Cherry red spot
Cherry-red spot refers to the reddish area at the center of macula surrounded by retinal opacification in certain disorders 1. Ruling out serious life-threatening or sight-threatening diseases is of paramount importance in a patient with a cherry-red spot at the macula. The management varies according to the cause. The retinal opacification may be due to various causes, including accumulation of different materials in the ganglion cells in storage disorders and retinal ischemia/infarction as in central retinal arterial occlusion (CRAO) 1. The cherry red spot occurs because the choroidal and retinal circulation is separate. The richly vascularized sub-foveal choriocapillaris remains perfused after central retinal arterial occlusion, but the surrounding retina becomes opaque due to edema as the tissue becomes ischemic 2.
Ruling out serious life-threatening or sight-threatening diseases is of paramount importance in a patient with a cherry-red spot at the macula. The management varies according to the cause. When the nurse practitioner, primary care provider, or emergency department physician encounters patients with vision alterations, referral to an ophthalmologist is a strong recommendation.
A cherry-red spot at the macula is a rare finding, and the epidemiology depends on the cause. Tay-Sachs disease is estimated to affect 1 in 320,000 newborns. The true epidemiology of central retinal arterial occlusion is unknown. A study estimated that acute central retinal arterial occlusion (duration less than 48 hours) affects approximately 0.85 per 100,000 per year or 1.13 per 10,000 outpatient visits 3.
Figure 1. Cherry red spot
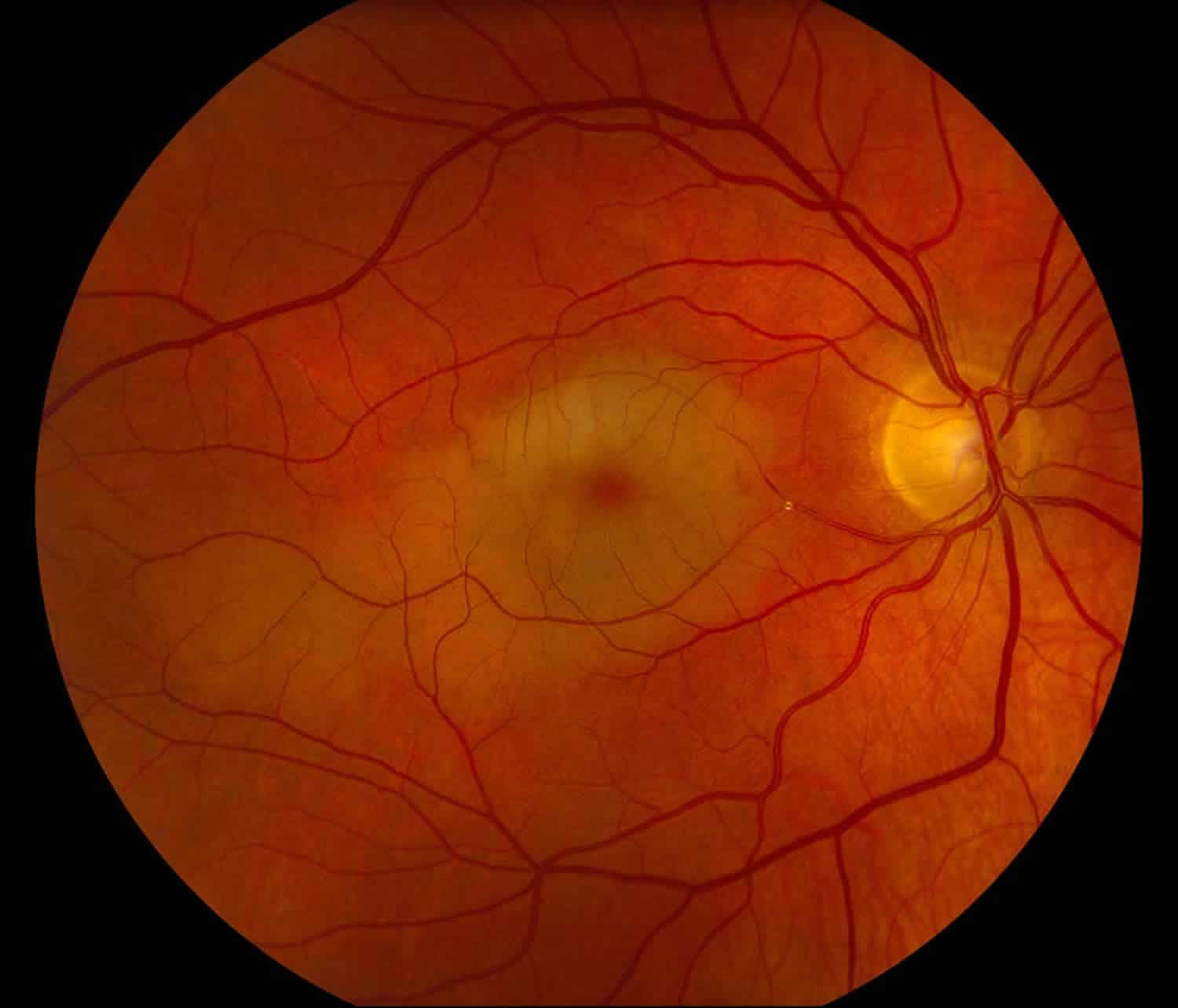
Footnote: An interesting case in which a cholesterol (Hollenhorst) plaque became lodged in an inferior branch of the central retinal artery that perfused both the superior and inferior macula. Unfortunately for this patient this resulted in significant central vision loss.
[Source 2 ]Causes of cherry red spot
The causes of the cherry-red spot include:
- Central retinal artery occlusion (CRAO) – The characteristic presentation of this disease is sudden onset unilateral visual loss in an elderly male or female. Usual cause in an embolism which blocks the central retinal artery. The embolus may be composed of cholesterol, fibrin-platelet, or calcium. The embolus usually originates from the carotid plaque and uncommonly from the heart or the aorta. Inflammatory conditions like giant cell arteritis (temporal arteritis), may also cause central retinal arterial occlusion, though branch retinal arterial occlusion (BRAO) is not a common feature in giant cell arteritis. Branch retinal arterioles do not have a continuous muscular coat or internal elastic lamina 4. Giant cell arteritis (temporal arteritis) involves only medium and large size arteries and not arterioles (like branch retinal arteriole). However, giant cell arteritis (temporal arteritis) might affect the cilioretinal artery, which is a branch of the posterior ciliary artery. On cursory examination, an examiner might mistake cilioretinal arterial occlusion in giant cell arteritis (temporal arteritis) for a branch retinal arterial occlusion. Other causes of central retinal artery occlusion include myxoma or vegetations of the cardiac valves, thrombophilic disorders, and retinal migraine. Other associations of central retinal arterial occlusion include dyslipidemia, diabetes, hypertension, smoking, and pyoderma gangrenosum 5. In an extensive series of 248 eyes of 240 patients with central retinal artery occlusion, the cherry-red spot was present on the initial examination in 90% of permanent central retinal arterial occlusion cases 6. Retinal whitening and cherry red spot are important manifestations of acute central retinal arterial occlusion. Other related situations causing a cherry-red spot include 7:
- Acute occlusion of the choroidal circulation causing outer retinal whitening and a cherry-red spot at the macula following cataract surgery (phacoemulsification)
- Intraorbital hemorrhage or mass may compromise the choroidal and retinal circulation and lead to a cherry-red spot at the macula
- Leukemic infiltrates of the optic nerve head may cause central retinal arterial occlusion and cherry red spot
- Optic nerve avulsion may cause a cherry-red spot at the macula
- Macular infarction due to various causes
- Tay-Sachs disease (GM2 gangliosidosis type 1, infantile amaurotic familial idiocy, B variant GM2 gangliosidosis): This is the most common type of gangliosidosis due to deficiency of the alpha subunit of hexosaminidase A on chromosome 15q23. The disease has an onset of the symptoms in the infancy. There is an abnormal accumulation of gangliosides in the brain and ganglion cell layer of the retina. The disease is fatal, and most affected children die within the first 2 to 5 years of age. This neurodegenerative disorder is inherited autosomal recessively. There is a developmental delay in the child. Other features include a cherry-red spot at the macula in both eyes, increased startle response, poor head control, seizures, dementia, apathy, hypotonia, deafness, blindness, and late hypertonia. Incidence is very high in Ashkenazi Jews at around 1 in 3500 to 4000. A cherry-red spot is present in approximately 90% of the patients affected with the disease.
- Sandhoff disease or GM2 gangliosidosis type 2 is due to mutation of the beta subunit of hexosaminidase at chromosome 5q13. Reports describe infantile, juvenile, and adults forms. It is inherited autosomal recessively. The cherry-red spot is present in a majority of cases. Clinical features are similar to Tay-Sachs disease.
- Commotio retinae: After blunt trauma, acutely retinal opacification may occur 8. When this whitening of the retina involves the macula, it is Berlin edema, though actual retinal thickening is not present; this causes a pseudo-cherry red spot.
- Niemann-Pick disease: Type A and B result from the deficiency of acid sphingomyelinase enzyme, which is a product of the SMPD1 gene (sphingomyelin phosphodiesterase-1) on chromosome 11p15.4. There is an accumulation of sphingomyelin in the tissues. The cause of Niemann-Pick disease type C1 and type D is a mutation in the NPC1 gene on chromosome 18q11. Niemann-Pick disease C2 is due to a mutation in the NPC2 gene at chromosome 14q24.3. Both NPC1 and NPC2 genes are involved in the transport of lipids (LDL cholesterol or low-density lipoprotein cholesterol) in endosomes and lysosomes.
- Type A disease (classic infantile) has an early onset and severe neurodegeneration. Hepatosplenomegaly may be present in early infancy, and there may be a failure to thrive and suboptimal gain of weight. The developmental milestones may be normal till the first birthday, after which there may be psychomotor regression or progressive loss of mental abilities and motor performance. Interstitial lung disease may be noted, and death is usually within early childhood. Most of the affected children have a cherry-red spot at the macula.
- Type B, usually (visceral), has a normal function of the central nervous system (CNS). Disease onset is usually in the mid-childhood, and presentation is similar to but less severe than type A. Features include short stature, hepatosplenomegaly, recurrent pulmonary infections, and thrombocytopenia. Some patients may have mixed features of type A, and B. Cherry-red spot at the macula is noted in around 1/3rd cases.
- Type C (subacute/juvenile) has slowly progressive disease and moderate CNS damage. Features include ataxia, speech and swallowing difficulty, vertical supranuclear gaze palsy, dystonia, interstitial lung disease, intellectual decline, and seizures.
- Type D (Nova Scotia form noted in individuals from Nova Scotia of Canada who have an Acadian ancestor) is associated with late-onset disease but may lead to severe CNS involvement with time. Currently, it classifies as the same condition as type C.
- GM1 gangliosidosis type 1 (Generalized gangliosidosis or Landing disease) results from the deficiency of beta-galactosidase at chromosome 3p22.3. A cherry-red spot in the macula occurs in around 50% cases.[6] There are three variants:
- Type I or infantile form is characterized by a severe disease with onset within the first year of life. Facial features are coarse with gingival hypertrophy. Other findings include developmental regression, increased startle response, skeletal anomalies, hepatosplenomegaly, intellectual disability, seizures, a cherry-red spot at the macula, corneal clouding, and death within early childhood.
- Type II or the late infantile or juvenile form presents within 18 months or 5 years, respectively. The severity is less than type I and the cherry-red spot at the macula or visceromegaly or facial features like type I is usually not seen.
- Type III or adult or chronic form is the mildest form. Features include dystonia and vertebral anomaly. The age of onset and life expectancy may be variable.
- Sialidosis or mucolipidosis type 1 or alpha-neuraminidase deficiency or sialidase deficiency or lipomucopolysaccharidosis presents with ataxia, movement disorders, nystagmus, and myoclonic seizures. There is a deficiency of neuraminidase 1 (NEU1) or sialidase (SIAL1) at chromosome 6p21.33. Intellect is usually normal, and somatic features are generally not seen. Two types have been described according to the age of onset and severity of the disease:
- Sialidosis type I or cherry-red spot myoclonus syndrome is characterized by onset in the 2nd or 3rd decade, gait disturbances, myoclonus, ataxia, tremor, a cherry-red spot at the macula, and seizures. The intellect and life expectancy are usually not affected, though the patient eventually needs wheelchair assistance with time. Almost all the patients have the cherry-red spot at the macula 9.
- Sialidosis type II causes early and more severe disease. The variants include:
- Congenital- There is either stillbirth or death soon after birth. Features include ascites, hydrops fetalis, coarse facial characteristics, dysostosis multiplex, and hepatosplenomegaly.
- Infantile – Features are similar but milder than the congenital form. There may be short stature, gingival hyperplasia, widely spaced teeth, myoclonus, a cherry-red spot at the macula, deafness, and intellectual decline. The patients may survive into adolescence.
- Juvenile form- This is the mildest variety, and life expectancy varies among the patients. Along with other features, angiokeratoma of the skin is present.
- Farber lipogranulomatosis – Ceramidase or acid ceramidase (chromosome 8p22) is deficient in this autosomal recessive disorder. The typical triad consists of painful swollen joints, subcutaneous nodules (lipogranulomas), and weak cry or hoarse voice due to laryngeal nodules. Other features are a developmental delay, visceromegaly, respiratory involvement, and neurological features, including quadriplegia, seizures, and myoclonus.
- Metachromatic leukodystrophy – There is a deficiency of arylsulfatase A or cerebroside sulfatase or sulfatide sulfatase on chromosome 22q13 in this autosomal recessive disease. There is an abnormal collection of sulfatides in cells producing myelin resulting in progressive destruction of white matter (leukodystrophy). There is a progressive decline in intellectual and motor functions, peripheral neuropathy resulting in diminished sensations, hearing loss, incontinence, loss of speech and mobility, blindness, unresponsiveness, and eventual death. The name comes from the color of the deposited sulfatide granules on histopathological examination. A cherry-red spot at the macula is occasionally a finding 9.
- Galactosialidosis (Goldberg Cotlier syndrome or neuraminidase deficiency with beta-galactosidase deficiency or cathepsin A deficiency): This is an autosomal recessive disorder due to a mutation in the CTSA gene (chromosome 20q13.12) coding for cathepsin A. The disease characteristically presents with seizures, mental retardation, growth retardation, dysostosis multiplex, coarse facies, short stature, skin hemangiomas, mitral and atrial valvular disease, and deafness. Ocular features include a cherry-red spot at the macula, corneal clouding, and conjunctival telangiectasia.
- Hyperalimentation causing hyperlipidemia
- Retinitis involving the central retina- including progressive outer retinal necrosis, subacute sclerosing panencephalitis 10
- Toxicity of various drugs may also cause a cherry-red spot. These drugs include 11:
- Quinine
- Carbon monoxide
- Dapsone poisoning associated with hemolytic anemia and peripheral neuropathy
- Methanol
- Intravitreal gentamicin
The association between Gaucher disease and cherry red spots at the macula is questionable.
Cherry red spot pathophysiology
The macula is characterized histologically by an area with more than one layer of ganglion cell layers, and the ganglion cell layer is thick at the macula. In diseases causing loss of transparency (whitening or opacification) of the inner retina, the reddish color of the vascular choroid and pigmentation of retinal pigment epithelium (RPE) is not seen through the opacified retina. However, foveola is devoid of inner retinal layer. The retinal layers present at the foveola are (from inside outwards)- internal limiting membrane, outer nuclear layer, external limiting membrane, photoreceptor layer, and retinal pigment epithelium. Inner retinal layers are absent in mature foveola. The vascular supply of the fovea is from the choroid, and in the occlusion of retinal vessels, it does not get hampered. Thus foveola, the thinnest part of the central retina, does not lose its transparency in inner retinal ischemia.
Thus, when there is inner retinal opacification, the reddish color of the vascular choroid and the retinal pigment epithelium is still seen through the foveola, which is surrounded by an area of the white/opacified retina; this gives rise to the typical cherry-red spot. The size of the cherry-red spot depends on the size of the foveola.
As the color of choroid and retinal pigment epithelium varies with racial variation, the color of foveola or the central dot may change according to race. A true cherry-red spot presents in whites. The color of foveola was brown, causing cherry brown spot in a Canadian aboriginal child with Sandhoff disease and black in a patient of east Indian race creating a cherry black spot in Sandhoff disease 12. Thus an alternate name of ‘perifoveal white patch’ has been suggested 12.
Therefore for cherry-red spot to be seen, the vascularity of choroid needs to be intact. In cases of ophthalmic artery occlusion, there is a compromise to the vascular supply to retina, choroid, and optic nerve head circulation. There is retinal opacification without a cherry red spot and severe vision loss, which might cause no perception of light or inaccurate projection of rays.
The opacification of inner retinal may be due to:
- Retinal ischemia due to occlusion of the central retinal artery – The retinal whitening is typically most prominent in the posterior pole, and the opaque retina obscures the details of the choroidal vessels. The whitening of the retina becomes less prominent a little peripheral to the arcades, which may be related to the reduced inner retinal thickness at the peripheral retina. There is a cessation of axoplasmic transport and opacification of the ganglion cell layer and the nerve fiber layer in central retinal arterial occlusion.
- Deposition or accumulation of material in the ganglion cell layer – In Tay-Sachs disease and other lipid storage disorders (sphingolipidoses), there is a progressive abnormal excessive intracellular accumulation of various glycolipids/phospholipids in the ganglion cell layer leading to opacification of the inner retina. The foveola is devoid of the ganglion cell layer and hence remains transparent and allows transmission of the color of the choroid and the retinal pigment epithelium. However, with the increasing age, the damaged ganglion cells are lost, and the cherry-red spot becomes less prominent. In such cases, there is consecutive optic atrophy and pallor of the optic disc due to the degeneration of the ganglion cell layer and nerve fiber layer.
Sphingolipidoses are disorders of lysosomal metabolism, which involve conjugated products of ceramide and sugar or phospholipids. Sphingolipidoses include gangliosidosis, Niemann-Pick disease, Farber disease, and metachromatic leukodystrophy.
Cherry red spot symptoms
Central retinal artery occlusion usually presents with sudden onset vision loss in the elderly. Often, there is a relative afferent pupillary defect in the affected eye. Fundus examination reveals a whitened opaque retina at the posterior pole blocking the visibility of choroidal vessels. The foveola remains transparent, giving rise to the cherry-red spot. Other features include fragmentation of blood column in retinal vessels (“box-carring”), which may be noted to move sluggishly on prolonged observation. An embolus within the central retinal artery or its branches may be present. In the late phase, retinal thinning, pigmentary changes at the fovea, and optic atrophy may occur.
In lipid storage disorders, there might be evidence of neurodegeneration and visceromegaly. In the early phase, there is a perifoveal white donut-like area around the fovea with a cherry-red spot appearance. In the late phase, there is atrophy of inner retinal layers, the disappearance of the cherry-red spot, and optic atrophy. Family history and examination of family members are of paramount importance.
Cherry red spot diagnosis
Fundus fluorescein angiography shows delayed arterial circulation, a slowly progressing front edge of the dye, and late filling of the retinal veins in central retinal artery occlusion. There may be an abrupt ending to the flow of the dye at the location of an embolus in the central retinal artery or its branches. In lipid storage disorders, fundus fluorescein angiography may show an area of hypofluorescence (block fluorescence) 360 degrees around the foveola 13.
The optical coherence tomography (OCT) in central retinal arterial occlusion shows hyperreflectivity of inner retinal layers and retinal thickening. In lipid storage disorders, there is hyperreflectivity of the nerve fiber layer and the ganglion cell layer on OCT. In commotio retinae, OCT usually does not show an increased thickness of the retina. Still, there may be hyperreflectivity of the inner segment-outer segment (IS-OS) junction with or without defects in inner segment-outer segment junction, external limiting membrane or cone outer segment tips line 14.
Autofluorescence may show hyper autofluorescence around the fovea in sialidosis 15. In central retinal arterial occlusion, there is hypo autofluorescence in the area of retinal whitening 16.
In suspected lipid storage disorder, evaluation for enzyme deficiency, and multisystem involvement is essential.
Cherry red spot treatment
The management of the cherry red spot depends on the cause.
For central retinal arterial occlusion, the options for management when the present patients early include ocular massage, anterior chamber paracentesis, reduction of intraocular pressure by systemic and topical medications, hyperbaric oxygen therapy, and intraarterial thrombolysis.
For various storage diseases, the management requires a team of various specialties. Generalized pearls for the management of such patients include avoiding/reducing intake of specific molecules, enzyme replacement therapy, and symptomatic management.
References- Tripathy K, Patel BC. Cherry Red Spot. [Updated 2019 Oct 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539841
- Hollenhorst plaque with branch retinal artery occlusion causing a cherry red spot. https://webeye.ophth.uiowa.edu/eyeforum/atlas/pages/Hollenhorst-plaque-BRAO-cherry-red-spot.htm
- Rumelt S, Dorenboim Y, Rehany U. Aggressive systematic treatment for central retinal artery occlusion. Am. J. Ophthalmol. 1999 Dec;128(6):733-8.
- Hayreh SS. Acute retinal arterial occlusive disorders. Prog Retin Eye Res. 2011 Sep;30(5):359-94.
- Tripathy K, Mazumdar S, Sarma B. Central retinal arterial occlusion in a patient with pyoderma gangrenosum. Indian J Ophthalmol. 2018 Jul;66(7):1019-1021.
- Hayreh SS, Zimmerman MB. Fundus changes in central retinal artery occlusion. Retina (Philadelphia, Pa.). 2007 Mar;27(3):276-89.
- Tripathy K, Kumawat B, Chawla R, Sharma YR, Bypareddy R. Acute Vision Loss Due to Central Retinal Arterial Occlusion, Partial Optic Nerve Avulsion, and Hemorrhage “Spurting Out” from Optic Disc after Blunt Trauma. J Ophthalmic Vis Res. 2017 Jul-Sep;12(3):351-352.
- Tripathy K, Chawla R. Extensive commotio retinae involving peripheral retina. Natl Med J India. 2017 Jul-Aug;30(4):242.
- Suvarna JC, Hajela SA. Cherry-red spot. J Postgrad Med. 2008 Jan-Mar;54(1):54-7.
- Tripathy K, Chawla R, Mittal K, Farmania R, Venkatesh P, Gulati S. Ophthalmic examination as a means to diagnose Subacute Sclerosing Panencephalitis: an optical coherence tomography and ultrawide field imaging evaluation. Eye Vis (Lond). 2017;4:1.
- Abhayambika K, Chacko A, Mahadevan K, Najeeb OM. Peripheral neuropathy and haemolytic anaemia with cherry red spot on macula in dapsone poisoning. J Assoc Physicians India. 1990 Aug;38(8):564-5.
- Ospina LH, Lyons CJ, McCormick AQ. “Cherry-red spot” or “perifoveal white patch”? Can. J. Ophthalmol. 2005 Oct;40(5):609-10.
- Heroman JW, Rychwalski P, Barr CC. Cherry red spot in sialidosis (mucolipidosis type I). Arch. Ophthalmol. 2008 Feb;126(2):270-1.
- Ahn SJ, Woo SJ, Kim KE, Jo DH, Ahn J, Park KH. Optical coherence tomography morphologic grading of macular commotio retinae and its association with anatomic and visual outcomes. Am. J. Ophthalmol. 2013 Nov;156(5):994-1001.e1
- Zou W, Wang X, Tian G. Fundus autofluorescence and optical coherence tomography of a macular cherry-red spot in a case report of sialidosis. BMC Ophthalmol. 2016 Mar 22;16:30.
- Mathew R, Papavasileiou E, Sivaprasad S. Autofluorescence and high-definition optical coherence tomography of retinal artery occlusions. Clin Ophthalmol. 2010 Oct 21;4:1159-63.





{kind=link}