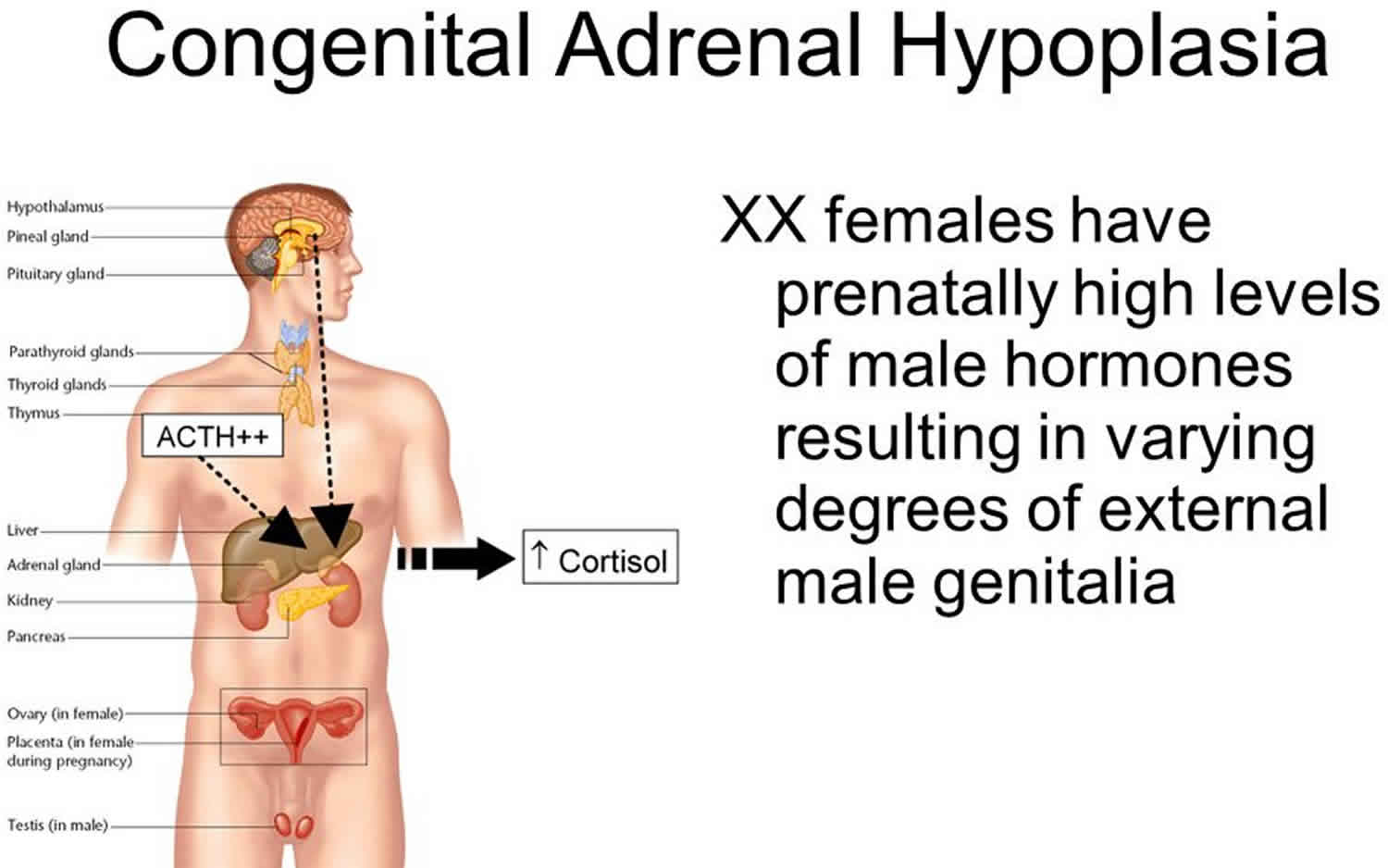
Adrenal hypoplasia
Adrenal hypoplasia is a condition in which there is underdevelopment or hypotrophy of the adrenal cortex due to several clinical conditions. Adrenal hypoplasia can divide into two categories 1:
- Primary adrenal hypoplasia: Primary adrenal hypoplasia is caused by the adrenal hypoplasia congenita or intrinsic defect during adrenal gland development. Adrenal hypoplasia congenita also known as congenital adrenal hypoplasia, involves hypofunctional and hypertrophic adrenal glands, which are caused by defects during differentiation of the fetal adrenal glands. It occurs in many forms 2.
- Secondary adrenal hypoplasia: Secondary adrenal hypoplasia results from pituitary or hypothalamic dysfunction. These conditions cause a decreased synthesis and secretion of adrenocorticotropic hormone (ACTH), leading to inadequate adrenal development and adrenal dysfunction because of the reduced adrenal stimulus 3.
Adrenal hypoplasia can occur 4:
- Secondary to defects in transcription factors involved in pituitary development;
- Defects in ACTH synthesis and secretion;
- As a primary defect in the development of the adrenal gland;
- As part of rare syndromes associated with adrenal hypoplasia/aplasia, which are inherited in an autosomal recessive or autosomal dominant manner;
- In the context of chromosomal abnormalities.
Early diagnosis and management are crucial because of the life-threatening nature of the condition. Depending on the cause, adrenal crisis may occur in early infancy or could insidiously develop over the course of childhood or adolescence 4. Moreover, some of these conditions previously thought to occur only in childhood, may also be diagnosed later in adulthood and present with variable phenotypes, including isolated infertility or disorders of sex differentiation. The clinical manifestations of primary adrenal insufficiency result from deficiency of all adrenocortical hormones (aldosterone, cortisol, androgens). The acute presentation can be precipitated by physiologic stress, such as surgery, trauma, or an intercurrent infection. Patients may present with signs and symptoms of complete adrenal insufficiency, usually early in life, including hypoglycemic convulsions, hyponatremia, hyperkalemia, metabolic acidosis or later with hyperpigmentation, vomiting and poor weight gain. It should be remembered, that the most common cause of primary adrenal insufficiency in children is congenital adrenal hyperplasia due to 21-hydroxylase deficiency and can be excluded by measuring baseline or ACTH-stimulated 17-hydroxyprogesterone levels in serum 4. Screening for autoimmune Addison disease includes detection of 21-hydroxylase antibodies. Males with negative 21-hydroxylase antibodies should be tested for adrenoleukodystrophy measuring very–long-chain fatty acids concentrations in plasma. The presence of alacrima (reduced or absent ability to secrete tears) in patients with primary adrenal insufficiency should raise suspicion for Triple A syndrome, whereas the combination of primary adrenal insufficiency and hypogonadotropic hypogonadism in a male patient point towards X-linked adrenal hypoplasia congenita. To date, molecular genetic testing is commercially available for the identification of several genes involved in adrenal hypoplasia syndromes. The early identification of these diseases can have important prognostic and therapeutic implications for patients with respect to surveillance for associated conditions, initiation of early treatment or screening of family members who are at risk.
Adrenal insufficiency is potentially life threatening, thus treatment should be initiated as soon as the diagnosis is confirmed, or sooner if the patient presents in adrenal crisis. Therapy consists of life-long replacement therapy with glucocorticoids and mineralocorticoids. Hypogonadism or other associated disorders should be treated appropriately. Screening of family members for the disease or carrier status may also be indicated and can be critical for family planning. When a monogenic cause of adrenal failure is identified, genetic counseling is indicated.
Figure 1. Adrenal glands
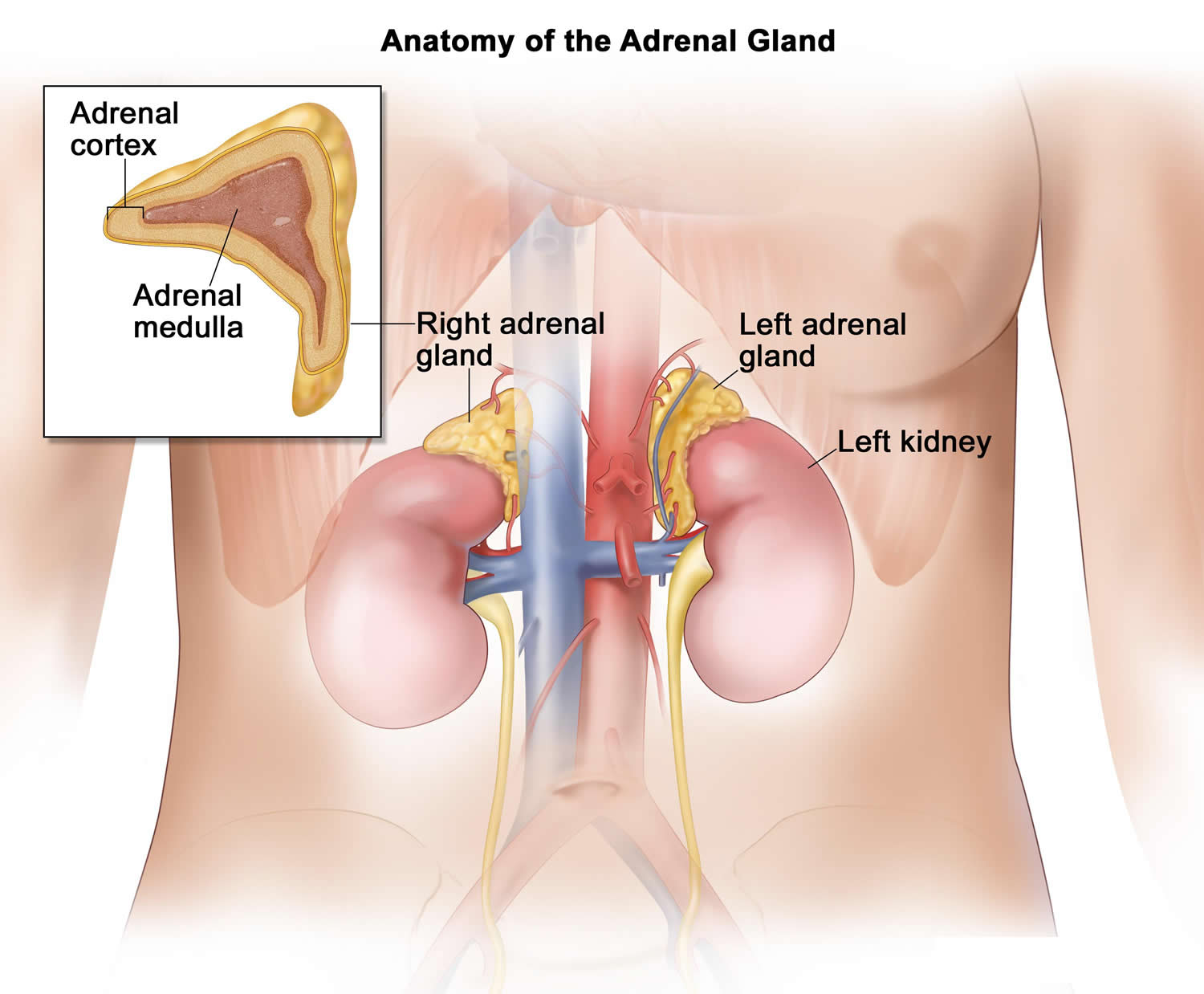
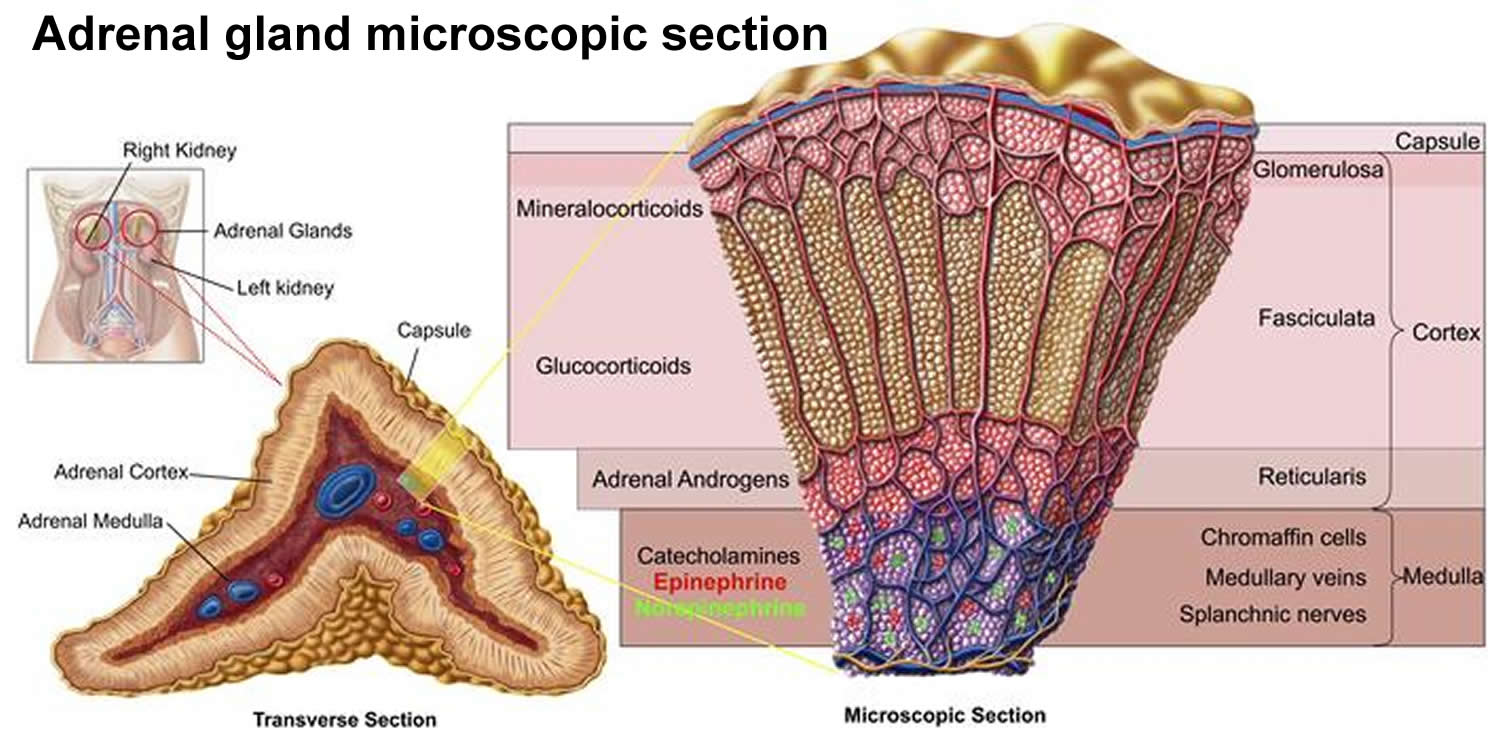
Figure 2. Adrenal gland functions
Congenital adrenal hypoplasia
Congenital adrenal hyperplasia also called adrenal hypoplasia congenita, is a group of genetic disorder in which the adrenal gland produces an overabundance of certain male hormones called androgens. As a result, a child with congenital adrenal hyperplasia is born with a reproductive tract that is not normally developed, and the child is neither “fully” male nor female. If your child has congenital adrenal hyperplasia, it also means that her adrenal glands don’t produce enough of the vital hormones cortisol and aldosterone. As a result, her body produces too much androgen.
When there is not enough cortisol and aldosterone, the body will make too much of certain male steroid hormones called androgens. The overabundance of androgens is responsible for the ambiguous genitalia in females born with adrenal hypoplasia congenita.
Congenital adrenal hyperplasia is one of the most common disorders of sexual differentiation, which describes a wide range of diseases in which development of the sex organs does not occur as it normally would.
Congenital adrenal hyperplasia is also the most common cause of ambiguous genitalia, a term which describes children whom you cannot tell by looking at their genitals whether they’re male or female.
In normal pattern of sexual development, the adrenal gland and hormones function like this:
- The adrenal glands and hormones:
- Adrenal glands, which are located on the top of each kidney, are responsible for releasing three different types of steroid hormones:
- glucocorticoids, which help modulate your sugar metabolism. The glucocorticoid involved in congenital adrenal hyperplasia is called cortisone.
- mineralocorticoids, which modulate your fluids and electrolytes. The mineralocorticoid involved in congenital adrenal hyperplasia is called aldosterone.
- sex steroid hormones, which aid in the formation of sex organs. The adrenal gland releases the male hormone androgen (testosterone) and the female hormone estrogen.
- Normally, the adrenal gland secretes the hormones directly into the blood stream, in three equally prominent metabolic pathways. This balance allows for the normal production of androgen and estrogen.
- Adrenal glands, which are located on the top of each kidney, are responsible for releasing three different types of steroid hormones:
- Hormones and congenital adrenal hyperplasia
- In congenital adrenal hyperplasia, an enzyme deficiency blocks the pathways to the glucocorticoid and mineralocorticoid, impairing the body’s ability to produce cortisol and aldosterone.
- Too little cortisol. The adrenal glands of infants born with congenital adrenal hyperplasia cannot make enough of the hormone cortisol. This hormone affects energy levels, blood sugar levels, blood pressure, and the body’s response to stress, illness, and injury.
- Too little aldosterone. In about 75% of cases, infants born with congenital adrenal hyperplasia cannot make enough of the hormone aldosterone, which helps the body maintain the proper level of sodium (salt) and water and helps maintain blood pressure.
- The overabundance of androgen leads to the virilization (masculinization) of a female fetus. This is responsible for the ambiguous genitalia in females born with this condition.
- While masculinization of a male fetus is possible, it’s less noticeable.
- In congenital adrenal hyperplasia, an enzyme deficiency blocks the pathways to the glucocorticoid and mineralocorticoid, impairing the body’s ability to produce cortisol and aldosterone.
Congenital adrenal hyperplasia can also cause imbalances in the hormone adrenaline, which affects blood sugar levels, blood pressure, and the body’s response to stress 5, 6.
The hormone imbalances in most cases of congenital adrenal hyperplasia (about 95%) are caused by too little of a substance called 21-hydroxylase. The adrenal glands need 21-hydroxylase to make proper amounts of hormones. This type of congenital adrenal hyperplasia is sometimes referred to as 21-hydroxylase deficiency. In congenital adrenal hyperplasia due to 21-hydroxylase deficiency, the adrenal glands cannot make enough cortisol or aldosterone. In addition, the glands make too much androgen. People with 21-hydroxylase deficiency also may not produce enough adrenaline.
About 5% of cases of congenital adrenal hyperplasia are caused by deficiency in a substance similar to 21-hydroxylase, called 11-hydroxylase. This type of congenital adrenal hyperplasia is sometimes referred to as 11-hydroxylase deficiency. In congenital adrenal hyperplasia due to 11-hydroxylase deficiency, the adrenal glands make too little cortisol and too many androgens. This type of congenital adrenal hyperplasia does not result in aldosterone deficiency.
Other very rare types of congenital adrenal hyperplasia include 3-betahydroxy-steroid dehydrogenase deficiency, lipoid congenital adrenal hyperplasia, and 17-hydroxylase deficiency.
Congenital adrenal hyperplasia occurs among all races. The worldwide incidence of classical 21-hydroxylase-deficient congenital adrenal hyperplasia is 1 in 15,000 to 20,000 births. Approximately 70% of the affected infants have the salt-wasting form, and 30% have the simple virilizing form 7.
Congenital adrenal hyperplasia can be categorized as classic or nonclassic types based on severity:
- Classic congenital adrenal hyperplasia is more severe than the nonclassic form. It can be life threatening in newborns if it is not diagnosed. Classic congenital adrenal hyperplasia can be caused by either 21-hydroxylase or 11-hydroxylase deficiency. Classic congenital adrenal hyperplasia occurs in about one of every 15,000 births worldwide 8.
- Nonclassic congenital adrenal hyperplasia is sometimes called late-onset congenital adrenal hyperplasia. It is a milder form of the disorder that usually is diagnosed in late childhood or early adolescence. Sometimes, people have nonclassic congenital adrenal hyperplasia and never know it. This form of congenital adrenal hyperplasia is almost always caused by 21-hydroxylase deficiency. Nonclassic congenital adrenal hyperplasia is among the most common genetic disorders. It occurs in about one of every 1,000 people, but could occur in as many as 1 in 20 people in some communities or specific ethnic groups such as Ashkenazi Jews and Hispanics 9. It is more common in Ashkenazi Jews, Hispanics, Italians, and Yugoslavians 5.
Congenital adrenal hyperplasia key points:
- Congenital adrenal hyperplasia is one of the most common disorders of sexual differentiation and the most common cause of ambiguous genitalia in newborns.
- Congenital adrenal hyperplasia affects one in 5,000 to 15,000 babies in the United States and Europe. congenital adrenal hyperplasia affects newborns of both genders equally.
- Females born with adrenal hypoplasia congenita are typically born with an enlarged clitoris, but with normal internal reproductive structures.
- Males born with adrenal hypoplasia congenita have normal genitals at birth, but may become more muscular or develop pubic hair and a deeper voice well before puberty.
- Females with congenital adrenal hyperplasia need hormone replacement therapy, and in some cases, reconstructive surgery. Males with congenital adrenal hyperplasia typically only require medical follow-up.
- With medical management, surgical management and psychosocial support, children with congenital adrenal hyperplasia can lead full and healthy lives and enjoy a normal life expectancy.
Can congenital adrenal hyperplasia be prevented?
Even if they’re not affected by congenital adrenal hyperplasia, all parents should seek genetic counseling before conceiving a child. Although no testing can be done at that point, the doctor can look at your family’s medical history to see if you might have an increased risk for having a child with congenital adrenal hyperplasia.
How will congenital adrenal hyperplasia affect my child?
Congenital adrenal hyperplasia has a more noticeable impact on girls than boys. This is because congenital adrenal hyperplasia causes the body to produce testosterone and testosterone is a hormone that causes masculine attributes.
Males will require medical management, but they won’t need hormone replacement therapy, surgery or medications. Girls who have corrective genital surgery may need further cosmetic surgery later in life. When they become sexually active, they’re more likely than are women who have not had genital surgery to experience sexual problems, such as pain during intercourse.
Will my child be able to function sexually?
Yes. With the proper surgical correction, children ought to be able to function sexually in a reasonably normal way.
Will my child be able to have children?
Depending on the genetics of their condition, some people with congenital adrenal hyperplasia may be able to have children. However, people with congenital adrenal hyperplasia have a lower fertility rate than those who do not have the condition.
Congenital adrenal hypoplasia causes
Congenital adrenal hyperplasia is caused by changes (mutations) in one of several genes. These changes lead to deficiencies in 21-hydroxylase or, less commonly, 11-hydroxylase. Both of these are chemicals called enzymes. The adrenal glands need these enzymes to make proper amounts of the hormones: cortisol, aldosterone, androgens, and adrenaline.
21-hydroxylase deficiency is the most common cause of congenital adrenal hyperplasia (more than 90% of the cases) 10.
21-hydroxylase (CYP21, P450c21) is a cytochrome e P-450 enzyme located in the endoplasmic reticulum. It hydroxylates 17-hydroxyprogesterone to 11-deoxycortisol, a precursor of cortisol and also hydroxylates progesterone to deoxycorticosterone, a precursor of aldosterone. Both hormones (cortisol and aldosterone) are deficient in the most-severe, ’salt-wasting’ form of congenital adrenal hyperplasia 11.
Due to the loss of 21-hydroxylase enzyme function, patients with a 21-hydroxylase deficiency cannot synthesize cortisol efficiently, and as a result, the ACTH levels are high, leading to hyperplasia of the adrenal cortex and overproduction of cortisol precursors. Some of these precursors are used for the synthesis of sex steroids, which may cause signs of androgen excess, including ambiguous genitalia in newborn girls and rapid postnatal growth in both sexes. Aldosterone deficiency may lead to salt wasting with consequent failure to thrive, hypovolemia, and shock.
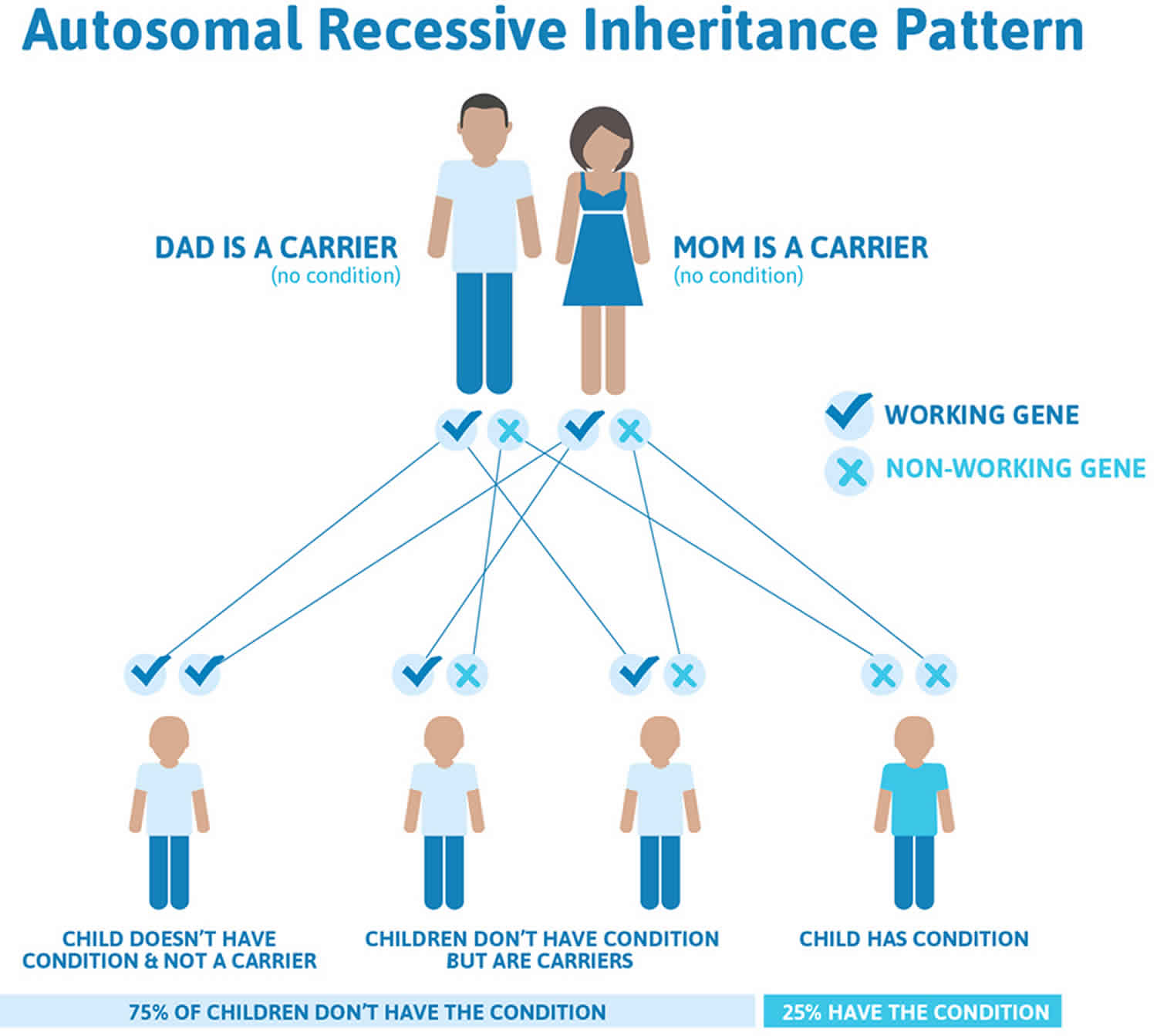
Congenital adrenal hypoplasia inheritance pattern
The genes for congenital adrenal hyperplasia are passed down from parents to their children. In general, people have two copies of every gene in their bodies. They receive one copy from each parent. For an infant to have congenital adrenal hyperplasia, both copies must have an error that affects an adrenal-gland enzyme.
Congenital adrenal hyperplasia is an example of an autosomal recessive disorder:
- Autosomal means the gene is not on the X chromosome or Y chromosome.
- Recessive means that both copies of the gene must have the error for the disease or disorder to occur.
If both parents have congenital adrenal hyperplasia, all of their children will also have it. If each parent carries one affected gene and one normal gene (called a “carrier”), there is a one-in-four chance of their child having congenital adrenal hyperplasia (see Figure 3).
Anyone with congenital adrenal hyperplasia, or from a family in which congenital adrenal hyperplasia has been diagnosed, should consider genetic counseling. Genetic counselors discuss all options for having a child. They explain the risks and benefits of each option.
The genes for congenital adrenal hyperplasia are passed down from parents to their children. In general, people have two copies of every gene in their bodies. They receive one copy from each parent. For an infant to have congenital adrenal hyperplasia, both copies must have an error that affects an adrenal-gland enzyme.
Women with congenital adrenal hyperplasia can get pregnant. In some of the women, high levels of androgens disrupt the regular release of the egg from the ovary, a process known as ovulation. Some women also have irregular menstrual cycles. These problems can make it more difficult to get pregnant. These women often can be helped with medicines. Women with congenital adrenal hyperplasia who want to become pregnant can meet with a reproductive endocrinologist. This is a health care provider who specializes in fertility issues.
Women with congenital adrenal hyperplasia who become pregnant should continue taking their medications.
Men with congenital adrenal hyperplasia can father children. The main challenges for these men are low testosterone (a hormone important for male fertility and sexual function), and growths in the testicles called adrenal rest tissue. These problems can cause reduced sperm production. These issues tend to occur when hormone imbalances are not well controlled with medicines. Men who wish to father children should take all medicines as directed. A health care provider may recommend that males with congenital adrenal hyperplasia who have gone through puberty get an ultrasound of the testicles. The ultrasound provides a picture of the inside of the testicle and can help a health care provider detect abnormal growths. Future ultrasounds can be compared with the original to quickly identify any problems.
Figure 3. Congenital adrenal hyperplasia autosomal recessive inheritance pattern
Congenital adrenal hypoplasia symptoms
At birth, boys usually appear to be unaffected and do not start to show symptoms until the first few years of life. Girls may display ambiguous genitalia, such as an enlarged clitoris and labia that resemble a scrotum. The internal reproductive organs (ovaries, uterus, fallopian tubes) are not affected by the disorder.
Boys may appear to enter puberty as early as 2 to 3 years old. The symptoms of this may include:
- deep voice
- early appearance of pubic and armpit hair
- enlarged penis
- small testes
- well-developed muscles
Girls may show the following changes:
- abnormal menstrual periods
- failure to menstruate
- ambiguous genitalia, often appearing more male than female
- deep voice
- early appearance of pubic and armpit hair
- excessive hair growth and facial hair
Symptoms in infants may include:
- weight loss
- vomiting
- failure to thrive (inability to grow and gain weight, especially in infants and toddlers)
- dehydration
- vomiting
If congenital adrenal hyperplasia is not treated, a child’s features will become more masculine as the child continues to grow.
Other complications may include:
- adrenal crisis, a life-threatening condition that occurs when there is not enough cortisol.
- short height as an adult, even if growth was rapid during childhood
- side effects of steroid medications used as treatment
- tumors of the testes can occur in adult men
- high blood pressure
- low blood sugar
Classic congenital adrenal hyperplasia
Symptoms of classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency (95% of classic congenital adrenal hyperplasia cases) can be grouped into two types according to their severity: salt wasting and simple virilizing (also called non-salt wasting).
Symptoms of classic congenital adrenal hyperplasia due to 11-hydroxylase deficiency (5% of classic congenital adrenal hyperplasia cases) are similar to those of simple virilizing congenital adrenal hyperplasia. About two-thirds of people with classic 11-hydroxylase deficiency also have high blood pressure (hypertension) 12.
Salt-wasting congenital adrenal hyperplasia:
Salt-wasting congenital adrenal hyperplasia is the severe form of classic 21-hydroxylase deficiency. In this type of congenital adrenal hyperplasia, the adrenal glands make too little aldosterone, causing the body to be unable to retain enough sodium (salt). Too much sodium is lost in urine (thus the name, “salt-wasting”). If undiagnosed, symptoms of classic salt-wasting congenital adrenal hyperplasia appear within days or weeks of birth and, in some cases, death occurs.
Symptoms may include:
- Dehydration
- Poor feeding
- Diarrhea
- Vomiting
- Heart rhythm problems (arrhythmias)
- Low blood pressure
- Very low blood sodium levels
- Low blood glucose
- Too much acid in the blood, called metabolic acidosis
- Weight loss
- Shock, a condition where not enough blood gets to the brain and other organs. Shock in infants with salt-wasting is called adrenal crisis. Signs include confusion, irritability, rapid heart rate, and/or coma.
Even when carefully treated, children with salt-wasting congenital adrenal hyperplasia are still at risk for adrenal crises when they become ill or are under physical stress. The body needs more than the usual amount of adrenal hormones during illness, injury, or physical stress. This means a child with congenital adrenal hyperplasia must be given more medication during these times to prevent an adrenal crisis.
Salt-wasting congenital adrenal hyperplasia also involves symptoms caused by low cortisol and high androgens. These symptoms may include:
- In female newborns, external genitalia can be ambiguous, i.e., not typical female appearing , with normal internal reproductive organs (ovaries, uterus, and fallopian tubes)
- Enlarged genitalia in male newborns
- Development of certain qualities called virilization in boys or girls before the normal age of puberty, sometimes as early as age 2 or 3. This is a condition characterized by:
- Rapid growth
- Appearance of pubic and armpit hair
- Deep voice
- Failure to menstruate, or abnormal or irregular menstrual periods (females)
- Well-developed muscles
- Enlarged penis (males)
- Unusually tall height as children, but being shorter than normal as adults
- Possible difficulties getting pregnant (females)
- Excess facial hair (females)
- Early beard (males)
- Severe acne
- Benign testicular tumors and infertility (males)
Simple virilizing (non-salt wasting) congenital adrenal hyperplasia:
Approximately 25% of patients with classic 21-hydroxylase deficiency present with simple virilization without salt wasting. Simple virilizing congenital adrenal hyperplasia is the moderate form of classic 21-hydroxylase deficiency. This type of congenital adrenal hyperplasia involves less severe aldosterone deficiency. Therefore, there are no severe or life-threatening sodium-deficiency symptoms in newborns. Like salt-wasting congenital adrenal hyperplasia, simple virilizing congenital adrenal hyperplasia involves too little cortisol and too much androgen. Female newborns have ambiguous genitalia and young children display virilization.
Without newborn screening, affected boys are diagnosed in childhood when signs of androgen excess develop. Later diagnosis is associated with greater difficulty in achieving hormonal control, and short stature.
Nonclassic congenital adrenal hyperplasia
Almost all cases of nonclassic or late-onset form of congenital adrenal hyperplasia are caused by a mild 21-hydroxylase deficiency, occurring in 0.1% to 0.2% in the general white population and 1% to 2% among Ashkenazi Jews. Most symptoms of nonclassic congenital adrenal hyperplasia are related to increased androgens. Symptoms can show up in childhood, adolescence, or early adulthood.
Females with the nonclassic form may be compound heterozygotes with a classic mutation and variant allele or heterozygotes with two variant alleles, allowing 20% to 60% of normal enzymatic activity.
Compound heterozygote females have a less severe phenotype, and clinical presentation varies. Females may present at any age but usually not younger than 6 months. Heterozygote females may have mild biochemical abnormalities but no clinically important endocrine disorder.
Patients with nonclassic form, have normal levels of cortisol and aldosterone at the expense of mild to moderate overproduction of sex hormones precursors. Newborn screening can detect nonclassic cases, but most are missed because of relatively low baseline levels of 17 hydroxyprogesterone.
Hirsutism is the single most common symptom at presentation, followed by oligomenorrhea and acne. Thus, nonclassic 21-hydroxylase deficiency and polycystic ovarian syndrome may present in similar ways.
Symptoms of nonclassic congenital adrenal hyperplasia can include:
- Rapid growth in childhood and early teens but shorter height than both parents
- Early signs of puberty
- Acne
- Irregular menstrual periods (females)
- Fertility problems (in about 10% to 15% of women)
- Excess facial or body hair in women
- Male-pattern baldness (hair loss near the temples)
- Enlarged penis (males)
- Small testicles (males)
Some people have nonclassic congenital adrenal hyperplasia and never know it because the symptoms are so mild 13, 14.
Congenital adrenal hyperplasia complications
Complications of congenital adrenal hyperplasia are common. If the patient does not get enough glucocorticoids, he or she can develop adrenal insufficiency and further virilization in the virilizing forms. If a patient receives excessive glucocorticoids, he or she can develop growth failure, obesity, striae, hypertension, hyperglycemia, and cataracts.
The complications of excess mineralocorticoid administration include hypertension and hypokalemia.
Aldosterone deficiency may lead to salt wasting with consequent failure to thrive, hypovolemia, and shock.
Congenital adrenal hypoplasia diagnosis
The screening workup should include:
- Newborn screening programs check for 21-hydroxylase deficiency
- 17-hydroxyprogesterone will be very high (usually greater than 1000 ng/dL) in a patient with the classic form
- Hyperkalemia, hyponatremia, low aldosterone, and high plasma renin activity, particularly the ratio of plasma renin activity to aldosterone, are markers of impaired mineralocorticoid synthesis
- An ACTH stimulation test should be performed to evaluate adrenal function and differentiate among the various potential enzymatic defects. Administration of 0.25 mg of cosyntropin (a synthetic ACTH) provides a pharmacologic stimulus to the adrenal glands, maximizing hormone secretion.
- A full adrenal profile, including measurement of 17-hydroxyprogesterone (17-OHP), cortisol, deoxycorticosterone, 11-deoxycortisol, 17-hydroxypregnenolone, dehydroepiandrosterone (DHEA), and androstenedione, should be obtained immediately before and 60 minutes after cosyntropin administration.
- Nomograms are available for interpreting the results
- In an infant with ambiguous genitalia, do karyotype to establish the chromosomal sex
- The pelvic ultrasound should be done to check for uterus or associated renal anomalies
- A bone age study is helpful in patients with precocious pubic hair
- In patients with signs of acute adrenal failure, CT of the adrenal glands can be done to exclude adrenal hemorrhage
- Urogenitography for defining the anatomy of the internal genitalia
During Pregnancy
If a woman already has a child with congenital adrenal hyperplasia and becomes pregnant with the same partner, her fetus has a one in four chance of having congenital adrenal hyperplasia. For this reason, prenatal testing can be done for some forms of congenital adrenal hyperplasia. A health care provider checks for the disorder by using techniques called amniocentesis or chorionic villus sampling 14.
- Amniocentesis. This involves inserting a needle into the womb, through the abdomen, to withdraw a small amount of fluid from the sac that surrounds the fetus. The procedure is usually done between the 15th and 20th week of pregnancy.
- Chorionic villus sampling. This is similar to amniocentesis. A health care provider inserts a needle into the womb, either through the abdomen or the cervix, and extracts a small piece of tissue from the chorionic villi (the tissue that will later become the placenta). This procedure is usually done between the 10th and 12th week of pregnancy.
After a health care provider takes a sample using one of these techniques, he or she will perform a genetic test on the sample. This test will reveal whether the fetus has a gene change that causes congenital adrenal hyperplasia.
Parents may also choose to wait until birth to have the newborn tested. Talking to their health care providers may help parents identify the option that is right for them.
At Birth
All U.S. states have neonatal screening for congenital adrenal hyperplasia 15. Infants who test positive need to have follow-up testing done to confirm the diagnosis. If, for some reason, the neonatal screening is negative but there is high suspicion for congenital adrenal hyperplasia (such as ambiguous genitalia), further evaluation is also indicated.
Positive Newborn Screen
- A positive newborn screening test for congenital adrenal hyperplasia must be confirmed by a second plasma sample (17-hydroxyprogesterone), and serum electrolytes should be measured.
- After the confirmatory blood sample is obtained, treatment doses of glucocorticoid and mineralocorticoid should be initiated in all infants in whom congenital adrenal hyperplasia is a consideration, to prevent the potentially life-threatening manifestations of an adrenal crisis.
- If the physician chooses not to initiate treatment while awaiting confirmatory steroid hormone measurements, serum electrolytes should be measured daily.
- A pediatric endocrinologist should manage these patients.
Later in Life
Newborns do not show symptoms of nonclassic congenital adrenal hyperplasia, and the test done on newborns does not detect nonclassic congenital adrenal hyperplasia. Nonclassic congenital adrenal hyperplasia is diagnosed in childhood or adulthood, when symptoms appear. To diagnose nonclassic congenital adrenal hyperplasia, a health care provider may:
- Ask whether family members have congenital adrenal hyperplasia.
- Do a physical exam.
- Take blood and urine to measure hormone levels.
- Do a genetic test to determine if the patient has the gene change that causes congenital adrenal hyperplasia.
An X-ray can help to diagnose congenital adrenal hyperplasia in children. Because some children with congenital adrenal hyperplasia grow too quickly, their bones will be more developed than normal for their age 16.
Congenital adrenal hypoplasia treatment
Treatments for congenital adrenal hyperplasia include medication and surgery as well as psychological support 17, 5, 18, 14.
Acute Adrenal Crisis
- Is a medical emergency
- Initial management should be fluid resuscitation: Intravenous (IV) bolus of isotonic sodium chloride solution (20 mL/kg). Repeated boluses may be needed.
- Administer dextrose if the patient is hypoglycemic and must be rehydrated with fluid after the bolus dose to prevent hypoglycemia
- Stress doses of hydrocortisone (100 mg/m2 per day) are vital in the management and should be given as early as possible, concomitant with IV fluid treatment.
- Central access and vasopressors, along with higher glucose concentrations, may be required in profoundly ill patients.
- Life-threatening hyperkalemia may require additional therapy with potassium-lowering resin, IV calcium, insulin, and bicarbonate.
Medication
Classic congenital adrenal hyperplasia
Newborns with classic congenital adrenal hyperplasia should start treatment very soon after birth to reduce the effects of congenital adrenal hyperplasia. Classic congenital adrenal hyperplasia is treated with steroids that replace the low hormones.
- Infants and children usually take a form of cortisol called hydrocortisone.
- Adults take hydrocortisone, prednisone, or dexamethasone, which also replace cortisol.
- Patients with classic congenital adrenal hyperplasia also take another medicine, fludrocortisone, to replace aldosterone.
- Eating salty foods or taking salt pills may also help salt-wasters retain salt 19.
The body needs more cortisol when it is under physical stress. Adults and children with classic congenital adrenal hyperplasia need close medical attention and may need to take more of their medication during these times. They may also need more medication if they:
- Have an illness with a high fever.
- Undergo surgery.
- Sustain a major injury.
People who have classic congenital adrenal hyperplasia need to wear a medical alert identification bracelet or necklace. To alert medical professionals in case of an emergency, the bracelet or necklace should read: “adrenal insufficiency, requires hydrocortisone.” Adults or parents also need to learn how to give an injection of hydrocortisone if there is an emergency.
Patients with classic congenital adrenal hyperplasia need to take medication daily for their entire lives. If a patient stops taking his or her medication, symptoms will return.
The body makes different amounts of cortisol at different times in life, so sometimes a patient’s dose of medication may be too high or too low. Taking too much medication to replace cortisol can cause symptoms of Cushing’s syndrome. These include:
- Weight gain
- Slowed growth
- Stretch marks on the skin
- Rounded face
- High blood pressure
- Bone loss
- High blood sugar
It is important to alert the health care provider if these symptoms appear so that he or she can adjust the medication dose.
Nonclassic congenital adrenal hyperplasia
People with nonclassic congenital adrenal hyperplasia may not need treatment if they do not have symptoms. Individuals with symptoms are given low doses of the same cortisol replacing medication taken by people with classic congenital adrenal hyperplasia.
Symptoms of nonclassic congenital adrenal hyperplasia that signal that the patient may need treatment are:
- Early puberty
- Excess body hair
- Irregular menstrual periods (females)
- Infertility
It may be possible for patients with nonclassic congenital adrenal hyperplasia to stop medication as adults if their symptoms go away.
Surgery
Classic congenital adrenal hyperplasia
Girls who are born with ambiguous external genitalia usually have surgery. For example, surgery is necessary if changes to the genitals have affected urine flow. Surgery to make the genitals look more female also can be done.
The Endocrine Society, which supports hormonal research and clinical practice, recommends that this feminizing surgery be considered during infancy. If it is done, the group recommends choosing an experienced surgeon who practices in a center that sees many congenital adrenal hyperplasia cases 20.
The Congenital Adrenal Hyperplasia Research, Education & Support (CARES) Foundation strongly recommends delaying surgery until:
- The child is medically stable,
- Parents are fully informed of the risks and benefits, and
- A surgeon with expertise in this type of procedure is found.
Parents should also find a psychologist, social worker, or other mental health professional to support them in their decision making. It is important to find an experienced mental health provider whose expertise includes working with children who have congenital adrenal hyperplasia and their special needs 21.
Nonclassic congenital adrenal hyperplasia
Girls with nonclassic congenital adrenal hyperplasia have normal genitals, so they do not need surgery.
Congenital adrenal hypoplasia prognosis
As long as your child remains on hormone replacement therapy, she will most likely lead a healthy normal life. Women with congenital adrenal hyperplasia have the potential to function normally as females from a sexual standpoint. However, have a lower fertility rate than females without congenital adrenal hyperplasia.
Children who have congenital adrenal hyperplasia often are tall in early childhood, but ultimately are short in adulthood. Recent data suggest that patients born with congenital adrenal hyperplasia are about 10 cm shorter than their parentally based targets. Advanced bone age and central precocious puberty due to androgen excess causing early epiphyseal fusion are the primary factors. In addition, the treatment of congenital adrenal hyperplasia with glucocorticoids can suppress growth and diminish the final height. Experimental treatment with growth hormone and luteinizing hormone-releasing hormone analog (to hold off puberty) are reported to lead to an average height gain of 7.3 cm 22.
The influence of prenatal sex steroid exposure on personality is controversial. More consistent evidence regarding the effects of androgens comes from gendered play activities of young children.
Most children with congenital adrenal hyperplasia manifest normal neuropsychological development. Moreover, despite a tendency toward male gender role behavior and homoerotic fantasy, most girls with congenital adrenal hyperplasia identify as females and exhibit a heterosexual preference. Both male and female patients are fertile but have reduced fertility rates. This consequence is due to biological, psychological, social, and sexual factors.
Bone density is reported to be normal in most patients. The prevalence of metabolic abnormalities such as obesity, insulin resistance, dyslipidemia, and the polycystic ovarian syndrome has been reported to be high due to the diseases themselves or glucocorticoid treatment
This disease and treatment complications and long-term consequences are challenging for practitioners. Multiple subspecialty professionals should be involved in management. Gene therapy shows the potential for a congenital adrenal hyperplasia cure.
Adrenal hypoplasia causes
Adrenal hypoplasia can be due to adrenal hypoplasia congenita (congenital adrenal hypoplasia). Affected patients present with adrenocortical insufficiency in infancy and childhood. Research has identified four forms of congenital adrenal hypoplasia 1:
- The X-linked form is the result of a mutation or deletion of the nuclear receptor protein DAX-1 gene (also called the NROB1 gene) located on the X-chromosome 23. NROB1 gene encodes for this DAX-1 protein, which is thought to be important in the development and function of the hypothalamic-pituitary-adrenal axis. This form is usually associated with hypogonadotropic hypogonadism 24. There have also been suggestions that this chromosomal aberrancy could be a part of a contiguous chromosomal deletion of a large segment on the X-chromosome carrying the NROB1 gene. Such a large deletion can additionally result in Duchenne muscular dystrophy and glycerol kinase deficiency.
- The autosomal recessive form involves a gene that codes for steroidogenic factor 1 (SF-1). This gene is on chromosome 9q33 25. This form is also associated with hypogonadotropic hypogonadism. Another discovered form of the disease is associated with metaphysical dysplasia, intrauterine growth retardation, and genital abnormalities.
- IMAGE syndrome (i.e, intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, genital anomalies), SERKAL syndrome (rare autosomal form) 26. Few other rare syndromes and mutations can lead to adrenal hypoplasia and aplasia.
- Autosomal recessive ACTH resistance syndrome, such as triple-A syndrome and familial glucocorticoid deficiency present with ACTH insensitivity.
Secondary adrenal hypoplasia can arise from pituitary or hypothalamic dysfunction. These include transcription factor defects in pituitary development, pituitary adenomas, POMC defect, and convertase 1 (PCSK1) enzyme defect. Other causes of secondary adrenal hypoplasia are isolated ACTH deficiency, diseases of pituitary development, and central nervous system developmental anomalies. Adrenal hypoplasia can occur due to chronic exogenous glucocorticoid treatment. The resultant reduced ACTH action leads to adrenal hypofunction and hypoplasia due to a low stimulus 3.
Adrenal hypoplasia symptoms
The most difficult aspect of adrenal hypoplasia is clinical suspicion because signs and symptoms can be insidious and subtle.
Adrenal hypoplasia clinical presentation includes:
- Congenital adrenal hypoplasia most commonly presents in the neonatal period but may not become apparent until later in childhood.
- Patients often present in crisis with dehydration, hyponatremia, hyperkalemia, hypotension, or hypoglycemia.
- Patients with adrenal hypoplasia secondary to intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, genital anomalies (IMAGE syndrome) association have a history of intrauterine growth retardation. Males have genital abnormalities.
Physical examination
- Patients may demonstrate hyperpigmentation from increased serum concentrations of adrenocorticotropic hormone (ACTH).
- Signs of dehydration are often present.
- Hypotension and symptoms of neuroglycopenia may be present.
- Testes are undescended in many patients; micropenis may be seen in subjects with hypogonadotropic hypogonadism. Hypospadias or cryptorchidism may be seen in patients with IMAGe association.
- Hearing loss may be an associated finding 27.
A case study by Karsli et al 28 reported two cases of adrenal hypoplasia presenting with chronic respiratory distress.
Adrenal insufficiency
The hallmark of adrenal hypoplasia is adrenal insufficiency, which is a decrease in adrenal gland function. Patients often present in infancy or childhood, but there are reports of adult-onset cases. Females are mostly carriers and are generally unaffected. Undiagnosed patients usually present with adrenal crisis, but adrenal crisis can also occur in patients who are taking glucocorticoid replacement therapy. Patients usually present early in infancy or childhood with the salt-wasting crisis. Non-specific symptoms of adrenal insufficiency like feeling unwell, nausea, vomiting, lethargy, fatigue, anorexia, feeding difficulties, poor weight gain, and abdominal pain are present early in life or slightly later. The patient may have a fever, convulsions, and altered consciousness. These varied symptoms can cause misdiagnosis in these children. There is hypoglycemia, hyperkalemia, hyponatremia, and metabolic acidosis. Patients have associated mineralocorticoid deficiency and can present with hypotension, hypovolaemia, dehydration, and shock.
Hypogonadotropic hypogonadism
Apart from adrenal insufficiency, affected males can lack male sex hormones, which can lead to hypogonadotropic hypogonadism, underdeveloped reproductive tissues, cryptorchidism, and infertility (azoospermia). Patients may have central precocious puberty.
- Short height can occur due to associated growth hormone deficiency.
- Adrenal hypoplasia can rarely present as respiratory symptoms, such as chronic respiratory distress.
- Triple-A syndrome causes esophageal achalasia, alacrima (absence of tears), and various types of central, peripheral, and autonomic neurological defects.
- Other unusual presentations include excessive pigmentation, hearing loss, and monosomy.
- Various other rare syndromes and mutations that can cause adrenal hypoplasia/aplasia can present with cardiac defects, central nervous system malformations, pulmonary hypoplasia, polydactyly, and other anomalies 29.
Adrenal hypoplasia diagnosis
Laboratory studies
When adrenal insufficiency is suspected, promptly obtain the following laboratory values:
- Electrolytes
- Blood sugar
- Serum adrenocorticotropic hormone (ACTH)
- Plasma-renin activity (PRA)
- Serum cortisol
- Aldosterone
- 17-hydroxyprogesterone
- High-resolution karyotype
When hyponatremia or hyperkalemia are found, a spot urine test or a 24-hour urine test for sodium, potassium, and creatinine (along with simultaneous serum sodium concentrations and creatinine concentrations) determine whether inappropriate natriuresis is occurring (fractional excretion of sodium >1% in the face of hyponatremia). This occurs with mineralocorticoid deficiency when renal function is otherwise normal. A plasma renin activity–to–aldosterone ratio of more than 30 is suggestive of inadequate mineralocorticoid production.
- In X-linked congenital adrenal hypoplasia with adrenal insufficiency, lab findings show electrolyte disturbances, including hyponatremia, hyperkalemia, hypoglycemia, and metabolic acidosis.
- Adrenal functions show a high ACTH level and low or baseline cortisol level.
- There is increased plasma renin activity, decreased aldosterone level, and no response to high dose Cosyntropin stimulation.
- The 17-OH-progesterone level is normal or low.
- Androgen secretion is also impaired, and there are low androstenedione and low testosterone levels.
- Growth hormone deficiency may be present. Gonadotropin-releasing hormone (GnRH) test may show impaired gonadotropin secretion.
Random serum cortisol concentrations must be interpreted within the context in which they were obtained. For example, in a healthy individual, an 8:00 am serum cortisol concentration higher than 10 mcg/dL makes adrenal insufficiency unlikely.
- A serum cortisol concentration less than 18 mcg/dL in a sick and stressed patient is highly suggestive of adrenal insufficiency.
- A serum cortisol concentration less than 18 mcg/dL in the presence of an elevated serum ACTH concentration and plasma renin activity confirms adrenal insufficiency. Serum cortisol less than 18 mcg/dL obtained 30-60 minutes following cosyntropin is confirmatory 30.
- These guidelines do not apply to premature infants and infants with low birth weight who have lower cortisol secretion and, most likely, decreased cortisol binding to carrier proteins 31. The diagnosis of adrenal insufficiency in premature infants remains problematic.
Measure a panel of adrenal cortical hormones either with or without prior cosyntropin stimulation to exclude the various forms of congenital adrenal hyperplasia.
A cosyntropin stimulation test can confirm the diagnosis of adrenocortical insufficiency.
- Controversy surrounds whether the best dose of cosyntropin is the standard dose (250 mcg for an adult) or the low dose (1 mcg or 0.5 mcg/m2), which some have advocated as more sensitive for central adrenal insufficiency. A meta-analysis suggested that the low dose cosyntropin stimulation test is superior, but the difference was small 32. This issue remains unresolved in the pediatric age group.
- Because dilution of cosyntropin to 1 mcg is cumbersome and prone to error, and because all the above doses are probably supraphysiologic, the author generally uses the standard dose or empirically adjusts the dose for patient size (25 mcg for an infant, 50 mcg for a young child, 100 mcg for an older child, and 250 mcg for an adolescent or adult).
- When a patient’s serum cortisol response to cosyntropin is subnormal but his or her serum ACTH level is not elevated, the possibility of central adrenal insufficiency should be considered. Other indications of pituitary dysfunction such as prior glucocorticoid exposure (suggesting a suppressed hypothalamic-pituitary-adrenal axis) or evidence of other pituitary dysfunction are helpful clues suggesting ACTH deficiency.
- In central adrenal insufficiency, 3 days of stimulation with ACTH produces a normal cortisol response, indicating intact adrenal glands and implying that the initial low cortisol response to cosyntropin was related to chronic ACTH deficiency. ACTH gel (ACTHar Gel) is administered at 25 U/m2 every 12 hours for 3 days. Plasma cortisol levels should increase to more than 40 mcg/dL in response. This procedure is now seldom performed
The standard ovine corticotropin-releasing hormone (CRH) stimulation test (1 mcg/kg over 1 min) may be helpful in the differential diagnosis of adrenal insufficiency.
- A lack of a 2-fold increase in serum ACTH concentration indicates pituitary dysfunction. A 2-fold or greater rise in ACTH levels without a concomitant rise in serum cortisol to more than 18-20 mcg/dL implies primary adrenal insufficiency 33.
- Ovine CRH is difficult to obtain, and this test is mainly done for research purposes.
In the most common form of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency, serum 17-hydroxyprogesterone is markedly elevated.
Consider adrenoleukodystrophy in older boys with evidence of adrenal insufficiency. Both males and females may be affected with autoimmune Addison disease, another important diagnostic consideration.
- Adrenal leukodystrophy is also X-linked and can be diagnosed by demonstrating elevated concentrations of very–long-chain fatty acids (>24 carbon) in serum.
- Autoimmune Addison disease is confirmed by the demonstration of antiadrenal antibodies in the serum.
Genetic screening
Precise genetic screening is advisable when adrenal hypoplasia is suspected. Identify the carrier status of women in the family 34.
Karyotype, fluorescent in situ hybridization (FISH) or microarray analysis may reveal the gene deletion involving DAX1.
Prenatal diagnosis is possible 35.
Imaging studies
Adrenal imaging, including CT and MRI, are necessary. Imaging may show small size adrenal glands. An ultrasound of adrenals may demonstrate small for age adrenal glands.
A detailed gastroenterological and neurological evaluation should take place.
Rarely, the patient may present with respiratory distress or other respiratory symptoms. Adrenal hypoplasia should be taken into account if a child is having respiratory symptoms along with electrolyte disturbances, and hypoglycemia. In congenital adrenal hypoplasia, due to DAX1 mutation, elevated 11-deoxycortisol levels have been noticed if checked very early in life.
Schirmer test is a recommended procedure in Triple-A syndrome 36.
Adrenal hypoplasia treatment
It is essential to differentiate between adrenal insufficiency due to primary adrenal hypoplasia or secondary adrenal hypoplasia because treatment gets tailored according to the deficient hormones. In primary adrenal hypoplasia, all the adrenal gland hormones that are synthesized by adrenals, i.e., cortisol, aldosterone, and sex steroids, are deficient. In secondary adrenal hypoplasia, aldosterone replacement is not necessary since it is under the regulation of the renin-angiotensin-aldosterone system. Only ACTH controlled hormones, i.e., cortisol and sex steroids, are decreased.
Emergency treatment of an adrenal crisis in adrenal hypoplasia should focus on correcting metabolic and electrolytic disturbances, including the correction of hypovolaemia and hypoglycemia. If the patient is hypotensive, aggressive fluid resuscitation, as well as, glucocorticoid treatment may be needed 29.
X-linked congenital adrenal hypoplasia gets treated with glucocorticoid, mineralocorticoid, and salt supplementation starting at two weeks of life. Symptoms usually resolve by two months of age, and ACTH and plasma renin activity return to normal. Management involves life-long glucocorticoid replacement therapy. According to guidelines, all adrenal hypoplasia patients who are receiving treatment should keep steroid emergency cards. Hydrocortisone is the preferred steroid as it has mineralocorticoid activity. However, if ordering a cosyntropin stimulation test to confirm the diagnosis, dexamethasone can be given before the test without any interference of results.
Growth hormone therapy (GnRH analogs) should start in patients with growth hormone deficiency.
Testosterone replacement is required when there is delayed puberty during adolescence to ensure normal development of secondary sexual characteristics and bone mineralization.
Medical care
- Patients with adrenal hypoplasia are generally hypovolemic and may be hypoglycemic; therefore, initial therapy should consist of intravenous normal saline and dextrose.
- If hypotensive, a bolus dose of 20 mL/kg of isotonic intravenous fluid over the first hour may be necessary to restore blood pressure. This can be repeated if the blood pressure remains low.
- Once samples for serum electrolytes, blood sugar, cortisol, 17-hydroxyprogesterone, and adrenocorticotropic hormone (ACTH) concentrations are obtained, treat the patient with glucocorticoids. This therapy is based on suspicion of adrenal insufficiency because it may be life preserving.
- A cosyntropin stimulation test confirms the diagnosis of adrenocortical insufficiency.
- Dexamethasone may be given prior to the cosyntropin without interfering with the results of the test because acute administration of dexamethasone does not interfere with the cortisol response or with the cortisol assay. Otherwise, hydrocortisone is preferable because of its mineralocorticoid activity.
Acute therapy
For a patient with suspected but unproved adrenal insufficiency, dexamethasone is best used to correct the glucocorticoid deficiency. This allows immediate procession to a cosyntropin stimulation test for confirming diagnosis. If a cosyntropin stimulation test is not planned, give stress doses of hydrocortisone (50-75 mg/m² or 1-2 mg/kg) intravenously as an initial dose and followed by 50-75 mg/m²/d intravenously in 4 divided doses. Hydrocortisone may be given intramuscularly if no intravenous access is available but works less quickly. Comparable stress doses of methylprednisolone are 10-15 mg/m2 and of dexamethasone 1-1.5 mg/m² intravenously or intramuscularly.
Methylprednisolone and dexamethasone have negligible mineralocorticoid effects. Therefore, if the patient is hypovolemic, hyponatremic, or hyperkalemic, large doses of hydrocortisone (even double or triple the stress doses mentioned above) are preferred. At the present time, no parenteral form of mineralocorticoid is available in the United States. If the patient has good gastrointestinal function, fludrocortisone (0.1-0.2 mg orally) may be given to replace aldosterone deficiency.
In hypotensive patients, normal saline (ie, 0.9% NaCl) must be administered by rapid intravenous infusion over the first hour followed by a continuous infusion. A reasonable amount to restore intravascular volume is 450 mL/m² or 20 mL/kg of normal saline intravenously over the first hour, followed by 3200 mL/m²/day or 200 mL/kg/100 kcal of estimated resting energy expenditure as normal saline or 0.45% NaCl in subsequent hours. Dextrose must also be provided. If the patient is hypoglycemic, 2-4 mL/kg of D10W corrects it. D5W must be provided to prevent further hypoglycemia or to prevent hypoglycemia from occurring if the patient is not hypoglycemic. Potassium is generally not needed in the acute situation, especially because patients with adrenal hypoplasia are often hyperkalemic.
Chronic medical therapy
In growing children with adrenal insufficiency, chronic glucocorticoid replacement must be balanced to prevent symptoms of adrenal insufficiency, while still allowing the child to grow at a normal rate and prevent symptoms of glucocorticoid excess. The dose must be tailored to each patient but generally runs in the range of 7-20 mg/m²/d of hydrocortisone orally in 2-3 divided doses. Hydrocortisone is available as tablets of 5 mg, 10 mg, and 20 mg. Hydrocortisone is recommended in the pediatric population because of its lower potency, which permits easier titration of appropriate doses. In large patients, prednisone or even dexamethasone may be substituted.
- The estimated equivalency is 1 mg prednisone = 4 mg hydrocortisone and 1 mg dexamethasone = 50 mg hydrocortisone, but this varies from patient to patient.
Patients with congenital adrenal hypoplasia also have mineralocorticoid deficiency and, therefore, must be provided with fludrocortisone (0.1-0.2 mg/d). Provide infants with NaCl (2-5 g/day oral) to counteract salt wasting. The dose of glucocorticoid is adjusted clinically (absence of symptoms of glucocorticoid deficiency or excess and normal growth).
In the author’s experience, plasma adrenocorticotropic hormone (ACTH) concentrations are of little help in adjusting doses of glucocorticoid in patients with primary adrenal insufficiency. Symptoms of salt craving, blood pressure, plasma renin activity, and electrolytes are helpful in adjusting the dose of fludrocortisone. Salt craving and an elevated plasma renin activity suggest the need for a larger dose of fludrocortisone, whereas elevated blood pressure or suppressed plasma renin activity suggests the need for a lower dose of fludrocortisone.
Stress and illness
One of the important physiological responses to stress is an increase in cortisol production mediated by ACTH. Patients with adrenal insufficiency, of whatever etiology, are unable to mount this response and must be provided with stress doses of glucocorticoids. In patients with minor illness (fever < 100.4 °F [<38°C]) administer at least double the dose of hydrocortisone. In patients with more severe illness (fever >100.4 °F [>38°C]), administer triple the dose of glucocorticoids. If the patient is vomiting or listless, give parenteral glucocorticoids (hydrocortisone 50-75 mg/m² intramuscularly or intravenously or equivalent of methylprednisolone or dexamethasone).
Because hydrocortisone succinate has a short duration of action, the dose must be repeated every 6-8 hours until the patient is well. Cortisone acetate and hydrocortisone acetate both have a longer duration of action (up to 24 h) but are often difficult to obtain in the United States. All patients with adrenal insufficiency must have injectable glucocorticoid available, and the caretaker must be instructed in its use and importance.
Hydrocortisone suppositories may be tried in patients or families who cannot administer injectable glucocorticoids. However, absorption is less predictable.
No contraindications to glucocorticoid or mineralocorticoid replacement are recognized when it is needed, and few adverse drug-to-drug interactions occur.
Patients on physiologic replacement doses of glucocorticoids may receive live virus immunizations.
Surgical care
Surgery is not necessary in the management of congenital adrenal hypoplasia; however, a patient requiring surgery must be covered with stress doses of glucocorticoids during the perioperative period.
The following recommendations are empiric rather than evidence-based:
- Administer 50-75 mg/m² hydrocortisone intramuscularly or intravenously on call prior to surgery.
- During the procedure, treat the patient with additional hydrocortisone. This may be accomplished with either a hydrocortisone drip of 2-4 mg/m2/h, or as an additional bolus of 10-25 mg/m2 intravenously every 6 hours throughout the procedure.
- Continue hydrocortisone in the immediate postoperative period.
- On the second and third postoperative day, the dose of hydrocortisone can be decreased by 50% each day, to a minimum of the patient’s usual daily requirement, provided no complications exist and the patient is recovering well.
- By the fourth postoperative day, the usual daily dose of steroids may be resumed if the patient is recovering well. If complications occur, stress doses of glucocorticoids must be continued.
- Fludrocortisone may be held on the day of surgery and while the patient is receiving stress doses of hydrocortisone because this high dose should provide ample mineralocorticoid effect.
- If the patient is unable to take fludrocortisone by mouth in the postoperative period, stress doses of hydrocortisone may be continued for a longer period to provide adequate mineralocorticoid activity.
Diet
- Patients should not be on a sodium-restricted or fluid-restricted diet.
- Patients should have ample access to salt since patients are deficient in aldosterone secretion and, therefore, are generally salt wasters.
- Monitor and restrict caloric intake if excess weight gain occurs on therapy because glucocorticoids stimulate appetite and weight gain.
Adrenal hypoplasia prognosis
Adrenal insufficiency in adrenal hypoplasia is a potentially life-threatening condition. Prognosis of untreated congenital adrenal hypoplasia is poor if the disorder is unrecognized or untreated, and death is a common outcome. The majority of the patients (35%) having adrenal insufficiency dies from cardiovascular disease, while 15% die from infections 29. With proper treatment and compliance, patients can live a normal life span without limitations.
Hypogonadotropic hypogonadism is nearly certain to develop secondary to DAX1 mutations or deletions. Infertility is common.
If the gene for muscle dystrophin is absent as a contiguous gene deletion, Duchenne muscular dystrophy results, and the prognosis is poor.
References- Saleem F, Baradhi KM. Adrenal Hypoplasia. [Updated 2020 Jun 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549779
- Kempná P, Flück CE. Adrenal gland development and defects. Best Pract Res Clin Endocrinol Metab. 2008 Feb. 22(1):77-93.
- Kyritsi EM, Sertedaki A, Charmandari E, Chrousos GP. Familial or Sporadic Adrenal Hypoplasia Syndromes. In: Feingold KR, Anawalt B, Boyce A, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000.
- Kyritsi EM, Sertedaki A, Charmandari E, et al. Familial or Sporadic Adrenal Hypoplasia Syndromes. [Updated 2018 Oct 22]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279132
- NIH Clinical Center. Patient education: Facts about CAH. https://www.cc.nih.gov/ccc/patient_education/pepubs/cah.pdf
- Texas Department of State Health Services. (2001). Congenital adrenal hyperplasia: A handbook for parents. http://www.dshs.texas.gov/newborn/hand_cah.shtm
- Rama Chandran S, Loh LM. The importance and implications of preconception genetic testing for accurate fetal risk estimation in 21-hydroxylase congenital adrenal hyperplasia (CAH). Gynecol. Endocrinol. 2019 Jan;35(1):28-31.
- National Adrenal Diseases Foundation. CAH: Diagnosis. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/
- National Adrenal Diseases Foundation. CAH: Nonclassical. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/
- Burdea L, Mendez MD. 21 Hydroxylase Deficiency. [Updated 2020 Mar 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK493164
- Doleschall M, Török D, Mészáros K, Luczay A, Halász Z, Németh K, Szücs N, Kiss R, Tőke J, Sólyom J, Fekete G, Patócs A, Igaz P, Tóth M. [Steroid 21-hydroxylase deficiency, the most frequent cause of congenital adrenal hyperplasia]. Orv Hetil. 2018 Feb;159(7):269-277.
- Medscape Reference. (2011). C-11 Hydroxylase deficiency. http://emedicine.medscape.com/article/117012-overview
- National Adrenal Diseases Foundation. (n.d.). Adrenal diseases—Congenital adrenal hyperplasia (CAH): The facts you need to know. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/
- Screening, Technology and Research in Genetics (STAR-G) Project. Genetic fact sheet for parents: Congenital adrenal hyperplasia. http://www.newbornscreening.info/Parents/otherdisorders/CAH.html
- Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, Meyer-Bahlburg HFL, Miller WL, Murad MH, Oberfield SE, White PC. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018 Nov 01;103(11):4043-4088.
- National Library of Medicine, Medline Plus. Congenital adrenal hyperplasia. https://medlineplus.gov/ency/article/000411.htm
- National Library of Medicine, MedLine Plus. Congenital adrenal hyperplasia. https://medlineplus.gov/ency/article/000411.htm
- The Hormone Foundation. (2010). Patient guide to congenital adrenal hyperplasia. http://www.hormone.org/diseases-and-conditions/adrenal/congenital-adrenal-hyperplasia
- National Adrenal Diseases Foundation. CAH: Standard treatment. http://www.nadf.us/adrenal-diseases/congenital-adrenal-hyperplasia-cah/
- The Endocrine Society. (2010). Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society clinical practice guideline. Journal of Clinical Endocrinology & Metabolism, 95(9), 4133–4160.
- Congenital Adrenal Hyperplasia Research Education & Support Foundation. (2007). Surgery considerations for girls with classical CAH. http://www.caresfoundation.org/dosing/surgery/
- Bachelot A, Grouthier V, Courtillot C, Dulon J, Touraine P. MANAGEMENT OF ENDOCRINE DISEASE: Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: update on the management of adult patients and prenatal treatment. Eur. J. Endocrinol. 2017 Apr;176(4):R167-R181.
- Zhang Z, Feng Y, Ye D, Li CJ, Dong FQ, Tong Y. Clinical and molecular genetic analysis of a Chinese family with congenital X-linked adrenal hypoplasia caused by novel mutation 1268delA in the DAX-1 gene. J Zhejiang Univ Sci B. 2015 Nov. 16 (11):963-8.
- Loureiro M, Reis F, Robalo B, Pereira C, Sampaio L. Adrenal Hypoplasia Congenita: A Rare Cause of Primary Adrenal Insufficiency and Hypogonadotropic Hypogonadism. Pediatr Rep. 2015 Sep 28. 7 (3):5936.
- Achermann JC, Ito M, Ito M, et al. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999 Jun. 22(2):125-6.
- Bergada I, Del Rey G, Lapunzina P, et al. Familial occurrence of the IMAGe association: additional clinical variants and a proposed mode of inheritance. J Clin Endocrinol Metab. 2005 Jun. 90(6):3186-90.
- Lin L, Gu WX, Ozisik G, et al. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (SF1/Ad4BP, NR5A1) in children and adults with primary adrenal failure: ten years” experience. J Clin Endocrinol Metab. 2006 May 9.
- Karsli T, Sutter J, Shekhawat PS. X-linked Adrenal Hypoplasia Congenita Due to NR0B1 (DAX1) Deficiency Presenting as Severe Respiratory Distress in Near Term Infants. Pediatr Neonatol. 2016 Oct. 57 (5):444-445.
- Dineen R, Thompson CJ, Sherlock M. Adrenal crisis: prevention and management in adult patients. Ther Adv Endocrinol Metab. 2019;10:2042018819848218
- Lashansky G, Saenger P, Fishman K, et al. Normative data for adrenal steroidogenesis in a healthy pediatric population: age- and sex-related changes after adrenocorticotropin stimulation. J Clin Endocrinol Metab. 1991 Sep. 73(3):674-86.
- Heckmann M, Hartmann MF, Kampschulte B, et al. Cortisol production rates in preterm infants in relation to growth and illness: a noninvasive prospective study using gas chromatography-mass spectrometry. J Clin Endocrinol Metab. 2005 Oct. 90(10):5737-42.
- Kazlauskaite R, Evans AT, Villabona CV, et al. Corticotropin tests for hypothalamic-pituitary- adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab. 2008 Nov. 93(11):4245-53.
- Schurmeyer TH, Avgerinos PC, Gold PW, et al. Human corticotropin-releasing factor in man: pharmacokinetic properties and dose-response of plasma adrenocorticotropin and cortisol secretion. J Clin Endocrinol Metab. 1984 Dec. 59(6):1103-8.
- Loureiro M, Reis F, Robalo B, Pereira C, Sampaio L. Adrenal Hypoplasia Congenita: A Rare Cause of Primary Adrenal Insufficiency and Hypogonadotropic Hypogonadism. Pediatr Rep. 2015 Sep 28;7(3):5936.
- Peter M, Partsch CJ, Dorr HG, Sippell WG. Prenatal diagnosis of congenital adrenal hypoplasia. Horm Res. 1996 Jul. 46(1):41-5.
- Shima H, Yatsuga S, Nakamura A, Sano S, Sasaki T, Katsumata N, Suzuki E, Hata K, Nakabayashi K, Momozawa Y, Kubo M, Okamura K, Kure S, Matsubara Y, Ogata T, Narumi S, Fukami M. NR0B1 Frameshift Mutation in a Boy with Idiopathic Central Precocious Puberty. Sex Dev. 2016;10(4):205-209.





{kind=link}