Congenital hyperinsulinism
Congenital hyperinsulinism also called familial hyperinsulinism, persistent hyperinsulinemic hypoglycemia of infancy, infancy hyperinsulinemia hypoglycemia or islet cell dysregulation syndrome, is a rare genetic disorder that causes individuals to have abnormally high levels of insulin, a hormone produced by the beta cells of the pancreas that helps control blood sugar levels. Because of the high levels of insulin, people with congenital hyperinsulinism have frequent episodes of low blood sugar (hypoglycemia) that can even occur after eating. Unlike typical episodes of hypoglycemia, which occur most often after periods without food (fasting) or after exercising, episodes of hypoglycemia in people with congenital hyperinsulinism can also occur after eating. In babies and young children, these episodes are characterized by a lack of energy (lethargy), irritability, or difficulty feeding. Repeated episodes of low blood sugar increase the risk for serious complications such as breathing difficulties, seizures, intellectual disability, vision loss, brain damage, and coma 1. The severity and onset of these episodes varies, even among members of the same family. In about 60% of the cases, the episodes start within the first month of life and are very severe and difficult to manage. In other cases, the disease starts in childhood or later, and the symptoms are mild 2. Early diagnosis and treatment is important to prevent neurologic damage from hypoglycemia 3.
Pancreatic beta cells (comprising <2% of all pancreatic cells) synthesize, store, and secrete insulin. Beta cells are located within the islets of Langerhans.
There are several forms of congenital hyperinsulinism:
- Diazoxide-Responsive and Diffuse KATP congenital hyperinsulinism: In these types of congenital hyperinsulinism, potassium channels in the beta cell (named KATP channels), which regulate insulin secretion, do not work properly.
- Focal KATP congenital hyperinsulinism: In children with KATP defect with focal congenital hyperinsulinism, only an isolated, or focal, area of the pancreas is abnormal; the remainder of the pancreas is normal.
- GDH-congenital hyperinsulinism: In GDH-congenital hyperinsulinism, excess insulin secretion causes low blood glucose with fasting or when the child eats protein.
- Glucokinase congenital hyperinsulinism: In glucokinase congenital hyperinsulinism, all beta cells in the pancreas are affected. The beta cells are not able to turn off insulin secretion when the blood glucose is too low.
- HNF1a and 4a Defects: HNF1a and 4a defects are rare forms of hyperinsulinism that progress to diabetes in adolescence and adulthood.
- MCT-1 congenital hyperinsulinism: MCT-1 hyperinsulinism is rare form of congenital hyperinsulinism that is triggered by exercise.
- SCHAD-congenital hyperinsulinism: SCHAD-congenital hyperinsulinism is a type of congenital hyperinsulinism caused by a very rare disorder of fatty acid metabolism. In SCHAD-congenital hyperinsulinism all beta cells are affected; therefore the disorder is diffuse across the whole pancreas.
- UCP-2 congenital hyperinsulinism: UCP-2 hyperinsulinism is rare form of congenital congenital hyperinsulinism that seems to be transient, meaning it is not a permanent condition, and eventually resolves over time.
Congenital hyperinsulinism affects approximately 1 in 25,000 to 50,000 babies 2. Congenital hyperinsulinism is more common in certain populations, affecting up to 1 in 2,500 newborns 2.
Congenital hyperinsulinism is caused by mutations in at least 11 different genes, including ABCC8 (responsible for about 45 % of the cases), KCNJ11, GLUD1, GCK, HK1, HADH, HNF4A, HNF1A, SLC16A1, UCP2, and PGM1. Inheritance may be autosomal recessive or autosomal dominant 4. Some cases are caused by loss of genetic material in a region of chromosome 11 (11p15) that comes from the mother (maternal chromosome). According to the extent of abnormal beta cells, the disease can be focal (when abnormal beta cells are limited to 1 or a few areas in the pancreas) and diffuse (where the abnormal beta cells are spread throughout the pancreas) 4. The goal of treatment is to manage the hypoglycemia to prevent brain damage. Medications may include diazoxide, octreotide, and glucagon 5. Surgery to remove part of the pancreas might be required in severe cases 3. Genetic testing may help to guide the best treatment 4. Genetic counseling with regard to risk of recurrence may be appropriate. Techniques for prenatal diagnosis are currently limited to investigational use but may be available at some medical centers.
A nutritionist should provide dietary education and meal-planning assistance. Patients (if old enough) and family members should be taught how to use a home blood glucose monitor. They should also understand the signs and symptoms of hypoglycemia and how to treat this condition with rapid-acting oral carbohydrates and subcutaneous glucagon.
Family members must understand the importance of prompt treatment of hypoglycemia to prevent severe complications or death. Family members should be instructed to call the local emergency medical service if they are unable to treat a hypoglycemic episode or if the patient does not respond to treatment promptly. Family members should know the local emergency phone number. Patients should wear a medical identification bracelet.
Patients and family members should be reminded to carry medications, a glucose meter, a rapid-acting carbohydrate source, and glucagon when traveling. Families should carry sufficient supplies for several extra days in case of unexpected travel delays.
Patients who have undergone surgery, as well as their family members, should be reminded of the risk of future development of diabetes mellitus and the importance of long-term follow-up. Failure to educate families about this potential late complication could result in a delay of diagnosis of diabetes mellitus if it occurs.
Figure 1. Pancreas anatomy

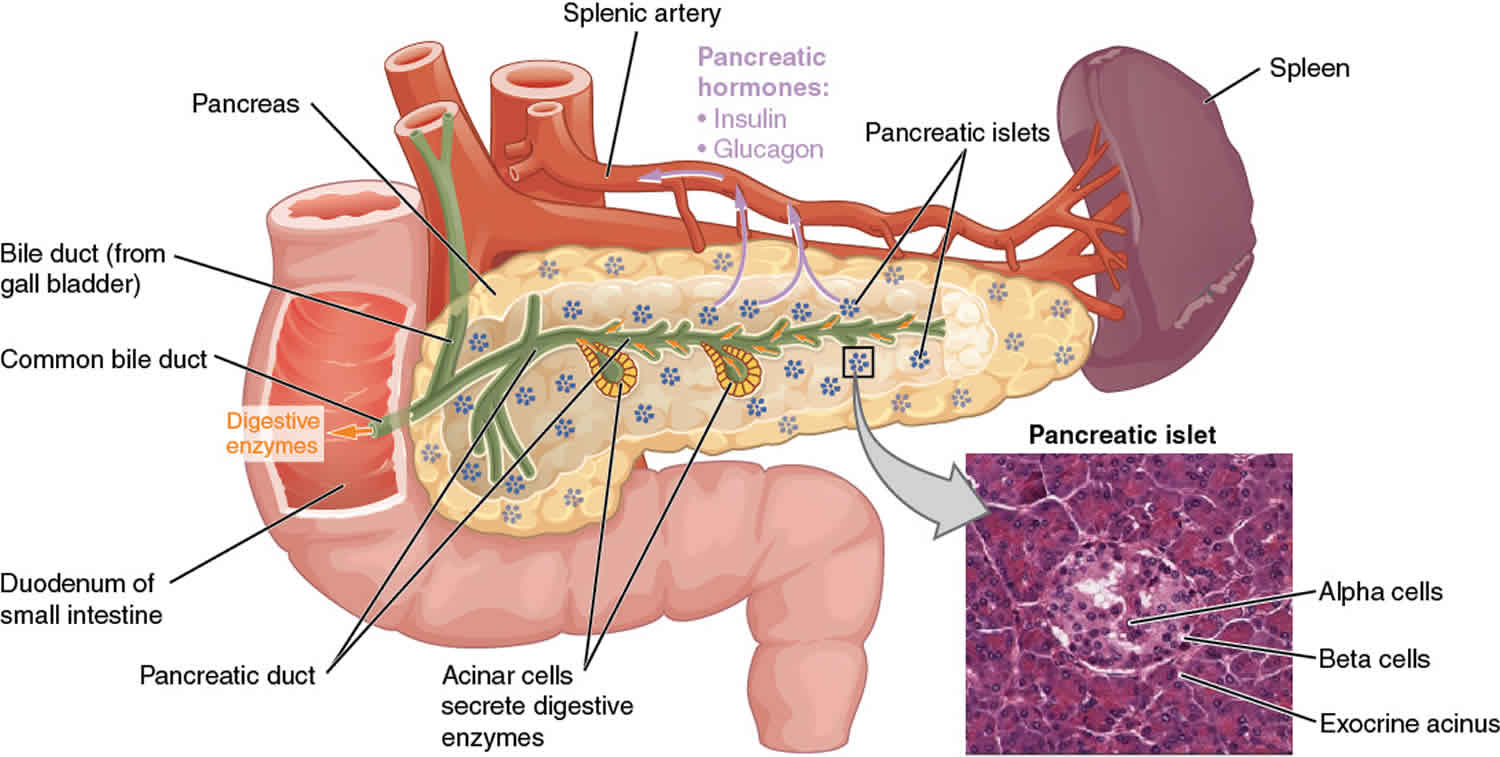
Congenital hyperinsulinism causes
Congenital hyperinsulinism is caused by mutations in genes that regulate the release (secretion) of insulin, which is produced by beta cells in the pancreas. Insulin clears excess sugar (in the form of glucose) from the bloodstream by passing glucose into cells to be used as energy.
Gene mutations that cause congenital hyperinsulinism lead to over-secretion of insulin from beta cells. Normally, insulin is secreted in response to the amount of glucose in the bloodstream: when glucose levels rise, so does insulin secretion. However, in people with congenital hyperinsulinism, insulin is secreted from beta cells regardless of the amount of glucose present in the blood. This excessive secretion of insulin results in glucose being rapidly removed from the bloodstream and passed into tissues such as muscle, liver, and fat. A lack of glucose in the blood results in frequent states of hypoglycemia in people with congenital hyperinsulinism. Insufficient blood glucose also deprives the brain of its primary source of fuel.
Mutations in at least nine genes have been found to cause congenital hyperinsulinism (see Table 1 below). Mutations in the ABCC8 gene are the most common known cause of the disorder. They account for this condition in approximately 40 percent of affected individuals. Less frequently, mutations in the KCNJ11 gene have been found in people with congenital hyperinsulinism. Mutations in each of the other genes associated with this condition account for only a small percentage of cases.
In approximately half of people with congenital hyperinsulinism, the cause is unknown.
Table 1. Congenital hyperinsulinism (familial hyperinsulinism) genes and distinguishing clinical features
| Gene | % of Congenital Hyperinsulinism Attributed to Pathogenic Variants in This Gene | Mode of inheritance | Distinguishing Clinical Features |
|---|---|---|---|
| ABCC8 3 | 40%-45% 6 | Autosomal recessive |
|
| Autosomal dominant |
| ||
| CACNA1D | 1 individual 7 |
| |
| GCK | <1% 8 | Autosomal dominant |
|
| GLUD1 | 5% 9 | Autosomal dominant | Hyperammonemia/hyperinsulinism syndrome
|
| HADH | <1% 10 | Autosomal recessive |
|
| HK1 | 1 family 11 | Autosomal dominant |
|
| HNF1A | Unknown | Autosomal dominant |
|
| HNF4A | 5% 12 | Autosomal dominant |
|
| PMM2 | 11 families 13 |
| |
| KCNJ11 3 | 5% 6 | Autosomal recessive |
|
| Autosomal dominant |
| ||
| SLC16A1 | Unknown | Autosomal dominant |
|
| UCP2 | 2% of diazoxide-responsive congenital hyperinsulinism 14 | Autosomal dominant |
|
| Unknown | 40% | NA |
Footnotes:
3. ABCC8- and KCNJ11-related congenital hyperinsulinism are also known as congenital hyperinsulinism-KATP.
5. A few exon or multiexon deletions have been reported in ABCC8.
6. In the Ashkenazi Jewish population, two ABCC8 founder variants, p.Phe1387del and c.3989-9G>A, are responsible for approximately 97% of congenital hyperinsulinism 16. ABCC8 founder variants present in the Finnish population include p.Val187Asp and p.Glu1506Lys 17.
Transient hyperinsulinism
Babies born small for gestational age, or prematurely, may develop hypoglycemia due to excessive insulin secretion. In addition, infants who experience fetal distress due to lack of oxygen to the brain may develop hypoglycemia. The cause of this inappropriate insulin secretion is unclear, but it can last a few days to months. Once recognized, this form of hypoglycemia is usually easy to treat. Many affected infants will not have hypoglycemia once they are fed every 3-4 hours. In the more severely affected children, intravenous glucose is needed to prevent hypoglycemia. Occasionally, drug therapy is required; in which case, diazoxide is usually a very effective treatment. Children with this form of hyperinsulinism have a fasting study done when medications have been weaned, to prove that the hyperinsulinism has resolved and therefore was transient. A small number of babies born to mothers with diabetes mellitus may have transient hypoglycemia. This tends to occur if the mother’s diabetes was not under good control. The mother’s high blood glucose levels are transmitted across the placenta to the fetus. The fetus compensates by secreting extra insulin. This step-up in insulin secretion does not cause hypoglycemia while the fetus is inside the mother, but after birth, the constant supply of high glucose from the placenta is gone and the blood sugar in the newborn falls precipitously. This form of hyperinsulinism should resolve within a few days with frequent feeding or in some cases intensive intravenous drip of glucose. Once the hypoglycemia resolves, it should never recur.
Persistent congenital hyperinsulinism
A number of different genetic defects causing congenital hyperinsulinism have been identified. In the past, before the different genetic forms of congenital hyperinsulinism were recognized, congenital hyperinsulinism was referred to by many names, including nesidioblastosis, islet cell dysregulation syndrome, idiopathic hypoglycemia of infancy, and persistent hyperinsulinemic hypoglycemia of infancy (PHcongenital hyperinsulinism). With the identification of the genes responsible for these disorders, the naming of the different forms of congenital hyperinsulinism has become more exact.
KATP-hyperinsulinism diffuse or focal disease
The KATP form of congenital hyperinsulinism was formerly known as “nesidioblastosis” or “persistent hyperinsulinemic hypoglycemia of infancy (PHHI)”. Neonates with this form of hyperinsulinism are frequently, although not always, larger than normal birth weight (many weigh above 9lbs) and present in the first days of life. It is called KATP-congenital hyperinsulinism because its genetic cause is due to defects in either of two genes that make up the potassium channel (called KATP channel) in the insulin secreting beta-cells of the pancreas. These two genes are the SUR1 gene (known as ABCC8) and the Kir6.2 gene (known as KCNJ11). Normally, when the beta cell senses that glucose levels are elevated, closure of the KATP channel triggers insulin secretion. When the KATP channel is defective, inappropriate insulin secretion occurs and causes hypoglycemia. Two forms of KATP-congenital hyperinsulinism exist: diffuse KATP-congenital hyperinsulinism and focal KATP-congenital hyperinsulinism. When these mutations are inherited in an autosomal recessive manner (one mutation in the gene inherited from each parent, neither of whom is affected) they cause diffuse disease, meaning every beta-cell in the pancreas is abnormal. Recently autosomal dominant mutations (a mutation in a single copy of the gene) have been found to cause diffuse disease. When a recessive mutation is inherited from the father and loss of heterozygosity for the maternal copy of the gene (loss of the mother’s unaffected gene from a few cells in the pancreas) occurs, a focal lesion arises. Abnormal beta cells are limited to this focal lesion and are surrounded by normal beta-cells.
Children with either form of KATP-congenital hyperinsulinism are identical in their appearance and behavior. They tend to have significant hypoglycemia within the first few days of life and require large amounts of glucose to keep their blood glucose normal. They may have seizures due to hypoglycemia. Diazoxide is often an ineffective treatment for these children because diazoxide works on the KATP channel and it cannot fix the broken channels. Octreotide given by injection every 6 to 8 hours or by continuous infusion may be successful (sometimes only in the short term). Glucagon may be given by intravenous infusion to stabilize the blood sugar as a temporary measure in the hospital setting. Some centers prefer the surgical approach. With the recent discovery of diffuse and focal KATP-congenital hyperinsulinism, attempts to differentiate these two forms are very important: surgical therapy will cure focal congenital hyperinsulinism but not diffuse congenital hyperinsulinism (see below).
GDH-congenital hyperinsulinism
GDH-congenital hyperinsulinism has also been known as the hyperinsulinism/hyperammonemia syndrome, leucine-sensitive hypoglycemia, and protein-sensitive hypoglycemia. GDH-congenital hyperinsulinism is caused by a mutation in the enzyme glutamate dehydrogenase (GDH). It is inherited in either an autosomal dominant manner or may arise as a sporadically new mutation in a child with no family history. Glutamate dehydrogenase (GDH) plays an important role in regulating insulin secretion stimulated by amino acids (especially leucine). Individuals with GDH-congenital hyperinsulinism develop hypoglycemia after eating a high protein meal or after fasting. Glutamate dehydrogenase (GDH)-congenital hyperinsulinism affected individuals can have significant hypoglycemia if they eat protein (for instance eggs or meat) without eating carbohydrate containing foods such as bread, juice or pasta. Glutamate dehydrogenase (GDH)-congenital hyperinsulinism is also associated with elevated blood concentrations of ammonia, which is derived from protein. Patients with GDH-congenital hyperinsulinism often present later than KATP channel congenital hyperinsulinism, typically, not until three to four months of age when they wean from low protein containing breast milk to infant formula. Others do not have recognizable hypoglycemia until they sleep overnight without a middle of the night feed or after they start higher protein-containing solid foods. In addition, GDH-congenital hyperinsulinism can be successfully treated with diazoxide and the avoidance of protein loads without carbohydrates. Most children with GDH-congenital hyperinsulinism will do very well once recognized, but if the diagnosis is delayed, they may also suffer brain damage from untreated hypoglycemia.
GK-congenital hyperinsulinism
This defect is inherited in an autosomal dominant fashion but can also arise sporadically. Glucokinase is the “glucose sensor” for the beta-cell. It tells the beta-cell how high the blood glucose is and when to secrete insulin. Glucokinase mutations that cause congenital hyperinsulinism instruct the beta-cell to secrete insulin at a lower blood glucose than is normal. Like GDH-congenital hyperinsulinism, GK-congenital hyperinsulinism can be treated with diazoxide, but sometimes, it may be severe and unresponsive to diazoxide.
Other forms of congenital hyperinsulinism
Other forms of congenital hyperinsulinism, responsive to diazoxide include: 1) congenital hyperinsulinism due to mutations in SCHAD, an enzyme that regulates GDH. Children with SCHAD congenital hyperinsulinism, are also protein-sensitive. 2) HNF4A and HNF1A congenital hyperinsulinism are caused by mutations in HNF4A and HNF1A, transcription factors that play an important role in the beta-cells. These mutations cause hyperinsulinism in infancy and familial diabetes (also known as MODY, or maturity onset diabetes of the young) later in life. 3) Exercise-induced hyperinsulinism is a rare form of congenital hyperinsulinism in which hypoglycemia is triggered by exercise.
Other forms of congenital hyperinsulinism are known to exist, but the genetic mutations are not yet well described. Their clinical features and response to therapy vary. congenital hyperinsulinism can also be associated with syndromes such as Beckwith Wiedemann syndrome, Kabuki syndrome, and Turner syndrome among others. In these cases, congenital hyperinsulinism is only one of the features that characterize the clinical picture.
Congenital hyperinsulinism inheritance pattern
Congenital hyperinsulinism can have different inheritance patterns, usually depending on the form of the condition. At least two forms of the condition have been identified. The most common form is the diffuse form, which occurs when all of the beta cells in the pancreas secrete too much insulin. The focal form of congenital hyperinsulinism occurs when only some of the beta cells over-secrete insulin.
Most often, the diffuse form of congenital hyperinsulinism is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Less frequently, the diffuse form is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
The inheritance of the focal form of congenital hyperinsulinism is more complex. For most genes, both copies are turned on (active) in all cells, but for a small subset of genes, one of the two copies is turned off (inactive). Most people with the focal form of this condition inherit one copy of the mutated, inactive gene from their unaffected father. During embryonic development, a mutation occurs in the other, active copy of the gene. This second mutation is found within only some cells in the pancreas. As a result, some pancreatic beta cells have abnormal insulin secretion, while other beta cells function normally.
Congenital hyperinsulinism symptoms
Congenital hyperinsulinism causes low plasma sugar (hypoglycemia). The symptoms of hypoglycemia in infants are often difficult to identify, as they can be similar to normal infant activities.
Common symptoms of hypoglycemia include:
- irritability
- sleepiness
- lethargy
- excessive hunger
- rapid heart rate
Most patients with congenital hyperinsulinism (persistent hyperinsulinemic hypoglycemia of infancy) present shortly after birth with symptoms of hypoglycemia (eg, hunger, jitteriness, lethargy, apnea, seizures). Older children, in addition to these symptoms, may also show sweating (diaphoresis), confusion, or unusual mood or behavior changes.
More severe symptoms, such as seizures and coma, can occur with a prolonged low plasma sugar or an extremely low plasma sugar. Common symptoms of hypoglycemia in older children include feelings of shakiness, weakness, tiredness, confusion, and rapid heart rate.
Hypoglycemia is persistent, requiring frequent or continuous glucose infusions or feedings to maintain adequate blood glucose levels.
Presenting symptoms of congenital hyperinsulinism reported in adults include confusion, headaches, dizziness, syncope, and loss of consciousness. The symptoms may be exacerbated by fasting and may improve after eating.
Doctors consider a normal plasma sugar to be >70 mg/dL. Anything less than 60 mg/dL is low, although severe symptoms due to hypoglycemia are not likely unless the plasma sugar is less than 50 mg/dL. Prolonged or severe low plasma sugar can cause seizures and permanent brain damage.
Congenital hyperinsulinism diagnosis
The diagnosis of congenital hyperinsulinism is based on history, laboratory findings, and genetic testing. Prompt diagnosis and establishment of effective treatment are essential to avoid neurologic damage.
Medical history
The history of the child is an important piece of the puzzle. This includes information such as when the low plasma sugars started, the timing of the low plasma sugars, whether the baby was born large for gestational age (LGA), any family history of low plasma sugar or unexplained infant deaths, seizures or SIDS (sudden infant death syndrome).
Laboratory findings
Blood tests drawn when the plasma sugar is less than 50 mg/dL are essential to the diagnosis of congenital hyperinsulinism. In congenital congenital hyperinsulinism, with a plasma sugar < 50, you will see suppressed ketones and free fatty acids, an elevated insulin level (although this may not always be captured), and a glycemic response to glucagon, with the plasma sugar rising more than 30 mg/dL when glucagon is administered.
It is important that these samples at the time of a critically low plasma sugar be obtained in an experienced, controlled environment, as you do not want a child to be kept with a critically low plasma sugar longer than is absolutely necessary to make the diagnosis.
The finding of nonketotic hypoglycemia in association with elevated insulin levels (>10 µU/mL) and normal levels of free fatty acid (FFA) supports the diagnosis of hyperinsulinism. The insulin-to-glucose ratio may range from 0.4-2.7 (normal, < 0.3). Sustained glucose use rates in excess of 10 mg/kg/min (evidenced by the need to administer intravenous [IV] glucose at a rate higher than 10 mg/kg/min to maintain normoglycemia) are consistent with exaggerated insulin activity and suggestive of congenital hyperinsulinism.
Cortisol and growth hormone levels are usually elevated in specimens taken during an episode of hypoglycemia (as an appropriate and normal response to hypoglycemia) and are usually within the reference range during periods of normoglycemia.
Serum metabolic screens, pH, lactate, and ammonia studies may be obtained to exclude other metabolic diseases. The results are expected to be within the reference range in cases of congenital hyperinsulinism. Urinary ketone, amino acid, and reducing-substance studies may be obtained to exclude other metabolic diseases. The results of these are also expected to be within the reference range in cases of congenital hyperinsulinism.
Hyperinsulinism with hyperammonemia and elevated levels of free fatty acid suggests a fatty acid oxidation disorder. Hyperinsulinism with hyperammonemia and normal levels of free fatty acid suggest the diagnosis of hyperinsulinism with hyperammonemia, a clinically and genetically distinct variant of congenital hyperinsulinism. Patients with this disorder usually respond very well to medical therapy alone and are much less likely to require surgical intervention. The hyperammonemia is mild and not symptomatic.
Imaging studies
Ultrasonography, computed tomography (CT), and magnetic resonance imaging (MRI) have been used to search for a focal mass in the pancreas; however, in many cases, the lesion is too small to be visible by such techniques. These forms of imaging cannot identify the diffuse form of congenital hyperinsulinism.
A newer imaging technique has been developed that uses positron emission tomography (PET) in conjunction with coregistered abdominal CT (see the image below) to distinguish between focal and diffuse disease and, in the case of focal disease, localize the lesions 18. This technique uses a novel isotope, fluorine-18 L-3,4-dihydroxyphenylalanine (18 F-L-DOPA), for which neuroendocrine cells have a high affinity.
A large series at Children’s Hospital of Philadelphia found that the technique had a specificity of 96%, sensitivity of 85%, and positive predictive value of 96% for diagnosing a focal lesion. When a focal lesion was identified by PET scan, the concordance between imaging and anatomic findings was 100% 19.
The 18 F-L-DOPA isotope remains investigational and is technically complex to prepare. It is currently available at only a few centers worldwide. However, because of the remarkable results seen in preliminary published trials, physicians treating patients with congenital hyperinsulinism should strongly consider consultation with an expert at one of these centers.
Portal and pancreatic venous sampling
Catheterization of the portal and pancreatic veins with venous sampling may help distinguish between focal and diffuse congenital hyperinsulinism. This procedure is well described in the pediatric population.
For venous sampling, a catheter is placed in the pancreatic venous system via a femoral vein or inserted by direct hepatic puncture to enter the portal vein. With the use of fluoroscopic guidance and IV contrast agents, the catheter is advanced into various pancreatic veins, and blood samples are taken to measure glucose, insulin, and C-peptide levels.
If a focal lesion is present, elevated insulin levels are expected in veins draining the area near the lesion, and insulin levels are expected to be within the reference range for congenital hyperinsulinism in other areas. If a diffuse lesion is present, insulin levels are expected to be high throughout the pancreatic venous bed.
In some cases, the results of this study are difficult to interpret, and correlation of results with pathologic findings remains imperfect. Pathologic examination remains the criterion standard for identification of focal disease. However, pancreatic venous sampling is one of few preoperative techniques available to identify focal lesions in patients in whom findings on conventional imaging are inconclusive.
Pancreatic venous sampling and intraoperative histologic studies should be strongly considered, because the identification of a focal lesion has profound implications for treatment and prognosis.
Intra-arterial calcium stimulation
A test using intra-arterial calcium stimulation has been employed in adults and, to a lesser extent, in children. In this test, a bolus of calcium gluconate is rapidly administered via a catheter in the celiac axis and the splenic, superior mesenteric, and gastroduodenal arteries. Blood samples are obtained through a catheter in the right hepatic vein before injection and at several intervals after injection. These blood samples are then tested for glucose, calcium, and insulin levels.
An excessive insulin response from calcium stimulation in a single artery suggests a focal lesion, and excessive poststimulation insulin secretion associated with all arteries suggests a diffuse form of hyperinsulinism.
Although pancreatic venous sampling has been studied more widely to date, especially in neonates, experience with intra-arterial calcium stimulation in children is increasing. Children’s Hospital of Philadelphia has reported on a large number of cases.
Histology
Two major pancreatic histologic types (“diffuse” and “focal”) have been described in individuals with congenital hyperinsulinism 1. A third histologic form (“atypical” or “mosaic”) has also been described, although the genetic etiology of this form of congenital hyperinsulinism has not yet been discovered 20:
- Diffuse involvement of beta cells throughout the pancreas. Seen in approximately 60%-70% of individuals, diffuse disease is characterized by essentially normal neonatal pancreatic architecture. All beta cells are affected and many have large nuclei, abundant cytoplasm, and histologic evidence of increased metabolic activity.
- Focal pancreatic adenomatous hyperplasia. Seen in approximately 30%-40% of individuals, focal changes involve a limited region of the pancreas, with the remainder of the tissue being both histologically and functionally normal. A focal lesion is the confluence of apparently normal islets. Focal lesions typically are not macroscopically visible; they differ from true adenomas, which can be identified on gross inspection of the pancreas. Beta cells outside the focal lesion have small nuclei and sparse cytoplasm-histologic evidence that they are suppressed and not actively producing and secreting insulin.
- Mosaic involvement of the pancreatic islets. Pancreatic histology reveals the coexistence of two types of islets: large islets with cytoplasm-rich beta cells and occasional enlarged nuclei alongside shrunken islets with beta cells exhibiting little cytoplasm and small nuclei. Large islets are mostly confined to a few lobules, suggesting that removal of these particular lobules by partial pancreatectomy could result in a cure [Sempoux et al 2011]. The genetic etiology of this form of congenital hyperinsulinism is not known.
Note: With the current availability of imaging and molecular genetic testing, diagnosis and management decisions do not require histology. Biopsy is not part of the initial evaluation, and the pancreatic histology is only known if surgical management is warranted.
Genetic testing
The DNA from a blood sample from the child with congenital congenital hyperinsulinism and each parent can be analyzed to screen for the mutations that cause the most common types of congenital hyperinsulinism. This can be done at commercial laboratories and should be considered in any child suspected to have congenital congenital hyperinsulinism.
Congenital hyperinsulinism treatment
Prompt treatment of hypoglycemia due to congenital hyperinsulinism is essential to prevent brain damage. Unlike other hypoglycemia-causing conditions in which alternative fuels, such as ketones or lactate, may be available for the brain during periods of hypoglycemia, congenital hyperinsulinism prevents the production of these fuels and leaves the brain without a source of energy.
Because congenital congenital hyperinsulinism causes dangerously low blood sugars as a result of excess insulin, the treatment for this condition is to try and maintain blood sugars greater than 70 mg/dL. There are two options for treatment of congenital congenital hyperinsulinism, medical therapy and surgical intervention. About 50 percent of children respond to medical therapy, while the other half require surgery for a partial or near total pancreatectomy.
Hypoglycemia can be treated by giving a carbohydrate-containing drink by mouth or if severe, by giving glucose through the vein or by injecting glucagon. A child with a feeding tube can have glucose given through the tube. The goal of treatment is to prevent hypoglycemia while the child has a normal feeding pattern for age with a little extra safety built in, e.g., a one year old who normally would not eat overnight for 10-12 hours should be able to fast for at least 14 -15 hours on a successful medical regimen. Medications used to treat congenital hyperinsulinism include diazoxide, octreotide, and glucagon.
Dietary intervention
- Frequent high-carbohydrate feedings, including formula supplemented with glucose polymer
- Nighttime continuous gastric drip containing glucose or glucose polymer
- Feeding gastrostomy to simplify the process of continuous nighttime feeding and to provide access for emergency home treatment of hypoglycemia
- Prolonged fasting of any sort should be avoided.
A diet of 3 meals and 3 snacks daily helps maintain adequate serum glucose levels. Patients should avoid fasting (eg, skipping meals and scheduled snacks), because hypoglycemia may develop quickly.
A high-protein, high-carbohydrate diet is preferred. The carbohydrates provide the most long-acting source of glucose to counter the continuous release of insulin, and concurrent protein helps prolong this effect.
Patients should always have access to a rapid-acting carbohydrate, such as glucose tablets, glucose gel, fruit juice, hard candy, or sugar cubes. Uncooked cornstarch may be used to provide additional carbohydrates, and it may be helpful in preventing fasting hypoglycemia during sleep if administered at bedtime 21.
Medical therapy
Diazoxide
Diazoxide, which binds to the ABCC8 subunit of the KATP channel, increases the channel’s probability of being open, resulting in membrane hyperpolarization and inhibition of insulin release. Some evidence suggests that diazoxide binding to mutated channels may correct abnormal protein folding and thus facilitate the transit of more channels to the membrane. The effective therapeutic dose varies from 5 to 15 mg/kg/day, but may be as high as 20 mg/kg/day in divided doses. Usually, if 15 mg/kg/day does not work, higher doses will not work. Diazoxide is given by mouth 2-3 times per day. A thiazide diuretic should be given along with diazoxide with doses >8-10 mg/kg/day to prevent fluid retention, which may be severe 22.
Diazoxide is generally effective for infants with stress-induced hyperinsulinism, infants with GDH-congenital hyperinsulinism or GK-congenital hyperinsulinism, and in a subgroup of infants whose basic defect is not known. Diazoxide often does not work in children with KATP-congenital hyperinsulinism. Side effects of diazoxide include fluid retention, a particular problem for the newborn who is receiving large amounts of intravenous glucose to maintain the blood glucose in the normal range. A diuretic medication is sometimes used with diazoxide in anticipation of such a problem. Diazoxide also causes excessive hair growth of the eyebrows, forehead, and back (referred to medically as hypertrichosis). This hair growth resolves several months after diazoxide therapy is stopped. Some patients choose to shave the hair occasionally and this does not intensify hair growth.
Octreotide
Somatostatin analogs (e.g., octreotide or lanreotide) suppress insulin secretion by binding to specific beta-cell receptors and initiating a number of intracellular signaling pathways.
Octreotide is a drug that also inhibits insulin secretion. It is administered by injection. It can be given periodically throughout the day by subcutaneous injection or may be administered continuously under the skin by a pump that is commonly used for insulin therapy in individuals with diabetes. The clinical efficacy of octreotide may be limited by the relatively short duration of inhibition of insulin secretion after subcutaneous bolus injection (~3 hours) and by the fact that these drugs also inhibit glucagon and growth hormone secretion, thus impairing hepatic glucose production. Octreotide is often very effective initially, but it may become less effective over time. In addition, more is not always better as the higher the dose (higher than 20 micrograms/kg/day), the less effective it may become. Careful attention to dosage (typically 10-15 μg/kg/day for octreotide) as well as the use of continuous subcutaneous injection using a portable pump greatly enhances clinical efficacy 22. Simultaneous treatment with glucagon may enhance efficacy. Side effects include alteration of gut motility, which may cause poor feeding. It may also cause gallstones and very rarely may produce hypothyroidism, and short stature. As with any injection, risks of pain, infection, and bruising exist. Additionally, octreotide is not currently recommended in neonates already at risk for necrotizing enterocolitis. There other drugs similar to octreotide that have a longer duration of action and can be used once a month, these include long-acting octreotide LAR 23 and lanreotide 24. These longer acting preparations are reserved for use in those patients that have responded to the short acting octreotide and are on a stable regimen and can be used after the hypoglycemia becomes manageable and stableusually after age three to four years.
Glucagon
Glucagon stimulates release of glucose from the liver and increases hepatic gluconeogenesis, helps prevent hypoglycemia. It is given through a vein or by injection under the skin or into the muscle. Glucagon can be used in cases of emergency when a child with congenital hyperinsulinism has low blood glucose levels and cannot be fed. It can also be given in the hospital as a continuous infusion through a vein. It is most effective as a holding therapy while the child is prepared for surgery.
Surgery
Once an individual is stabilized, a decision must be made as to the need for surgical intervention and the extent of such intervention. In some severe cases, even the most aggressive medical management fails to maintain plasma glucose concentration consistently within the safe limits (>60 mg/dL). In such individuals, surgery must be considered. Prior to surgical intervention, differentiation between focal and diffuse disease using one of the following techniques is important, as the surgical approach and the clinical outcome are quite different:
- Genetic studies, in certain circumstances, can be useful in differentiating focal from diffuse disease:
- Finding two recessive pathogenic variants or a single dominant pathogenic variant is diagnostic of diffuse disease.
- Finding a single recessive pathogenic variant on the maternal allele suggests diffuse disease; it is assumed that the other pathogenic variant on the paternal allele was missed because of technical limitations of the molecular genetic testing.
- Finding a single recessive pathogenic variant on the paternal allele is consistent with and highly suggestive of focal disease, although it cannot be considered diagnostic, as molecular genetic testing methods could have failed to detect a pathogenic variant on the maternal allele. In such individuals, further testing to diagnose and localize focal disease is indicated. Because of the large size of ABCC8 and KNCJ11, complete sequencing and analysis of all variants discovered is expensive and time-consuming and may not be completed in time to aid in clinical decision making for a severely ill individual. With the incorporation of modern sequencing techniques, rapid complete sequencing of all relevant genes is becoming feasible. In contrast, for a person from an ethnic group with a known founder variant (e.g., Ashkenazi Jews), targeted analysis for pathogenic variants can provide rapid and inexpensive clinically useful information.
- Fluoro-DOPA positron emission tomography (F-DOPA-PET) has been used successfully for the preoperative localization of focal lesions 25. While the scan itself is relatively easy to perform, the radiopharmaceutical is not readily available in many centers and the scan can be difficult to interpret, requiring extensive experience to obtain reliable results.
- Intraoperative histologic evaluation of a pancreatic biopsy in very experienced hands can be used to differentiate between diffuse congenital hyperinsulinism and a normal, suppressed pancreas in an individual with a focal lesion. Since intraoperative identification of the focal lesion can be very difficult or impossible, resection of the lesion is usually only possible if its location is determined preoperatively.
Individuals with diffuse disease require extensive (80%-95%) pancreatic resection (pancreatectomy) and are at risk for persistent hypoglycemia postoperatively and/or insulin-requiring diabetes mellitus later in childhood. Although the apparent risk for postoperative diabetes appears to be very low after limited pancreatectomy, very long-term follow up is not yet available on these individuals 26. Since focal lesions can only rarely be identified grossly at the time of surgery, perioperative diagnosis and localization of focal lesions is needed.
In children with focal KATP channel congenital hyperinsulinism can be cured by localized resection of the hyperplastic region (surgery to remove only the small part of the pancreas) that is affected is the procedure of choice. This requires a multidisciplinary team of endocrinologists, radiologists, pathologists and surgeons, specialized in the treatment of these children. Therefore it is generally only available in the major centers treating patients with congenital hyperinsulinism. The majority of patients with focal congenital hyperinsulinism will be cured or will not require any medical therapy after the surgery. This is in stark contrast to those with diffuse disease in whom medical therapy after surgery is the rule.
These surgeries are not curative and KATP-congenital hyperinsulinism children who have undergone such surgeries may continue to require frequent feeds and medications to prevent hypoglycemia. They also may need repeat surgeries. The hope with such surgery is to lessen the intense medical regimen that otherwise would be needed to protect the child from recurrent, severe hypoglycemia.
Islet cell autotransplantation
Some authors advocate cryopreservation of islet cells from the resected portion of the pancreas for possible future autotransplantation if the patient develops diabetes mellitus. In theory, this process would cure diabetes without the need for immunosuppression or risk of rejection, as is observed in pancreatic or islet cell allotransplants.
Since 1977, several centers have reported success using this approach in adults and children undergoing total pancreatectomy for severe pancreatitis or pancreatic tumors 27. Success in eliminating insulin requirements varies from 29 -100%; in small series, this approach has been reported to prevent the development of diabetes for at least 13 years 28.
No cases of islet cell autotransplantation in patients with congenital hyperinsulinism have been published to date. However, some patients (or their families, in the case of infants) have elected to have islet cell cryopreservation performed, in anticipation of future developments in this area.
Ethical, technical, and safety considerations related to this therapy have not been fully developed, but the concept appears promising, especially given the rapid progress being made in islet cell allotransplantation. Patients or their families should consider islet cell preservation for possible future autotransplantation, as the knowledge base continues to develop.
Long-term monitoring
In persons with clinically mild disease, episodes of subtle, undiagnosed hypoglycemia can cause permanent brain damage. Therefore, close monitoring and vigilance is just as critical in mild cases as it is in severe cases. Furthermore, in persons with mild disease and in those with severe disease in clinical remission, severe hypoglycemia may be precipitated by intercurrent viral illness. Thus, it is imperative that parents monitor glucose concentrations closely especially during intercurrent illness, even in the absence of symptomatic hypoglycemia. Identification of the genetic cause of congenital hyperinsulinism can help guide the frequency of blood glucose testing. Individuals who are diazoxide responsive and take their medication regularly will need less frequent glucose monitoring than non-diazoxide-responsive individuals. In the first few years of life, use of continuous glucose monitoring is recommended for children with very unstable glucose levels regardless of the genetic cause.
Patients should have regular follow-up visits with a pediatric endocrinologist to review blood glucose levels, diet, growth, and medication side effects. Patients should record blood glucose levels and bring these records to follow-up visits or use a home glucose meter with a memory that can be downloaded.
Home therapy must be individualized and usually involves 1 or more of the following:
- Oral diazoxide, possibly in conjunction with oral chlorothiazide
- Oral nifedipine: Nifedipine, which acts as an inhibitor of the voltage-dependent calcium channels present in the beta cell, inhibits insulin secretion by decreasing calcium influx 22. In vitro, this drug effectively suppresses insulin secretion depending on the pathogenic variant; however, in vivo side effects are usually dose limiting, and the drug is only rarely clinically effective.
- Subcutaneous octreotide
- Subcutaneous glucagon (for emergency use).
Activity
Regular activity should be encouraged, with appropriate precautions.
Patients or parents should always carry a supply of rapid-acting carbohydrate (eg, glucose tablets or gel, sugar cubes, fruit juice, hard candy) to use in case of hypoglycemia.
Patients should increase their carbohydrate intake when increased exertion is anticipated, such as before strenuous exercise.
Pregnancy management
Affected individuals who previously underwent near-total or sub-total pancreatectomy typically have insulin-requiring diabetes by the time they become pregnant. In this case, treatment is the same as for individuals with preexisting diabetes from any cause. There is little, if any, experience with pregnancy in individuals who were treated conservatively or who underwent limited pancreatectomy for focal congenital hyperinsulinism, as these treatments are relatively new. In this situation, close monitoring of glucose to detect both recurrent hypoglycemia and hyperglycemia is warranted. If hyperglycemia is documented, treatment should be instituted as for any woman with gestational diabetes. A fetus at risk for congenital hyperinsulinism should be monitored for size and weight. Excessive fetal weight gain during the last trimester of pregnancy increases the risk of obstetric complications and of cesarean delivery. In pregnant women with a history of congenital hyperinsulinism and gestational hyperglycemia due to prior surgical treatment, the fetus should be monitored as for any case of preexisting type 1, preexisting type 2, or gestational diabetes.
Congenital hyperinsulinism prognosis
Scientists know that with rapid diagnosis and appropriate treatment, keeping blood sugars >70 mg/dL, it is less likely that children with congenital congenital hyperinsulinism will have long-term effects of hypoglycemia.
If medical treatment can be safely maintained, glycemic control usually becomes easier with time, and most individuals treated medically enter clinical remission after several months or years of treatment 29. It is generally accepted that those individuals who respond well to medical treatment can be treated chronically without undue risk for long-term complications. Long-term follow up of medically treated individuals shows that some eventually develop glucose intolerance, which can be effectively managed with mild dietary restrictions 29.
With focal hyperinsulinism, 96.9 percent of children are cured with surgery. If a solitary focal lesion can be identified and excised, the patient usually maintains blood glucose levels within the reference range without the need for medication or continuous feedings 30.
Hypoglycemia often persists even after a 95-98% pancreatectomy. Hypoglycemia may be easier to control after partial pancreatectomy and may resolve months or years later or persist throughout life.
In a study of 101 patients, 50% of patients who underwent a 95% or greater pancreatectomy were cured (ie, they did not require medical or dietary treatment to maintain normoglycemia within the follow-up period of the study). The mean time from surgery to cure was 4.7 years 31. However, in some series, 40-63% of patients managed with medical therapy alone had late remission of hypoglycemia. Later onset of disease is correlated with a higher likelihood of being able to discontinue medical therapy.
Future development of diabetes mellitus
Patients who undergo partial pancreatectomy are at high risk for developing diabetes mellitus later in life 30. The risk of diabetes mellitus appears to increase with the extent of pancreatic resection; however, the risk is significant even with conservative surgical procedures.
In one series, 14% of children with diffuse lesions developed diabetes mellitus, regardless of the surgical procedure performed. The mean time from surgery to development of diabetes mellitus was 9.6 years 31. Because most series are limited by relatively short follow-up times, the lifetime incidence of diabetes mellitus is not well understood. Islet cell preservation and autotransplantation remain promising but untested therapies for patients who develop diabetes mellitus.
Diabetes mellitus is extremely rare after resection of focal lesions.
In a series of 3 patients treated without pancreatic resection, 2 developed impaired glucose tolerance, and one developed diabetes mellitus 32. All 3 patients had mutations of the ABCC8 (SUR1) gene. The significance of this small series is uncertain, but the results suggest that development of impaired glucose tolerance may be part of the underlying disease process and not solely due to surgical reduction in islet cell mass.
Education of the patient and family and long-term follow-up are essential to prevent delays in the diagnosis of disease recurrence, glucose intolerance, or diabetes mellitus.
Neurodevelopmental outcome
In some series, a high frequency of mental retardation, developmental delay, and nonhypoglycemic seizures has been observed. These findings are generally attributed to minimal brain damage from early hypoglycemic events, although the existence of these disorders as inherent comorbid conditions with congenital hyperinsulinism has not been fully excluded. Other series, usually in conjunction with medication studies, have shown normal developmental progress in patients with congenital hyperinsulinism.
Some data suggest that patients with early, severe disease treated with early, aggressive surgery have a better neurodevelopmental outcome. No comprehensive long-term studies of neurodevelopmental outcomes in patients with congenital hyperinsulinism are available, and the heterogeneity of the disease likely confounds many neurodevelopmental studies.
Permanent neurologic dysfunction (eg, seizures, developmental delay, focal neurologic deficits) or death secondary to severe, prolonged hypoglycemia may occur if congenital hyperinsulinism goes untreated or is inadequately treated.
References- Gillis D. Familial Hyperinsulinism. 2003 Aug 19 [Updated 2019 Mar 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1375
- Congenital hyperinsulinism. https://ghr.nlm.nih.gov/condition/congenital-hyperinsulinism
- Congenital Hyperinsulinism. https://emedicine.medscape.com/article/923538-overview
- Pathogenesis, clinical presentation, and diagnosis of congenital hyperinsulinism. https://www.uptodate.com/contents/pathogenesis-clinical-presentation-and-diagnosis-of-congenital-hyperinsulinism
- Congenital Hyperinsulinism. https://rarediseases.org/rare-diseases/congenital-hyperinsulinism
- Tornovsky S, Crane A, Cosgrove KE, Hussain K, Lavie J, Heyman M, Nesher Y, Kuchinski N, Ben-Shushan E, Shatz O, Nahari E, Potikha T, Zangen D, Tenenbaum-Rakover Y, de Vries L, Argente J, Gracia R, Landau H, Eliakim A, Lindley K, Dunne MJ, Aguilar-Bryan L, Glaser B. Hyperinsulinism of infancy: novel ABCC8 and KCNJ11 mutations and evidence for additional locus heterogeneity. J Clin Endocrinol Metab. 2004;89:6224–34.
- Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H, Hussain K, Ellard S. A. CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes. 2017;18:320–3.
- Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, Suchi M, Taub R, Grimsby J, Matschinsky FM, Stanley CA. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58:1419–27.
- Bahi-Buisson N, Roze E, Dionisi C, Escande F, Valayannopoulos V, Feillet F, Heinrichs C, Chadefaux-Vekemans B, Dan B, de Lonlay P. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev Med Child Neurol. 2008;50:945–9.
- Flanagan SE, Patch AM, Locke JM, Akcay T, Simsek E, Alaei M, Yekta Z, Desai M, Kapoor RR, Hussain K, Ellard S. Genome-wide homozygosity analysis reveals HADH mutations as a common cause of diazoxide-responsive hyperinsulinemic-hypoglycemia in consanguineous pedigrees. J Clin Endocrinol Metab. 2011;96:E498–502.
- Pinney SE, Ganapathy K, Bradfield J, Stokes D, Sasson A, Mackiewicz K, Boodhansingh K, Hughes N, Becker S, Givler S, Macmullen C, Monos D, Ganguly A, Hakonarson H, Stanley CA. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm Res Paediatr. 2013;80:18–27.
- Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, Siahanidou T, Banerjee I, Akcay T, Rubio-Cabezas O, Shield JP, Hussain K, Ellard S. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. 2010;162:987–92.
- Cabezas OR, Flanagan SE, Stanescu H, García-Martínez E, Caswell R, Lango-Allen H, Antón-Gamero M, Argente J, Bussell AM, Brandli A, Cheshire C, Crowne E, Dumitriu S, Drynda R, Hamilton-Shield JP, Hayes W, Hofherr A, Iancu D, Issler N, Jefferies C, Jones P, Johnson M, Kesselheim A, Klootwijk E, Koettgen M, Lewis W, Martos JM, Mozere M, Norman J, Patel V, Parrish A, Pérez-Cerdá C, Pozo J, Rahman SA, Sebire N, Tekman M, Turnpenny PD, Hoff WV, Viering DHHM, Weedon MN, Wilson P, Guay-Woodford L, Kleta R, Hussain K, Ellard S, Bockenhauer D. Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J Am Soc Nephrol. 2017;28:2529–39.
- Ferrara CT, Boodhansingh KE, Paradies E, Fiermonte G, Steinkrauss LJ, Topor LS, Quintos JB, Ganguly A, De Leon DD, Palmieri F, Stanley CA. Novel hypoglycemia phenotype in congenital hyperinsulinism due to dominant mutations of uncoupling protein 2. J Clin Endocrinol Metab. 2017;102:942–9.
- Laver TW, Weedon MN, Caswell R, Hussain K, Ellard S, Flanagan SE. Analysis of large-scale sequencing cohorts does not support the role of variants in UCP2 as a cause of hyperinsulinaemic hypoglycaemia. Hum Mutat. 2017;38:1442–4.
- Glaser B, Blech I, Krakinovsky Y, Ekstein J, Gillis D, Mazor-Aronovitch K, Landau H, Abeliovich D. ABCC8 mutation allele frequency in the Ashkenazi Jewish population and risk of focal hyperinsulinemic hypoglycemia. Genet Med. 2011;13:891–4.
- Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, Vauhkonen I, Kere J, Laakso M, Ashcroft F, Otonkoski T. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest. 2000;106:897–906.
- Hashimoto Y, Sakakibara A, Kawakita R, Hosokawa Y, Fujimaru R, Nakamura T, et al. Focal form of congenital hyperinsulinism clearly detectable by contrast-enhanced computed tomography imaging. Int J Pediatr Endocrinol. 2015. 2015 (1):20.
- Laje P, States LJ, Zhuang H, Becker SA, Palladino AA, Stanley CA, et al. Accuracy of PET/CT Scan in the diagnosis of the focal form of congenital hyperinsulinism. J Pediatr Surg. 2013 Feb. 48(2):388-93.
- Sempoux C, Capito C, Bellanné-Chantelot C, Verkarre V, de Lonlay P, Aigrain Y, Fekete C, Guiot Y, Rahier J. Morphological mosaicism of the pancreatic islets: a novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metab. 2011;96:3785–93.
- Maiorana A, Manganozzi L, Barbetti F, Bernabei S, Gallo G, Cusmai R, et al. Ketogenic diet in a patient with congenital hyperinsulinism: a novel approach to prevent brain damage. Orphanet J Rare Dis. 2015 Sep 24. 10 (1):120.
- Hussain K, Aynsley-Green A, Stanley CA. Medications used in the treatment of hypoglycemia due to congenital hyperinsulinism of infancy (HI). Pediatr Endocrinol Rev. 2004;2 Suppl 1:163–7.
- Le Quan Sang KH, Arnoux JB, Mamoune A, Saint-Martin C, Bellanné-Chantelot C, Valayannopoulos V, Brassier A, Kayirangwa H, Barbier V, Broissand C, Fabreguettes JR, Charron B, Thalabard JC, de Lonlay P. Successful treatment of congenital hyperinsulinism with long-acting release octreotide. Eur J Endocrinol. 2012;166:333–9.
- Modan-Moses D, Koren I, Mazor-Aronovitch K, Pinhas-Hamiel O, Landau H. Treatment of congenital hyperinsulinism with lanreotide acetate (SomatulineAutogel). J Clin Endocrinol Metab. 2011;96:2312–7.
- Mohnike K, Blankenstein O, Minn H, Mohnike W, Fuchtner F, Otonkoski T. [18F]-DOPA positron emission tomography for preoperative localization in congenital hyperinsulinism. Horm Res. 2008a;70:65–72.
- Beltrand J, Caquard M, Arnoux JB, Laborde K, Velho G, Verkarre V, Rahier J, Brunelle F, Nihoul-Fékété C, Saudubray JM, Robert JJ, de Lonlay P. Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care. 2012;35:198–203.
- Chinnakotla S, Bellin MD, Schwarzenberg SJ, Radosevich DM, Cook M, Dunn TB, et al. Total Pancreatectomy and Islet Autotransplantation in Children for Chronic Pancreatitis: Indication, Surgical Techniques, Postoperative Management, and Long-term Outcomes. Ann Surg. 2014 Feb 6.
- Robertson RP, Lanz KJ, Sutherland DE, Kendall DM. Prevention of diabetes for up to 13 years by autoislet transplantation after pancreatectomy for chronic pancreatitis. Diabetes. 2001 Jan. 50(1):47-50.
- Mazor-Aronovitch K, Landau H, Gillis D. Surgical versus non-surgical treatment of congenital hyperinsulinism. Pediatr Endocrinol Rev. 2009;6:424–30.
- Congenital Hyperinsulinism. https://emedicine.medscape.com/article/923538-overview#a6
- Lovvorn HN 3rd, Nance ML, Ferry RJ Jr, et al. Congenital hyperinsulinism and the surgeon: lessons learned over 35 years. J Pediatr Surg. 1999 May. 34(5):786-92; discussion 792-3.
- Gussinyer M, Clemente M, Cebrián R, Yeste D, Albisu M, Carrascosa A. Glucose intolerance and diabetes are observed in the long-term follow-up of nonpancreatectomized patients with persistent hyperinsulinemic hypoglycemia of infancy due to mutations in the ABCC8 gene. Diabetes Care. 2008 Jun. 31(6):1257-9.





{kind=link}