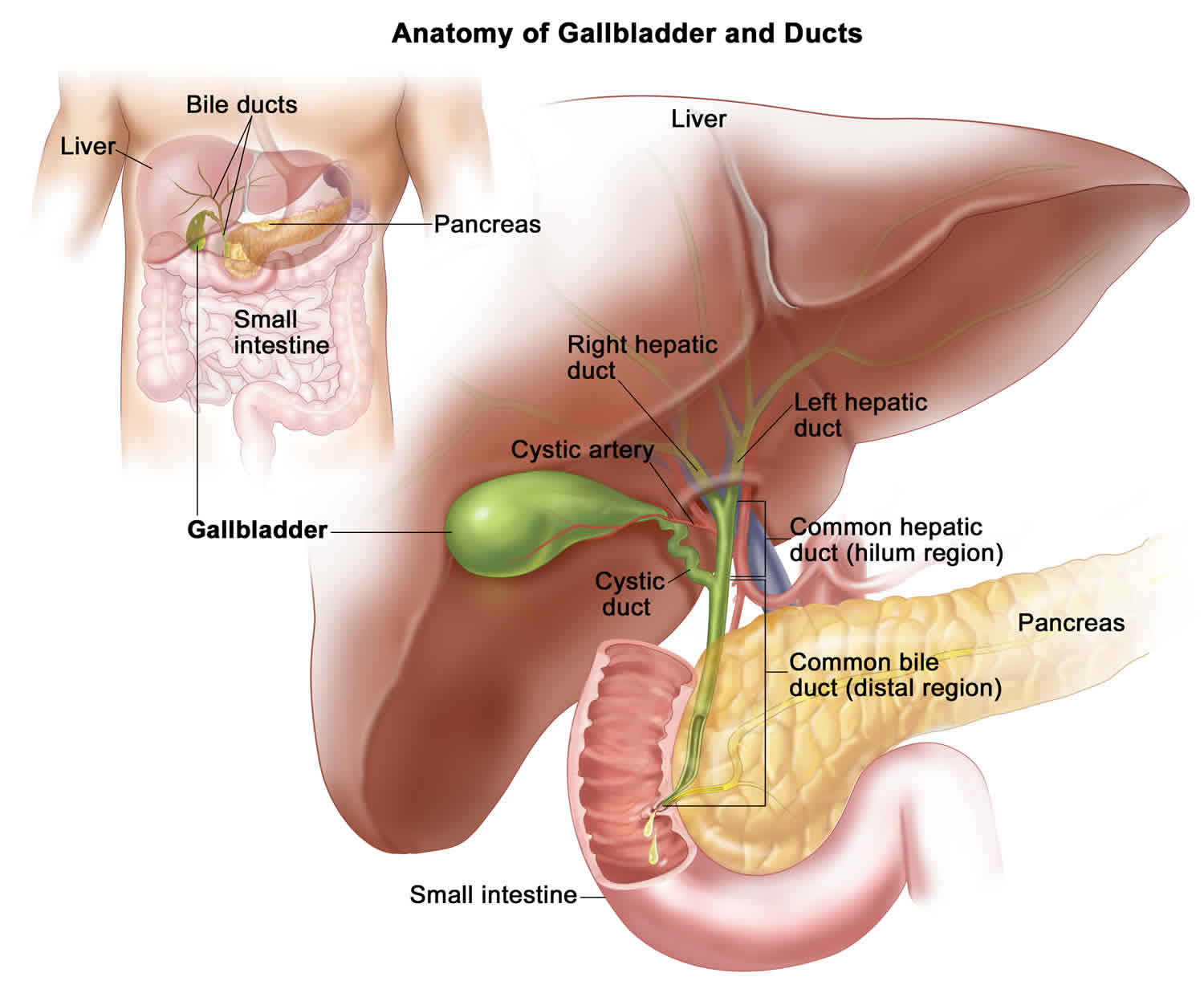
Caroli disease
Caroli disease is a rare inherited disorder characterize by an abnormal widening (dilatation) of the intrahepatic bile ducts (the ducts that carry bile from the liver) and renal cysts 1. According to the medical literature, there are two forms of Caroli disease. In most cases, the isolated or simple form is characterized by widening of the bile ducts (dilatation or ectasia). A second, more complex form is often called Caroli syndrome. The complex form or Caroli syndrome is associated with the presence of bands of fibrous tissue in the liver (congenital hepatic fibrosis) and high blood pressure in the portal artery (portal hypertension. Caroli syndrome, the second form of Caroli disease, is also often associated, in ways that are not well understood, with polycystic kidney disease, and, in severe cases, liver failure 1.
Caroli disease is a rare disorder that may occur as an isolated finding or associated with congenital hepatic fibrosis. Both forms of Caroli disease are characterized by abnormal widening of the ducts that carry bile from the liver (intrahepatic bile ducts). This widening may occur as a result of the formation of fluid-filled sacs or lumps (choledochal cysts) in the bile ducts.
Caroli syndrome, the second form of Caroli disease, is associated with abnormal formation bands of fibrous tissue in the portal area of the liver (congenital hepatic fibrosis). The portal area of the liver is where the portal vein and the hepatic artery enter the liver. The portal vein is the blood vessel that carries blood from the stomach, intestine, and spleen to the liver, and the hepatic artery is the blood vessel that carries blood away from the aorta. Caroli syndrome is also often associated with high blood pressure of the portal vein (portal hypertension) and liver abcess. In addition, Caroli syndrome may also be associated with polycystic kidney disease and, in severe cases, liver failure.
People affected by Caroli disease experience recurrent episodes of cholestasis, stone development in the bile ducts, and bacterial cholangitis 2. In addition to the symptoms of Caroli disease, people affected by Caroli syndrome may also experience liver fibrosis and portal hypertension (high blood pressure of the portal vein). In some cases, individuals with Caroli disease may appear to be more prone to developing certain benign tumors or malignancies (e.g., cholangiocarcinoma) than the general population.
Caroli disease and Caroli syndrome are very rare, with an estimated incidence of less than 1 case per 100,000 population. Caroli syndrome (ectasia of the large and small bile ducts with congenital hepatic fibrosis) is more common than Caroli disease (ectasia of only the large bile ducts) 3.
Although the underlying cause of Caroli disease and Caroli syndrome are poorly understood, they are thought to be genetic conditions. Caroli disease generally occurs sporadically in people with no family history of the condition; however, rare reports exist of autosomal dominant inheritance in association with autosomal dominant polycystic kidney disease. Caroli syndrome is generally inherited in an autosomal recessive manner and is frequently seen in association with autosomal recessive polycystic kidney disease.
Treatment for Caroli disease and Caroli syndrome is based on the signs and symptoms present in each person. For example, frequent episodes of bacterial cholangitis can be treated with antibiotics. Fat soluble vitamin supplementation may be recommended in people with cholestasis. In severe cases of cholestasis, surgery (called a lobectomy) may be required. Stones that develop within the bile ducts may be dissolved with ursodeoxycholic acid or may require surgical removal, if feasible. In those with portal hypertension, medications may be prescribed to prevent bleeding and surgery to reroute blood flow (called portosystemic shunting) may be recommended in severe cases 3.
People who have recurrent infections, especially those who also have complications related to portal hypertension, may require liver transplantation.
Caroli disease causes
Caroli disease is a birth defect distinguished by abnormal prenatal development of the bile duct in the liver. The exact cause is unknown. In most cases, the simple or isolated form of Caroli disease is believed to result from a spontaneous genetic change (mutation) that occurs for unknown reasons (sporadic). Researchers believe that this form is inherited as an autosomal dominant genetic trait. In contrast, the more complex form of Caroli disease appears to be inherited as an autosomal recessive genetic trait. The gene responsible for the more complex form of the disorder has been tracked to chromosome 6 (6p21.1-p12).
Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 6p21.1-p12” refers to a region between bands 21.1 and 12 on the short arm of chromosome 6. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All individuals carry 4-5 abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
Caroli disease symptoms
Caroli disease is a condition characterized by an abnormal widening of the intrahepatic bile ducts (the ducts that carry bile from the liver) and renal cysts. People affected by Caroli disease experience recurrent episodes of cholestasis which may be associated with abdominal pain and itching. This stagnation of the bile can also lead to the development of stones within the bile ducts (called intraductal lithiasis). Bacterial cholangitis occurs frequently which may be accompanied by fever and pain in the right upper abdomen 3.
The isolated or simple form of Caroli disease is characterized by frequent recurring inflammation of the bile ducts inside the liver. There may also be localized accumulation of pus (abscess), stones that develop inside the bile ducts (intraductal lithiasis), abdominal pain, and/or fever. In rare cases, individuals may exhibit yellowing of the skin, mucous membranes, and whites of the eyes (jaundice) and/or abnormal enlargement of the liver (hepatomegaly).
In addition to the symptoms outlined above, people affected by Caroli syndrome may also experience liver fibrosis and portal hypertension (high blood pressure of the portal vein). Portal hypertension can be associated with vomiting blood, bloody stools, and ascites. Caroli syndrome is often associated with an inherited condition called autosomal recessive polycystic kidney disease 3.
People affected by Caroli disease and Caroli syndrome may also have an increased risk of developing cholangiocarcinoma 4.
Caroli disease complications
Caroli disease complications include
- Simple type
- intrahepatic stone formation
- recurrent cholangitis that may lead to bacteremia and sepsis
- hepatic abscesses
- Periportal fibrosis type
- cirrhosis and portal hypertension
- hepatomegaly
- ascites
- varices
- There is an increased risk of cholangiocarcinoma, which develops in 7% of patients 5.
Caroli disease diagnosis
When the liver and spleen are unusually large (hepatomegaly and splenomegaly) and intermittent stomach pain is present, the doctor may ask for imaging studies (such as ultrasound and CT scans) to be done. The results of these studies may lead to a diagnosis of Caroli disease.
Caroli disease treatment
The mainstay of therapy is supportive and individualized according to the presentation. Cholangitis due to biliary obstruction is treated with antibiotics which cover gram-negative rods and anaerobic rods. Adequate biliary drainage may be required in the form of biliary stent placement through endoscopic retrograde cholangiopancreatography (ERCP) or interventional radiology-guided percutaneous trans-hepatic catheter placement. Percutaneous trans-hepatic catheter is usually more effective at draining intra-hepatic obstruction and patients sometimes require indwelling catheters with periodic flushing and changing of the catheter. Ursodeoxycholic acid is used to treat severe cholestasis. Since there are multiple cysts, the patient may develop recurrent episodes of cholangitis and lobectomy or liver transplant may be required in that case.
Once hepatic fibrosis sets in, it leads to portal hypertension and its sequelae of variceal bleeding and recurrent ascites which should be managed as they are when related to liver cirrhosis, for example, with non-selective beta-blockers, endoscopic band ligation, and diuretic therapy for ascites. Liver transplantation is the only definitive treatment available at this time. There are 3 indications for liver transplantation in Caroli disease: hepatic decompensation, recurrent cholangitis that is unresponsive to intervention, and development of focal adenocarcinoma 6.
Medical care
Ursodeoxycholic acid can decrease the frequency of Caroli disease complications due to cholelithiasis. Broad-spectrum antibiotic coverage, including anaerobic coverage, is indicated in cases of cholangitis. Patients with cholestasis should receive fat-soluble vitamin supplementation. Because patients with Caroli syndrome or Caroli disease are at an increased risk for cholangiocarcinoma, initial radiographic (ie, ultrasonography, MRI) and serologic (ie, CA19-9, CEA) screening should be performed.
Surgical care
Surgical treatment may be necessary for recurrent or refractory cholangitis. Obstructing stones can be removed and bile flow can be maintained by means of a hepaticojejunostomy or external drainage. In cases of localized stasis, lobectomy can be curative and can also reduce the risk of cholangiocarcinoma. Liver transplantation may be indicated in severe cases of refractory or chronic cholangitis, liver failure, or malignant transformation 7.
Liver transplantation represents an uncommon indication for Caroli disease. Excellent results have been reported, as shown by multicentric European and American registry reports 8.
Isolated or combined kidney and liver transplantations have become available for children with polycystic liver and kidney disease. Immunosuppression after isolated renal transplantation may lead to an increased number of episodes of cholangitis and worsening liver condition. Therefore, a combined liver-kidney transplantation may be required in children with established end-stage renal failure and advanced chronic liver disease 9.
Patients who have developed esophageal varices should receive prophylaxis with a nonselective beta-blocker and variceal endoscopic therapy. The shunting procedure can provide relief from portal hypertension because liver function may be well preserved.
Caroli disease prognosis
The long-term outlook for people with Caroli disease and Caroli syndrome is variable and determined by the frequency and severity of the episodes of cholangitis; the presence of associated diseases; and the increased risk of bile duct cancer 3.
Caroli’s disease life expectancy
Caroli disease life expectancy varies according to the extent of underlying genetic abnormality (gene deletion or mutation) and also the number of different organ systems involved. Recurrent biliary obstruction and fibrosis cause significant morbidity, and liver transplantation is the only definitive treatment at this time. Patients with Caroli disease or Caroli syndrome may have recurrent episodes of cholangitis and are also at risk for associated bacteremia and sepsis. Patients with Caroli syndrome or Caroli disease may have cholangitis and may also have complications of portal hypertension as is observed in congenital hepatic fibrosis. Caroli syndrome is associated with autosomal dominant polycystic kidney disease, and patients may have various degrees of renal cysts, interstitial fibrosis, and renal failure. Both Caroli disease and Caroli syndrome are associated with a risk of cholangiocarcinoma at a rate of 100 times that of the general population.
References- Caroli disease. https://rarediseases.org/rare-diseases/caroli-disease/
- Caroli disease. https://rarediseases.info.nih.gov/diseases/6002/caroli-disease
- Pediatric Caroli Disease. https://emedicine.medscape.com/article/927248-overview
- Liang JJ, Kamath PS. Caroli syndrome. Mayo Clin Proc. June 2013; 88(6):e59.
- Federle MP, Jeffrey RB, Woodward PJ et-al. Diagnostic Imaging: Abdomen, Published by Amirsys®. Lippincott Williams & Wilkins. (2009) ISBN:1931884714
- Lai Q, Lerut J. Proposal for an algorithm for liver transplantation in Caroli’s disease and syndrome: putting an uncommon effort into a common task. Clin Transplant. 2016 Jan;30(1):3-9.
- [Guideline] Murray KF, Carithers RL Jr. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology. 2005 Jun. 41(6):1407-32.
- Lai Q, Lerut J. Proposal for an algorithm for liver transplantation in Caroli’s disease and syndrome: putting an uncommon effort into a common task. Clin Transplant. 2016 Jan. 30 (1):3-9.
- Biliary Atresia and Neonatal Disorders of the Bile Ducts. Robert Wyllie, Jeffrey Hyams and Marsha Kay. Pediatric Gastrointestinal and Liver Disease. Fifth Edition. Elsevier; 2016. 1172-1173.


{kind=link}