advancing age, those of epinephrine tend to decrease 7. Plasma epinephrine levels and urinary epinephrine excretion also tend to be lower in obese than in lean people and in women than in men. Inconsistencies in the literature on these topics may reflect incomplete controls for demographic and metabolic factors, variable numbers of subjects, and interlaboratory differences in assay reliability 7.
Plasma epinephrine concentrations increase markedly and to a greater extent than do norepinephrine concentrations in response to hypoglycemia, hemorrhagic hypotension, asphyxiation, circulatory collapse, and distress, presumably reflecting relatively greater adrenomedullary hormonal than sympathetic neuronal activation 7. Even mild, asymptomatic hypoglycemia elicits larger increases in epinephrine than norepinephrine levels, and in the relatively benign form of circulatory failure represented by fainting, plasma epinephrine concentrations increase with smaller increases in plasma norepinephrine concentrations 26.
Tracer kinetic studies have demonstrated epinephrine spillover in the heart in severe exercise, panic attacks, and in some patients with essential hypertension 27. Although extra-adrenal epinephrine synthesis and phenylethanolamine-N-methyltransferase have been reported in the heart, it is likely that the epinephrine released in the heart is derived from uptake from the circulation.
Addison’s disease, usually due to an autoimmune inflammation of the adrenal glands involving mainly the adrenal cortex, includes impaired adrenal medullary secretion of epinephrine. The medulla is intact, but plasma levels of epinephrine are decreased 28. This occurs despite glucocorticoid replacement, indicating that the normal high intra-adrenal steroid levels are required for an adequate production of catecholamines in the human adrenal medulla. Epinephrine secretion is also impaired in secondary adrenocortical insufficiency in children with hypocorticotropic hypopituitarism, further supporting the importance of a local source of steroids for adrenal medullary release of catecholamines.
Patients with severe 21-hydroxylase deficiency have markedly decreased plasma concentrations of epinephrine, associated with incomplete formation of the adrenal medulla 29. These patients also have low plasma concentrations of metanephrine, consistent with decreased adrenal medullary stores of epinephrine.
As for norepinephrine in sympathetic nerves, under resting conditions, metabolism of epinephrine in the adrenal medulla takes place before the hormone enters the bloodstream. Since adrenomedullary chromaffin cells possess catechol-O-methyltransferase (COMT), metanephrine constitutes a major metabolite of epinephrine before its release into extracellular fluid, whereas in sympathetic nerves, which contain monoamine oxidase type A (MAO-A) but not catechol-O-methyltransferase (COMT), dihydroxyphenylacetic acid (DHPG) constitutes the main metabolite of norepinephrine before norepinephrine release into extracellular fluid.
The fate of epinephrine that enters the bloodstream differs quantitatively from that of norepinephrine. Epinephrine is a poorer substrate than norepinephrine for uptake-1 and a better substrate for uptake-2. It is also a better substrate than norepinephrine for catechol-O-methyltransferase (COMT). Because of these differences, extraneuronal uptake and O-methylation metabolize more of circulating epinephrine than norepinephrine.
Plasma metanephrine levels are roughly the same as plasma normetanephrine levels, even though plasma norepinephrine levels exceed epinephrine levels by about 5- to 10-fold. The relatively high metanephrine concentration results from a much greater rate of production of epinephrine than of norepinephrine in adrenomedullary chromaffin cells, metabolism of adrenomedullary catecholamines by catechol-O-methyltransferase (COMT), and a relatively high proportion of metabolism of circulating epinephrine by the same enzyme.
Figure 5. Epinephrine and norepinephrine
Norepinephrine and Epinephrine Removal and Metabolism
The binding of norepinephrine (NE) to its receptor depends on the concentration of norepinephrine (NE) in the vicinity of the receptor. If the nerve stops releasing norepinephrine (NE), then the norepinephrine concentration in the junctional cleft will decrease and norepinephrine will leave the receptor. There are several mechanisms by which the norepinephrine (NE) is removed from the intercellular (junctional) space and therefore from the postjunctional receptor:
- Most (~90%) of the norepinephrine (NE) is transported back into the nerve terminal by a neuronal reuptake transport system. This transporter is blocked by cocaine; therefore, cocaine increases junctional norepinephrine concentrations by blocking its reuptake and subsequent metabolism. (This is a major mechanism by which cocaine stimulates cardiac function and raises blood pressure.)
- Some of the junctional norepinephrine diffuses into capillaries and is carried out of the tissue by the circulation. Therefore, high levels of sympathetic activation in the body increase the plasma concentration of norepinephrine and its metabolites.
- Some of the junctional norepinephrine is metabolized within the extracellular space before reaching the capillaries.
- A small amount of norepinephrine (~5%) is taken up by the postjunctional tissue (termed “extraneuronal uptake”) and metabolized.
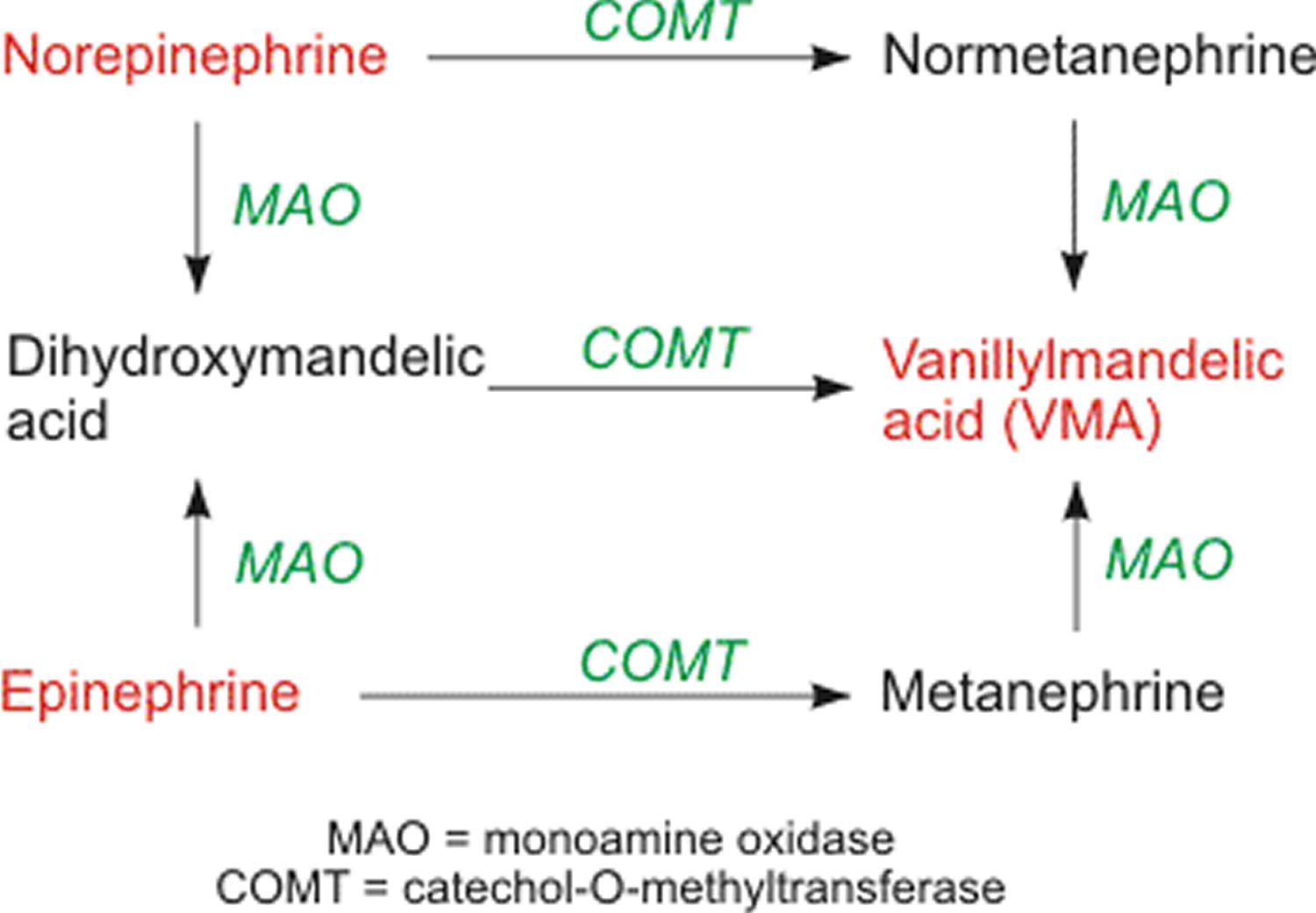
Norepinephrine and epinephrine is metabolized by catechol-O-methytransferase (COMT) and monoamine oxidase (MAO). The final product of these pathways is vanillylmandelic acid (VMA). This final product, along with its precursors normetanephrine and metanephrine, is measured in urine and plasma in the diagnosis of pheochromocytoma, which can cause severe hypertension and cardiac arrhythmias.
Figure 6. Norepinephrine and Epinephrine metabolism
Norepinephrine deficiency
Human norepinephrine deficiency (or dopamine β-hydroxylase (DBH) deficiency) is a rare congenital disorder of primary autonomic failure, in which neurotransmitters norepinephrine and epinephrine are undetectable 30. Dopamine beta-hydroxylase deficiency (norepinephrine deficiency) is characterized by lack of sympathetic noradrenergic function but normal parasympathetic and sympathetic cholinergic function 31. Affected individuals exhibit profound deficits in autonomic regulation of cardiovascular function that predispose to orthostatic hypotension, but apparently only subtle signs of central nervous system dysfunction 32. Although norepinephrine deficiency appears to be present from birth, the diagnosis is not generally recognized until late childhood when orthostatic hypotension becomes more severe 31. The combination of ptosis of the eyelids in infants and children, together with hypotension, is suggestive of the disease 31. In the perinatal period, norepinephrine deficiency (dopamine β-hydroxylase (DBH) deficiency) has been complicated by vomiting, dehydration, hypotension, hypothermia, and hypoglycemia requiring repeated hospitalization; children have reduced exercise capacity, perhaps because of hypotension engendered by physical exertion. By early adulthood, individuals have profound orthostatic hypotension, greatly reduced exercise tolerance, ptosis of the eyelids, and nasal stuffiness. Males experience retrograde or prolonged ejaculation. Presyncopal symptoms include dizziness, blurred vision, dyspnea, nuchal discomfort, and chest pain. Life expectancy is unknown, but some affected individuals have lived beyond 60 years 31.
The prevalence of dopamine β-hydroxylase deficiency is unknown. Only 21 affected individuals, all of western European descent, have been reported in the literature, suggesting that it is a rare disorder 31.
Diagnosis and testing for norepinephrine deficiency (dopamine β-hydroxylase deficiency)
The diagnosis of norepinephrine deficiency (dopamine β-hydroxylase deficiency) is based on clinical findings (including poor cardiovascular regulation, other autonomic dysfunction, and intact sweating), physiologic findings of autonomic function (which reveal failure of sympathetic noradrenergic and adrenomedullary function but intact vagal and sympathetic cholinergic function), and unique biochemical features (minimal or absent plasma norepinephrine and epinephrine AND a five- to tenfold elevation of plasma dopamine, a finding probably pathognomonic of norepinephrine deficiency). Norepinephrine deficiency is caused by biallelic pathogenic variants in dopamine β-hydroxylase.
Clinical findings:
- Poor cardiovascular regulation evident from supine, seated, and standing vital signs:
- A low-to-normal supine blood pressure and low or normal supine heart rate
- Systolic blood pressure <80 mm Hg in the upright position
- An inadequate compensatory rise in heart rate with standing
- Inability to stand motionless for more than one minute
- Other autonomic dysfunction evident from an ophthalmic examination:
- Ptosis in some individuals
- A marked decrease in intraocular pressure with standing [Phillips et al 2013]
- Somewhat small pupils that respond to light and accommodation but not to hydroxyamphetamine. Parasympatholytics dilate the pupils appropriately.
- A comprehensive history and physical examination (including neurologic exam) typically revealing:
- Intact sweating
- Skeletal and muscle findings in some affected individuals:
- Arched palate
- Hyperextensible joints
- Sluggish deep-tendon reflexes
- Mild facial-muscle weakness
- Hypotonic skeletal muscles
Laboratory Findings:
Plasma catecholamines. Biochemical features unique to dopamine β-hydroxylase deficiency:
- Minimal or absent plasma norepinephrine (NE) and epinephrine AND a five- to tenfold elevation of plasma dopamine (DA). This combination is probably pathognomonic of dopamine β-hydroxylase deficiency.
- Plasma norepinephrine concentration should be below the limits of detection (<25 pg/mL or 0.15 nmol/L)
- Plasma dopamine concentration is frequently higher than 100 pg/mL (0.65 nmol/L). One atypical individual who was not diagnosed until age 73 years was reported to have a plasma dopamine concentration of 10,000 pg/mL (67 nmol/L) 33.
- Although both baroreflex afferent and catecholamine release mechanisms are intact, dopamine is released in place of norepinephrine.
Note: (1) It is essential to assay both norepinephrine and dopamine and to use a procedure with high specificity for these catechols. (2) With some radioenzymatic methods for catecholamine determinations, a proportion of the dopamine may be erroneously measured as epinephrine 34.
The plasma dopamine concentration responds to various physiologic and pharmacologic stimuli as does norepinephrine in normal individuals:
A change from supine to upright posture doubles or triples the plasma dopamine concentration. This observation suggests that sympathetic nerves and reflex arcs are intact, but dopamine (rather than norepinephrine) is stored and released at the sympathetic synapse.
Establishing the Diagnosis
The diagnosis of dopamine β-hydroxylase deficiency is established in a proband with the following:
- Minimal or absent plasma concentration of norepinephrine and epinephrine with a five- to tenfold elevation of plasma dopamine on high performance liquid chromatography and electrochemical detection.
- Biallelic pathogenic variants in dopamine β-hydroxylase deficiency. Sequence analysis of dopamine β-hydroxylase deficiency should be pursued first, followed by deletion/duplication analysis if only one or no pathogenic variant is found.
A multi-gene panel that includes dopamine β-hydroxylase deficiency and other genes of interest may also be considered. The genes included and sensitivity of multi-gene panels vary by laboratory and over time.
Genetic counseling
Norepinephrine deficiency (dopamine β-hydroxylase deficiency) is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives is possible if both pathogenic variants in the family are known. If the pathogenic variants have been identified in an affected family member, prenatal testing for at-risk pregnancies is possible through laboratories offering either prenatal testing for the gene of interest or custom testing.
Norepinephrine deficiency (dopamine β-hydroxylase deficiency) treatment
For the most part, treatment for dopamine β-hydroxylase deficiency deficiency is supportive and directed at relieving orthostatic symptoms.
The treatment of choice is administration of L-threo-3,4-dihydroxyphenylserine (or droxidopa, marketed in the USA as Northera™). Droxidopa is converted directly to norepinephrine by L-aromatic amino acid decarboxylase, thereby bypassing dopamine β-hydroxylase deficiency (Figure 7).
Treatment of manifestations: Administration of L-threo-3,4-dihydroxyphenylserine (droxidopa) alleviates the orthostatic hypotension and other symptoms. Affected individuals do not respond as well to standard therapeutic approaches for autonomic failure. Surgery can correct ptosis. Sinus problems should be treated as needed.
- Administration of 100 to 500 mg droxidopa (L-threo-3,4-dihydroxyphenylserine) orally twice or three times daily increases blood pressure and concomitantly restores plasma norepinephrine to the normal range; however, urinary norepinephrine excretion exceeds normal levels.
- Although norepinephrine becomes detectable, plasma epinephrine concentration still remains below a detectable level.
- Droxidopa administration restores L-DOPA (levodopa) to within the normal range and reduces dopamine somewhat, but plasma concentration of dopamine and its metabolites remains somewhat elevated 35.
- This favorable alteration in catecholamines alleviates the orthostatic hypotension and restores function to a near-normal level. An affected female completed a marathon approximately five years after her diagnosis, while taking 1200 mg of droxidopa daily 36.
Prevention of primary manifestations: Droxidopa can improve the orthostatic hypotension and symptoms, but these recur if treatment is stopped.
Surveillance: Renal function (measurement of plasma creatinine and blood urea nitrogen [BUN] concentrations) is assessed every two years or more often if loss of renal function is evident. Plasma magnesium and potassium should also be assessed.
Agents/circumstances to avoid: Untreated individuals should avoid hot environments, strenuous exercise, standing motionless, and dehydration.
Pregnancy management: Routine blood pressure monitoring during pregnancy and delivery, with adjustment of droxidopa dosage as needed; extra doses of droxidopa may be required during delivery and dose adjustment may be required post partum.
At least two affected women have successfully given birth 37 following uncomplicated deliveries. Based on these experiences, it is recommended that affected pregnant women have their blood pressure monitored regularly throughout the pregnancy and delivery so that the droxidopa dose can be modified as needed. One or two extra doses of droxidopa should be available to be taken as needed at the time of delivery. Dose adjustment may also be required post partum. The effects of maternal droxidopa therapy on the developing fetus have not been studied in humans; however, studies on pregnant animals do not suggest an increased risk for malformations in offspring.
Figure 7. Synthesis of norepinephrine from dopamine or droxidopa
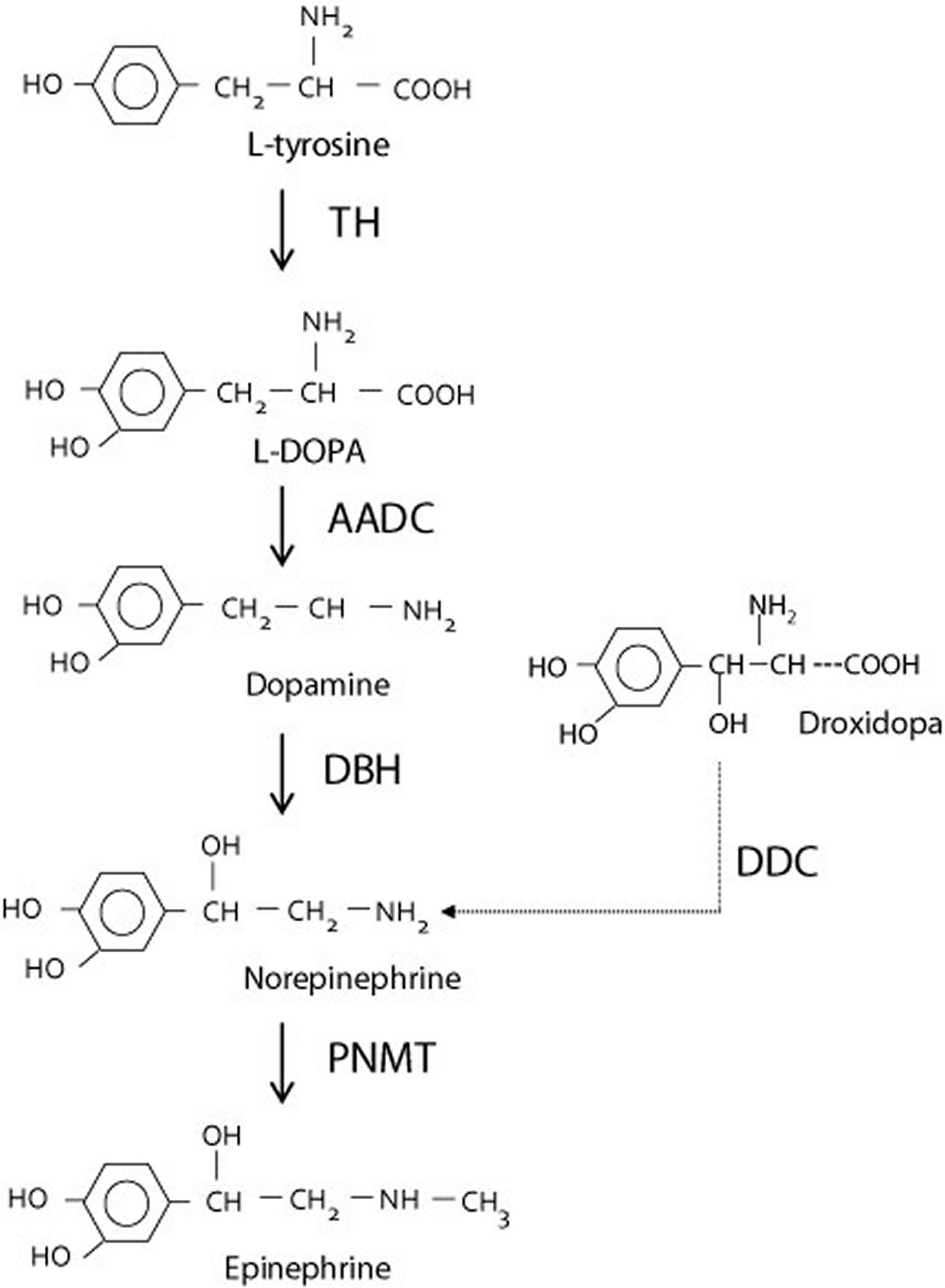
Abbreviations: TH = tyrosine hydroxylase; AADC = aromatic L-amino acid decarboxylase; DBH = dopamine β-hydroxylase deficiency; DDC = DOPA decarboxylase; PNMT = phenylethanolamine N-methyltransferase
[Source 31] References- Esler MD, Turner AG, Kaye DM, Thompson JM, Kingwell BA, Morris M, Lambert GW, Jennings GL, Cox HS, and Seals DR (1995) Aging effects on human sympathetic neuronal function. Am J Physiol 268: R278–R285
- Grossman E, Goldstein DS, Hoffman A, and Keiser HR (1991) Glucagon and clonidine testing in the diagnosis of pheochromocytoma. Hypertension 17: 733–741
- Goldstein DS, Levinson PD, Zimlichman R, Pitterman A, Stull R, and Keiser HR (1985) Clonidine suppression testing in essential hypertension. Ann Intern Med 102: 42–49.
- Robertson D, Goldberg MR, Tung CS, Hollister AS, and Robertson RM (1986) Use of α2 adrenoreceptor agonists and antagonists in the functional assessment of the sympathetic nervous system. J Clin Investig 78: 576–581.
- Charney DS, Heninger GR, and Breier A (1984) Noradrenergic function in panic anxiety. Effects of yohimbine in healthy subjects and patients with agoraphobia and panic disorder. Arch Gen Psych 41: 751–763.
- Goldstein DS and Holmes C (1997) Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exper Hypertens 19: 155–161.
- Sources and Significance of Plasma Levels of Catechols and Their Metabolites in Humans. Journal of Pharmacology and Experimental Therapeutics Vol. 305, Issue 3, 1 Jun 2003. http://jpet.aspetjournals.org/content/305/3/800.long
- Musacchio JM (2013). “Chapter 1: Enzymes involved in the biosynthesis and degradation of catecholamines”. In Iverson L. Biochemistry of Biogenic Amines. Springer. pp. 1–35. ISBN 1-4684-3171-4.
- Hamill RW, Shapiro RE, Vizzard MA (2012). “Peripheral Autonomic Nervous System”. In Robertson D, Biaggioni I, et al.. Primer on the Autonomic Nervous System. Academic Press. pp. 17–20. ISBN 978-0-12-386525-0.
- Schacter D, Gilbert D, Wegner D, Hood B (2011). Psychology: European Edition. Palgrave Macmillan. p. 93. ISBN 978-0-230-34367-2.
- Dartt DA. Neural Regulation of Lacrimal Gland Secretory Processes: Relevance in Dry Eye Diseases. Progress in retinal and eye research. 2009;28(3):155-177. doi:10.1016/j.preteyeres.2009.04.003. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3652637/
- Peripheral and central effects of circulating catecholamines. Compr Physiol. 2015 Jan;5(1):1-15. doi: 10.1002/cphy.c140007. https://www.ncbi.nlm.nih.gov/pubmed/25589262
- Dahlstroem A, Fuxe K (1964). “Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons”. Acta Physiologica Scandinavica. Supplementum. 232 (Supplement 232): 1–55. https://www.ncbi.nlm.nih.gov/pubmed/14229500
- Neuroendocrine control of body fluid metabolism. Physiol Rev. 2004 Jan;84(1):169-208. https://www.physiology.org/doi/abs/10.1152/physrev.00017.2003
- Rinaman L (February 2011). “Hindbrain noradrenergic A2 neurons: diverse roles in autonomic, endocrine, cognitive, and behavioral functions”. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 300 (2): R222–35. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3043801/
- Bruinstroop E, Cano G, Vanderhorst VG, Cavalcante JC, Wirth J, Sena-Esteves M, Saper CB (June 2012). “Spinal projections of the A5, A6 (locus coeruleus), and A7 noradrenergic cell groups in rats”. The Journal of Comparative Neurology. 520 (9): 1985–2001 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3508755/
- Sara SJ, Bouret S (2012). “Orienting and reorienting: the locus coeruleus mediates cognition through arousal”. Neuron. 76 (1): 130–41. doi:10.1016/j.neuron.2012.09.011 https://www.ncbi.nlm.nih.gov/pubmed/23040811
- Berridge CW, Schmeichel BE, España RA (2012). “Noradrenergic modulation of wakefulness/arousal”. Sleep Med Rev. 16 (2): 187–97. doi:10.1016/j.smrv.2011.12.003 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3278579/
- Sara SJ (2015). “Locus Coeruleus in time with the making of memories”. Curr. Opin. Neurobiol. 35: 87–94. doi:10.1016/j.conb.2015.07.004 https://www.ncbi.nlm.nih.gov/pubmed/26241632
- Pacak K, Baffi JS, Kvetnansky R, Goldstein DS, and Palkovits M (1998) Stressorspecific activation of catecholaminergic systems: implications for stress-related hypothalamic-pituitary-adrenocortical responses. Adv Pharmacol 42: 561–564
- Clausen T (1983) Adrenergic control of Na+-K+-homoeostasis. Acta Med Scand Suppl 672: 111–115.
- Eisenhofer G, Goldstein DS, Stull RW, Gold PW, Keiser HR, and Kopin IJ (1987) Dissociation between corticotrophin and catecholamine responses to isoprenaline in humans. Clin Exp Pharmacol Physiol 14: 337–341.
- Thompson JM, Wallin BG, Lambert GW, Jennings GL, and Esler MD (1998) Human muscle sympathetic activity and cardiac catecholamine spillover: no support for augmented sympathetic noradrenaline release by adrenaline co-transmission. Clinical Science (Wash DC) 94: 383–393.
- Mathe A and Knapp P (1969) Decreased plasma free fatty acids and urinary epinephrine in bronchial asthma. N Engl J Med 281: 234–238.
- Yoshimatsu H, Oomura Y, Katafuchi T, and Niijima A (1987) Effects of hypothalamic stimulation and lesion on adrenal nerve activity. Am J Physiol 253: R418–R424.
- Goldstein DS, Spanarkel M, Pitterman A, Toltzis R, Gratz E, Epstein S, and Keiser HR (1982) Circulatory control mechanisms in vasodepressor syncope. Am Heart J 104: 1071–1075.
- Esler M (2000) The sympathetic system and hypertension. Am J Hypertens 13: 99S–105S
- Bornstein S, Breidert M, Ehrhart-Bornstein M, Kloos B, and Scherbaum W (1995) Plasma catecholamines in patients with Addison’s disease. Clin Endocrinol 42: 215–218.
- Merke D, Chrousos G, Eisenhofer G, Weise M, Keil M, Rogol A, Van Wyk J, and Bornstein S (2001) Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N Engl J Med 343: 1362–1368
- Kim C-H, Leung A, Huh YH, et al. Norepinephrine Deficiency Is Caused by Combined Abnormal mRNA Processing and Defective Protein Trafficking of Dopamine β-Hydroxylase. The Journal of Biological Chemistry. 2011;286(11):9196-9204. doi:10.1074/jbc.M110.192351. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3059068/
- Robertson D, Garland EM. Dopamine Beta-Hydroxylase Deficiency. 2003 Sep 4 [Updated 2015 Oct 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1474
- Jepma M, Deinum J, Asplund CL, Rombouts SA, Tamsma JT, Tjeerdema N, Spapé MM, Garland EM, Robertson D, Lenders JW, Nieuwenhuis S. Neurocognitive function in dopamine-β-hydroxylase deficiency. Neuropsychopharmacology. 2011;36:1608–19.
- Despas F, Pathak A, Berry M, Cagnac R, Massabuau P, Liozon E, Galinier M, Senard JM. DBH deficiency in an elderly patient: efficacy and safety of chronic droxidopa. Clin Auton Res. 2010;20:205–7.
- Robertson D, Goldberg MR, Onrot J, Hollister AS, Wiley R, Thompson JG Jr, Robertson RM. Isolated failure of autonomic noradrenergic neurotransmission. Evidence for impaired beta-hydroxylation of dopamine. N Engl J Med. 1986;314:1494–7
- Biaggioni I, Robertson D. Endogenous restoration of noradrenaline by precursor therapy in dopamine-beta-hydroxylase deficiency. Lancet. 1987;2:1170–2
- Garland EM, Raj SR, Demartinis N, Robertson D. Case report: Marathon runner with severe autonomic failure. Lancet. 2005b;366 Suppl 1:S13.
- Scurrah NJ, Ross AW, Solly M. Peripartum management of a patient with dopamine beta-hydroxylase deficiency, a rare congenital cause of dysautonomia. Anaesth Intensive Care. 2002;30:484–6





{kind=link}