Copper deficiency
Copper deficiency or frank dietary copper deficiency is less common in healthy individuals because copper is a ubiquitous element and is present in almost all foods 1, 2, 3. Copper deficiency in the United States is believed to be relatively rare but has been described in the setting of zinc supplementation 4, myelodysplastic syndrome 5, use of total parenteral nutrition (TPN) 6 and chronic tube feeding 7 especially via jejunostomy 8 and in various malabsorption syndromes such as cystic fibrosis, celiac disease and in people who’ve had gastrectomy and roux-en-Y gastric bypass surgery for obesity and in infants exclusively fed cow-milk formulas 9. A rare X-linked genetic condition called Menkes kinky hair syndrome or Menkes disease is caused by a defect in the ATP7A gene 10, 11. The defect makes it hard for the body to properly distribute (transport) copper throughout the body. As a result, the brain and other parts of the body do not get enough copper, while it builds up in the small intestine and kidneys of affected infants. Menkes disease, which affects primarily males, is associated with seizures, delayed development, abnormal artery development in the brain, and unusual gray brittle kinky hair 11.
Based on studies in animals and humans, the effects of copper deficiency include anemia, hypopigmentation (a low amount of melanin in your skin), high blood cholesterol (hypercholesterolemia), connective tissue disorders, osteoporosis (condition in which bones become weak and brittle) and other bone defects, abnormal lipid metabolism, a lack of coordination or unsteadiness (ataxia), and increased risk of infection 12, 13, 14. Features of copper deficiency include hematologic abnormalities (anemia, leukopenia, neutropenia, and thrombocytopenia) and myeloneuropathy (spinal cord and peripheral nerves damage in the lower limbs); the latter is a rarer and often unrecognized complication of copper deficiency 15, 2, 9. A low copper level can affect the structure of bone, skin, hair, and blood vessels, and interfere with nerve function. Most importantly, the neurologic complications of copper deficiency may be irreversible, making early diagnosis and prompt treatment essential to successful outcomes.
One of the most common clinical signs of copper deficiency is an anemia that is unresponsive to iron therapy but is corrected by copper supplementation 16. It was hypothesized that this copper-deficiency anemia could result from defective iron mobilization due to decreased ceruloplasmin (CP) activity, yet individuals with inherited aceruloplasminemia (a rare inherited autosomal recessive genetic disorder in which a mutation in the ceruloplasmin (CP) gene leads to the absence or dysfunction of ceruloplasmin) do not always develop overt anemia 17. Ceruloplasmin (CP) plays a role in iron metabolism and carries more than 95% of the total copper in healthy human plasma 18. In copper-deficient pig, intestinal iron absorption is impaired but iron distribution among tissues or organs is normal 19, 20, 21. Low serum iron from reduced absorption is an unlikely cause of this anemia since intravenous provision of iron did not correct it 16. An alternative hypothesis is that copper-deficiency anemia is caused principally by impaired hemoglobin production and red blood cell proliferation, and a shortened red blood cell lifespan. These physiological processes are thus likely to require copper. Copper deficiency may also lead to neutropenia, which can increase susceptibility to infection. Copper depletion studies demonstrated that low copper might affect erythroid and myeloid cell lineages, supporting a role for copper in the regulation of blood cell proliferation and maturation 22, 23. More research is clearly needed to further define the mechanisms underlying copper deficiency-induced anemia and neutropenia 22, 24. Furthermore, osteoporosis and other abnormalities of bone development have been described in copper-deficient, low-birth-weight infants and young children. Less common features of copper deficiency may include impaired growth, depigmentation, and development of neurological pathologies 25, 26.
Copper is an essential trace mineral present in all body tissues that is essential for adequate use of iron and zinc, two other essential transition metals by your body 27. Copper is also a cofactor of proteins and enzymes called cuproenzymes (copper-dependent enzymes) that are involved in regulation of iron metabolism, formation of connective tissue, energy (ATP) production at the cellular level, oxygen transportation, formation of blood cells such as red blood cells, platelets, and white blood cells (hematopoiesis), the formation of new blood vessels (angiogenesis), neurohormone homeostasis, the production of melanin (the pigment that produces skin color), the function of the nervous system, brain development, aids in iron absorption, regulation of gene expression, immune system functioning, mitochondrial electron transport and free radical scavenging (redox reactions or oxidation-reduction reactions) 28, 29, 12, 16, 30. Furthermore, defense against oxidative damage depends mainly on the copper-containing superoxide dismutases 31, 32. Copper also helps keep the blood vessels, nerves, immune system, and bones healthy. Copper is vital to the normal function of the hematologic, vascular, skeletal, antioxidant, and neurologic systems 9, 16, 30. Copper is therefore essential for living cells. Both the accumulation of copper and copper deficiency are associated with brain dysfunction.
More recently, new evidence shows that copper is involved in the pathogenesis of neurodegenerative disorders. Copper is an essential transition metal. Copper participates in critical cuproenzymes preventing neurodegeneration and regulating neurotransmission. Via the ferroxidase activity of ceruloplasmin, copper is a metabolic regulator of the contents of iron in the CNS (central nervous system). An excess of free copper is directly involved to neurodegeneration. Wilson’s disease, Alzheimer’s disease and Parkinson’s disease are major neurodegenerative disorders associated with copper dyshomeostasis 33. Wilson disease is a rare inherited disorder that prevents your body from getting rid of extra copper, which can lead to excess storage of copper in the liver, brain, and other organs 34, 35. Copper excess (toxicity) can also occur when a person is exposed to and absorbs large amounts over a short period of time (acute exposure) or various amounts over a long period (chronic exposure).
Copper is found in many foods including nuts, chocolate, mushrooms, shellfish, whole grains, dried fruits, and liver. Drinking water may acquire copper as it flows through copper pipes, and food may acquire it when people cook or serve food in copper dishes. Normally, the body absorbs copper from food or liquids in the intestines, converts it to a non-toxic form by binding it to a protein, and transports it to the liver. The liver stores some of the copper and binds most of the rest to another protein called apoceruloplasmin to produce the enzyme ceruloplasmin. About 95% of the copper in the blood is bound to ceruloplasmin, and most of the rest is bound to other proteins such as albumin. Only a small amount is normally present in the blood in a free (unbound) state. The liver eliminates excess copper into the bile and it is removed from the body in the stool. Some copper is also eliminated in the urine.
The average daily intake of copper is approximately 1,400 mcg/day for men and 1,100 mcg/day for women, coming mainly from seeds, grains, shellfish, nuts, beans and liver 36, 12, 3, 37, 38, 39. The current recommended dietary intake of copper in the USA is 900 mcg/day 40, 41.
Copper is absorbed mainly from the duodenum and proximal jejunum, and partly from the stomach 42. Low dietary copper intake reduces immune response, which is not restored to normal levels even after several weeks of a high copper intake 43. Only small amounts of copper are typically stored in your body, and the average adult has a total body content of 50–120 mg copper 12, 3. Almost two-thirds of the body’s copper is located in the bone and muscle 12, 40.
Most copper is excreted in bile, and a small amount is excreted in urine. Total fecal losses of copper of biliary origin and nonabsorbed dietary copper are about 1 mg/day 12, 3. Copper levels in your body are homeostatically maintained by copper absorption from the intestine and copper release by the liver into bile to provide protection from copper deficiency and copper toxicity 40.
Copper status is not routinely assessed in clinical practice, and no biomarkers that accurately and reliably assess copper status have been identified 3. Human studies typically measure copper and cuproenzyme activity in plasma and blood cells because individuals with known copper deficiency often have low blood levels of copper and ceruloplasmin (an abundant cuproenzyme) 3. Serum copper and ceruloplasmin (CP) levels may fall to 30% of normal in cases of severe copper deficiency 16. However, plasma ceruloplasmin (CP) and copper levels can be influenced by other factors, such as estrogen status, pregnancy, infection, inflammation, and some cancers 3. Low serum copper level or hypocupremia is also observed in genetic disorders of copper metabolism, including Wilson disease and aceruloplasminemia (a rare inherited autosomal recessive genetic disorder in which a mutation in the ceruloplasmin (CP) gene leads to the absence or dysfunction of ceruloplasmin); however, neither disorder has been linked to low dietary copper intake 16.
Normal serum concentrations are 10–25 micromol/L (63.5–158.9 mcg/dL) for copper and 180–400 mg/L for ceruloplasmin (CP) 44.
The treatment for copper deficiency is not yet developed and depends on the underlying cause of the copper deficiency 15. However, it is easy to consume food containing high amounts of copper as a copper supplement. Commercially available pure cocoa contains approximately 4 mg copper per 100 g and is easy to administer to tube‐feeding patients because it is in powder form. Experts recommended other copper‐rich foods such as nuts, shellfish, or liver to patients with diabetes mellitus because almost all patients add sugar to suppress the bitterness of cocoa. Experts also used other copper‐rich foods for patients who could not continue oral cocoa intake due to diarrhea because cocoa contains fibers 45. For individuals with Menkes kinky hair syndrome or Menkes disease, there is no cure at this time, but treatment with parenteral copper histidinate (CuHis) can increase survival and lessen the neurological symptoms if initiated early, within approximately 28 days following birth 46.
What is copper?
Copper is a cofactor of proteins and enzymes (called cuproenzymes) involved in fundamental mechanisms, such as energy generation, oxygen transportation, hematopoiesis, cellular metabolism and signal transduction 28. Copper is therefore essential for living cells. Its metabolism is linked to the metabolism of iron and zinc, two other essential transition metals 47. The bulk of evidence that copper is involved in the pathogenesis of neurodegenerative disorders is now huge. Copper is an essential transition metal. Copper participates in critical cuproenzymes preventing neurodegeneration and regulating neurotransmission. Via the ferroxidase activity of ceruloplasmin, copper is a metabolic regulator of the contents of iron in the CNS (central nervous system). An excess of free copper is directly involved to neurodegeneration. Wilson’s disease, Alzheimer’s disease and Parkinson’s disease are major neurodegenerative disorders associated with copper dyshomeostasis 33.
Metabolism of copper
In the human body, most of the copper (Cu) is present as Cu+ (cuprous) and oxidized Cu2+ (cupric) compounds 33. Copper is therefore an intermediary for electron transfer in redox reactions. The oxidation states, Cu3+ and Cu4+, are uncommon 48. Copper is mainly absorbed in the duodenum and proximal jejunum, with a little bit of absorption occurring in the stomach and the distal portion of the small intestine 49. The human copper transport protein 1 (hCTR1), located at the level of enterocytes, transfers the ion following the reduction of dietary Cu2+ into Cu+. In hepatocytes, copper binds to metallothioneins, to reduced glutathione or to one of the copper chaperones regulating the traffic of intracellular copper (chaperone for superoxide dismutase 1; SOD1, which is the sole cytosolic cuproenzyme; COX17: chaperone for cytochrome C oxygenase; ATOX1 antioxidant-1: chaperone for the ATPases, ATP7A and ATP7B). The group of transmembrane copper transporters includes CTR1, ATP7A and ATP7B. ATP7A (expressed in the placenta, gut and nervous system) and ATP7B (expressed in the hepatocytes, where it exports copper into the bile and provides copper to nascent ceruloplasmin, and in the nervous system) are linked to the enzyme, tyrosinase, and the ceruloplasmin, respectively. In blood, about 65%–90% of the copper Cu2+ is bound to ceruloplasmin. The remaining 10–35 percent participate in exchanges with albumin, transcuprein, alpha 2 macroglobulin, and low-molecular-weight compounds 50.
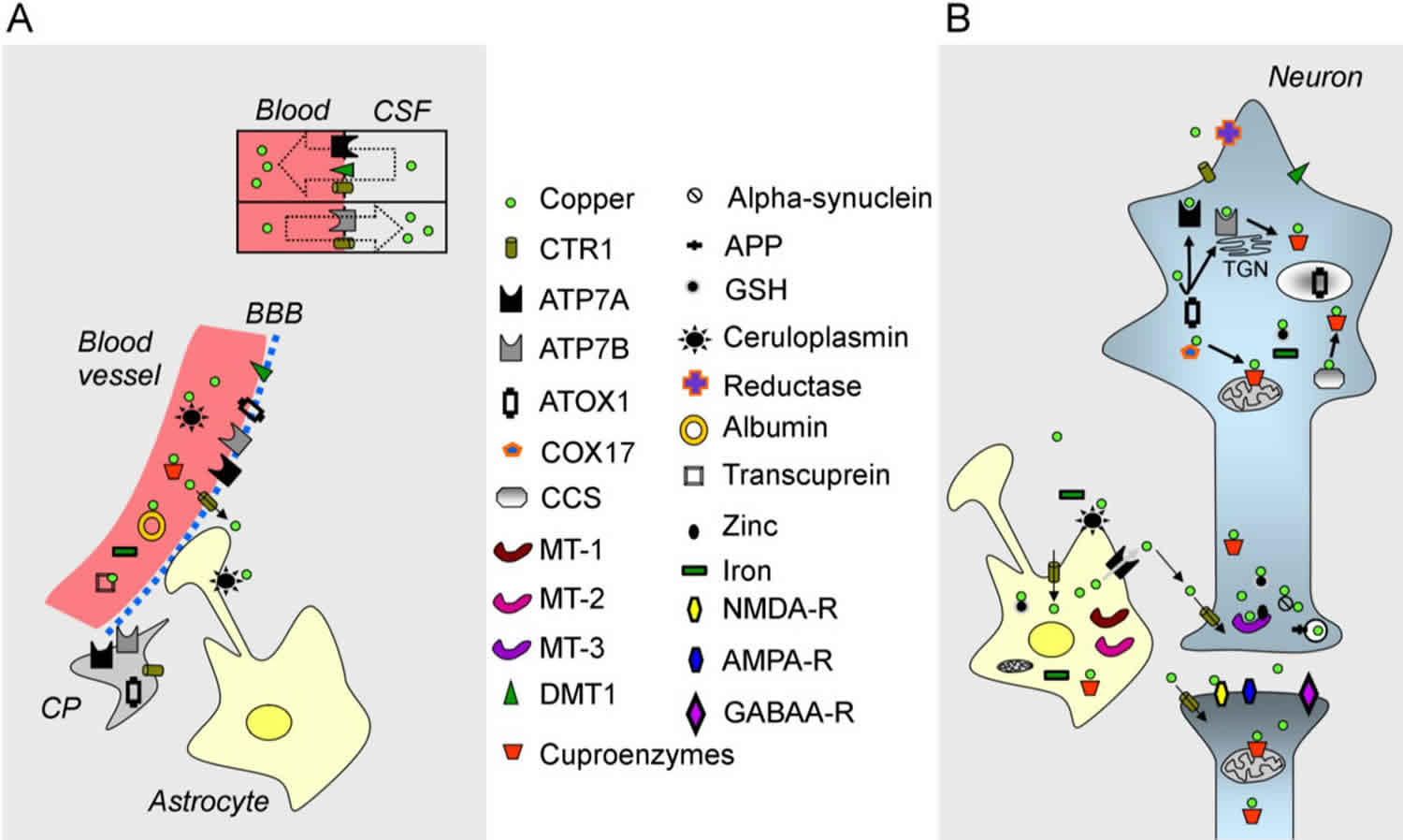
In terms of storage, the total amount of copper in an adult is estimated to be about 90–110 mg. The organs with the highest concentrations are the liver, brain, kidney and heart 51. Bones and skeletal muscles contain about 47% and 27% of the copper, respectively 52. Both the liver and brain contain about 8%–11% of the total body copper. Figure 1 illustrates the metabolism of copper in the brain. Brain concentrations range from 3.1 to 5.1 mg/g wet weight 53. Within the brain, the distribution of copper is heterogeneous. Concentrations are higher in the hippocampus, substantia nigra and locus coeruleus 48. Glial cells are enriched in copper as compared to neurons 48. The concentrations of copper in the synaptic cleft range from 0.2 to 1.7 µM. Some studies have shown concentrations higher than 200 µM 54. Concentrations in cerebrospinal fluid (CSF) are 100-fold lower as compared to plasmatic levels, and intra-neuronal concentrations are kept very low 55. Choroid plexus could serve as a storage compartment in the vicinity of the brain.
Copper is excreted endogenously in the saliva, the gastric fluids and intestinal liquids. Copper is excreted from the body either in a non-absorbed form or via the bile. The estimated amount of copper in feces is 1–1.5 mg/day. The quantities lost in urine, saliva and perspiration are even smaller.
Figure 1. Copper metabolism in the brain
Footnote:
(A) A blood vessel (in red on the left), the blood-brain barrier (BBB), the blood-CSF barrier (shown by a rectangle with two compartments), a choroid plexus (CP) and an astrocyte are illustrated;
(B) A neuron, a synapse and an astrocyte are shown. Copper is represented by a green circle. At the blood-CSF barrier, CTR1 (copper transporter 1), DMT1 (divalent metal transporter) and the ATPase, ATP7A, transport copper towards the blood, whereas the ATPase, ATP7B, and CTR1 transport copper in the opposite direction. Organelles and proteins involved in the cellular regulation of copper are represented. CTR1 is the main transporter for transferring copper within cells. Chaperones (ATOX1, COX17, CCS) deliver copper to the ATPases, ATP7A/ATP7B, and to cuproenzymes, including in the mitochondria. Metallothioneins (MT-1, MT-2, MT-3) exert a function as a buffer. Copper is also found in secretory granules.
Abbreviations: APP = amyloid precursor protein; GSH= glutathione; NMDA-R = NMDA receptor; AMPA-R = AMPA receptor; GABAA-R = GABAA receptor; TGN = trans-Golgi network.
[Source 33 ]Free Copper
The fact that cells contain one free copper ion or less highlights the tight regulation 56. The redox capacities of free copper and its ability to initiate the production of free radicals are two important features. Free copper is a catalyst of the Fenton reaction (Cu+ and H2O2 generate Cu2+ + OH− + OH). The hydrogen peroxide is transformed into the very reactive hydroxyl radical, which combines with nucleic acids, proteins and lipids. An increase in free copper is potentially harmful for brain circuits, despite the presence of metallothioneins and chaperones (see below).
Ceruloplasmin
Ceruloplasmin is a multicopper-containing protein mainly synthesized by the liver. Although the rate of synthesis is not influenced by copper intake, ceruloplasmin lacking bound copper has a shorter half-life 57. Ceruloplasmin plays important functions, acting as an iron oxidase, an amine oxidase, an antioxidant and a glutathione peroxidase 58. As a multicopper oxidase, ceruloplasmin reduces dioxygen, O2, to two water molecules. Ceruloplasmin is a scavenger of reactive oxygen species (ROS). It can be considered that the genuine link between copper metabolism and iron metabolism is mediated by ceruloplasmin. Its soluble form controls the oxidation of iron to be included into transferrin. A deficit in copper reduces the ferroxidase activity of ceruloplasmin (Fe2+ to Fe3+). Dietary and recycled iron are in the Fe2+ oxidation state, but iron is transported in serum by transferrin only as Fe3+ after its export by ferroportin 47. Iron itself contributes to the formation of reactive oxygen species (ROS). A deficit of dietary copper leads not only to an accumulation of iron in the liver, but also to an impaired distribution within the spinal cord (see zinc-induced myeloneuropathy in the next section). In the central nervous system (CNS), a glycosylphosphatidylinositol-linked ceruloplasmin bound to the cell membranes is the major isoform of this protein 59. Astrocytes can synthesize their own ceruloplasmin. This glial ceruloplasmin controls also the process of iron oxidation, which allows the clearance of iron from the CNS (central nervous system) 60. Overall, functional ceruloplasmin promotes the synthesis of proteins involved in iron efflux. The maintenance of the iron balance in the brain is thus closely linked to the metabolism of copper.
Copper and Synapses in the CNS
There is evidence that copper is enriched in synaptic levels and could even play a signaling role 61. Copper is found in secretary granules containing SOD3, in constitutive vesicles and in endosomes. Copper in the secretory pathway is released in a calcium-dependent manner. Copper interacts with glutamatergic and GABAergic synapses and modulates voltage-gated calcium channels 61. Stimulation of the NMDA receptor (NMDA-R) evokes the release of copper in hippocampal neurons and is associated with a repositioning of the ATP7A transporter towards the hyperactive sites 62. Copper interacts closely with the NMDA-R and may inhibit currents related to NMDA-R activation 63. Copper acts on the S-nitrosylation of the NMDA receptor 64. Copper also interacts with AMPA-R, but the IC50 for inhibition is higher (4.5 µM) as compared to the NMDA-R (0.27 µM) 65. The transition metal modulates also GABAA-R, with an impact on Cl− currents 66. The IC50 is 2.4 µM. It affects also the extra-synaptic GABA receptors 67. Overall, the acute effects of copper on AMPA-R and GABAA-R is inhibitory 48.
What is the function of copper?
Your body uses copper to carry out many important functions, including making energy, connective tissues, and blood vessels. Copper also helps maintain the nervous and immune systems, and activates genes 24, 68, 69, 70. Your body also needs copper for brain development. Physiologic functions of these copper-dependent enzymes, and the biochemical pathways in which they function, are outlined below.
Energy production
The copper-dependent enzyme cytochrome C oxidase (CCO) plays a critical role in cellular energy (ATP) production in mitochondria by catalyzing the reduction of molecular oxygen (O2) to water (H2O), thereby generating an electrical gradient that is required for ATP production 26. Redox-active copper contained within the cytochrome C oxidase (CCO) enzyme complex is required for the electron transfer reactions that are critical for its function.
Connective tissue formation
Another cuproenzyme, lysyl oxidase (LOX), is required for the cross-linking of collagen and elastin fibers, which is essential for the formation of strong and flexible connective tissue. Lysyl oxidase (LOX) function is critical for bone formation and maintenance of connective tissue in the heart and blood vessels 25.
Iron metabolism
Multi-copper oxidases (MCOs) are copper-dependent ferroxidases that function in iron homeostasis. Multi-copper oxidases (MCOs) oxidize ferrous iron (Fe2+) to the ferric (Fe3+) form, which enables binding to transferrin (the main iron carrier) in the blood, thus allowing iron transport to sites of utilization (e.g., the bone marrow). The multi-copper oxidases (MCOs) include: (1) ceruloplasmin (CP), which contains 60%-95% of plasma copper; (2) a membrane-bound form of ceruloplasmin (CP) (GPI-CP), expressed in brain and other tissues; and (3) the membrane-bound ferroxidases hephaestin (HEPH) and zyklopen, which function in the intestine and placenta, respectively 71, 72. Ceruloplasmin (CP) knockout (Cp-/-) mice accumulate excess iron in their liver but have normal copper status 73, 74. Similarly, humans with aceruloplasminemia, who lack functional ceruloplasmin (CP), display iron overload in liver, brain, and retina but have no observable defects in copper homeostasis 75. Moreover, absorption of dietary iron and iron mobilization from storage sites (e.g., liver) are impaired in copper deficiency, when ceruloplasmin (CP) and membrane-bound ferroxidases hephaestin (HEPH) activity is reduced, further supporting a role for the multi-copper oxidases (MCOs) in iron metabolism 76.
Central nervous system function
Several physiological processes within the brain and nervous system, including neurotransmitter synthesis and formation and maintenance of myelin, depend upon catalysis mediated by cuproenzymes. Dopamine beta-hydroxylase, for example, catalyzes the conversion of dopamine to the neurotransmitter norepinephrine 77. Also, cytochrome C oxidase (CCO) is required for the biosynthesis of phospholipids, which are critical structural components of the myelin sheath 25.
Melanin biosynthesis
The cuproenzyme tyrosinase (TYR) is required for the biosynthesis of melanin in melanocytes, which is critical for normal pigmentation of hair, skin, and eyes 25. Low tyrosinase (TYR) activity most likely explains the absence or loss of pigmentation in the hair (achromotrichia) seen in copper-deficient laboratory and agricultural animals, and the depigmentation noted in severely copper-depleted patients with Menkes disease.
Antioxidation
Superoxide dismutase (SOD) functions as an antioxidant by catalyzing the conversion of reactive oxygen species, such as the superoxide anion (O2-) and the hydroxyl radical (•OH), to hydrogen peroxide (H2O2), which is subsequently reduced to water by other antioxidant systems 78. Two forms of superoxide dismutase (SOD) contain copper: copper/zinc SOD (SOD1), which is expressed in most cells, including red blood cells; and extracellular SOD (EcSOD), which is highly expressed in the lungs and found at lower levels in plasma 25. Also, ceruloplasmin (CP) has antioxidant properties relating to iron metabolism. The ferroxidase activity of ceruloplasmin (CP) may prevent ferrous iron (Fe2+) from participating in harmful free-radical-generating reactions via Fenton chemistry 78.
Regulation of gene expression
Copper-related gene expression pathways seem to be mainly regulated at a post-translation level, in some cases via protein trafficking-related mechanisms that respond to intracellular copper levels 79. Cytosolic copper may also influence mRNA expression levels of specific genes, in a dose-dependent manner, implicating possible transcriptional regulation 80, 81, 82. For example, intracellular copper may alter the redox state of cells and thus induce oxidative stress, which can activate signal transduction pathways that increase the expression of genes encoding proteins involved in the detoxification of reactive oxygen species 83.
Immune system function
Copper is known to play several important roles in the development and maintenance of immune system function, including innate and adaptive immunity 84. Neutropenia is a clinical sign of copper deficiency in humans. Adverse effects of insufficient copper on immune system function appear most pronounced in infants. For example, infants with Menkes disease, a genetic copper-deficiency disorder, suffer from frequent and severe infections 85, 86. Moreover, in a study of 11 malnourished infants with evidence of copper deficiency, the ability of white blood cells to engulf pathogens increased significantly after one month of copper supplementation 87. Moreover, 11 men on a low-copper diet (660 mcg/day of copper for 24 days and 380 mcg/day for another 40 days) showed an impaired monocyte proliferative response in an ex vivo immune challenge assay 43. Mechanistic studies also support a role for copper in the innate immune response to bacterial and viral infections 88, 89. Severe copper deficiency thus has adverse effects on immune system function; however, whether marginal copper insufficiency impairs immunity in humans has not been established.
How much copper do I need?
The amount of copper you need each day depends on your age. Average daily recommended amounts are listed below in micrograms (mcg).
Intake recommendations for copper and other nutrients are provided in the Dietary Reference Intakes (DRIs) developed by the Food and Nutrition Board (FNB) at the National Academies of Sciences, Engineering, and Medicine 40. Dietary Reference Intake (DRI) is the general term for a set of reference values used for planning and assessing nutrient intakes of healthy people. These values, which vary by age and sex, include:
- Recommended Dietary Allowance (RDA): Average daily level of intake sufficient to meet the nutrient requirements of nearly all (97%–98%) healthy individuals; often used to plan nutritionally adequate diets for individuals.
- Adequate Intake (AI): Intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an RDA.
- Estimated Average Requirement (EAR): Average daily level of intake estimated to meet the requirements of 50% of healthy individuals; usually used to assess the nutrient intakes of groups of people and to plan nutritionally adequate diets for them; can also be used to assess the nutrient intakes of individuals.
- Tolerable Upper Intake Level (UL): Maximum daily intake unlikely to cause adverse health effects.
According to the National Health and Nutrition Examination Survey (NHANES), the mean dietary intake of copper in the US is 1100 mcg/day for adult women and 1300 mcg/day for adult men, levels that exceed the established Recommended Dietary Allowance (RDA) for copper for adults of 900 mcg/day (see Table 1) 90.
Table 1 lists the current Recommended Dietary Allowance (RDA) for copper 40. For infants from birth to 12 months, the Food and Nutrition Board established an Adequate Intake (intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an RDA). for copper that is equivalent to the mean intake of copper in healthy, breastfed infants.
Table 1. Recommended Dietary Allowances (RDAs) for Copper
| Life Stage | Recommended Amount |
|---|---|
| Birth to 6 months | 200 mcg |
| Infants 7–12 months* | 220 mcg |
| Children 1–3 years | 340 mcg |
| Children 4–8 years | 440 mcg |
| Children 9–13 years | 700 mcg |
| Teens 14–18 years | 890 mcg |
| Adults 19 years and older | 900 mcg |
| Pregnant teens and women | 1,000 mcg |
| Breastfeeding teens and women | 1,300 mcg |
Footnote: *Adequate Intake (AI) = intake at this level is assumed to ensure nutritional adequacy; established when evidence is insufficient to develop an Recommended Dietary Allowance (RDA).
[Source 30 ]Food Sources of Copper
Many foods contain copper. You can get recommended amounts of copper by eating a variety of foods, including the following 91:
- Beef liver and shellfish such as oysters
- Nuts (such as cashews), seeds (such as sesame and sunflower), and chocolate
- Wheat-bran cereals and whole-grain products
- Potatoes, mushrooms, avocados, chickpeas, and tofu
The estimated copper content of some foods that are relatively rich in copper is listed in Table 2.
For more information on the nutrient content of foods, search USDA’s FoodData Central.
Typical diets in the United States meet or exceed the copper Recommended Dietary Allowance (RDA). Mean dietary intakes of copper from foods range from 800 to 1,000 mcg per day for children aged 2–19 36. In adults aged 20 and older, average daily intakes of copper from food are 1,400 mcg for men and 1,100 mcg for women. Total intakes from supplements and foods are 900 to 1,100 mcg/day for children and 1,400 to 1,700 mcg/day for adults aged 20 and over.
Table 2. Selected Foods Copper Content
| Food | Micrograms (mcg) per serving | Percent DV* |
|---|---|---|
| Beef, liver, pan fried (3 ounces) | 12400 | 1378 |
| Oysters, eastern, wild, cooked, 3 ounces | 4850 | 539 |
| Baking chocolate, unsweetened, 1 ounce | 938 | 104 |
| Potatoes, cooked, flesh and skin, 1 medium potato | 675 | 75 |
| Mushrooms, shiitake, cooked, cut pieces, ½ cup | 650 | 72 |
| Cashew nuts, dry roasted, 1 ounce | 629 | 70 |
| Crab, Dungeness, cooked, 3 ounces | 624 | 69 |
| Sunflower seed kernels, toasted, ¼ cup | 615 | 68 |
| Turkey, giblets, simmered, 3 ounces | 588 | 65 |
| Chocolate, dark, 70%-85% cacao solids, 1 ounce | 501 | 56 |
| Tofu, raw, firm, ½ cup | 476 | 53 |
| Chickpeas, mature sees, ½ cup | 289 | 32 |
| Millet, cooked, 1 cup | 280 | 31 |
| Salmon, Atlantic, wild, cooked, 3 ounces | 273 | 30 |
| Pasta, whole wheat, cooked, 1 cup (not packed) | 263 | 29 |
| Avocado, raw, ½ cup | 219 | 24 |
| Figs, dried, ½ cup | 214 | 24 |
| Spinach, boiled, drained, ½ cup | 157 | 17 |
| Asparagus, cooked, drained, ½ cup | 149 | 17 |
| Sesame seeds, ¼ cup | 147 | 16 |
| Turkey, ground, cooked, 3 ounces | 128 | 14 |
| Cereals, Cream of Wheat, cooked with water, stove-top, 1 cup | 104 | 12 |
| Tomatoes, raw, chopped, ½ cup | 53 | 6 |
| Yogurt, Greek, plain, lowfat, 7-ounce container | 42 | 5 |
| Milk, nonfat, 1 cup | 27 | 3 |
| Apples, raw, with skin, ½ cup slices | 17 | 2 |
Footnote: *DV = Daily Value. The U.S. Food and Drug Administration (FDA) developed Daily Values (DVs) to help consumers compare the nutrient contents of foods and dietary supplements within the context of a total diet. The Daily Value (DV) for copper is 0.9 mg (900 mcg) for adults and children age 4 years and older 92. The FDA does not require food labels to list copper content unless copper has been added to the food. Foods providing 20% or more of the DV are considered to be high sources of a nutrient, but foods providing lower percentages of the DV also contribute to a healthful diet.
[Source 30 ]Copper supplements
Copper is available in many multivitamin or multimineral supplements, in supplements that contain only copper, and in other dietary supplements in combination with other ingredients. Copper in dietary supplements is often in the forms of cupric oxide, cupric sulfate, copper amino acid chelates, and copper gluconate. To date, no studies have compared the bioavailability of copper from these and other forms 93. The amount of copper in dietary supplements typically ranges from a few micrograms to 15 mg (about 17 times the Daily Value [DV] for copper) 30.
Supplementation of adults with 10 mg/day of cupric gluconate for 12 weeks did not cause copper toxicity 94. Importantly, however, higher copper intakes could be detrimental in some at-risk (unknown) individuals. Copper supplementation of infants should be approached with caution since homeostatic regulators of copper absorption and excretion are not fully developed, thus increasing the potential for copper toxicosis. From a clinical perspective, copper overload most frequently presents in biliary atresia, biliary cirrhosis and in Wilson’s disease patients, and as such, individuals suffering from these conditions should avoid taking supplemental copper.
According to an analysis of data from the 2009–2012 National Health and Nutrition Survey (NHANES), 6% to 15% of adults aged 19 and older who do not take dietary supplements containing copper have copper intakes below the Estimated Average Requirement (average daily level of intake estimated to meet the requirements of 50% of healthy individuals; usually used to assess the nutrient intakes of groups of people and to plan nutritionally adequate diets for them; can also be used to assess the nutrient intakes of individuals) 95. In those who do use supplements, rates of adults with intakes below the copper Estimated Average Requirement (EAR) range from 2.2% to 7.2% 95.
Should I be taking copper supplements or trying to get more copper in my diet?
In most cases, a regular diet satisfies the body’s requirements for copper. Talk to your healthcare provider before taking any supplements or changing your diet.
Can copper be harmful?
Yes, copper can be harmful if you get too much. Getting too much copper on a regular basis can cause liver damage, abdominal pain, cramps, nausea, diarrhea, and vomiting 44. More serious signs of acute copper toxicity include severe liver damage, kidney failure, coma, and death 16. Copper toxicity is rare in healthy individuals who do not have a hereditary copper homeostasis defect (e.g., Wilson’s disease, a rare genetic disorder). Wilson’s diseaseis a rare, autosomal recessive disease, which is caused by a mutation in ATP7B, leads to abnormally high tissue levels of copper as a result of defective copper clearance 96. People with Wilson’s disease can develop neurologic and liver damage that can result in cirrhosis 12. Patients can also develop acute hepatitis, hemolytic crisis, and liver failure. Lifelong copper chelation therapy or high doses of zinc can prevent permanent organ damage in these patients.
Copper toxicity has been reported in people who consume water containing high levels of copper as a result of stagnant water in copper-containing pipes and fixtures as well as copper alloys in water distribution systems and household plumbing that allow copper to leach into water 44, 97. The Environmental Protection Agency has established a recommended upper limit for copper in public water systems of 1.3 mg/L 97, 98 and by the World Health Organization (2 mg/liter) 99.
The daily upper limits (ULs) for copper include intakes from all sources—food, beverages, and supplements—and are listed below in micrograms (mcg). The Food and Nutrition Board has established Tolerable Upper Intake Level (maximum daily intake unlikely to cause adverse health effects) for copper from food and supplements for healthy individuals based on levels associated with liver damage 44. The Tolerable Upper Intake Levels (ULs) do not apply to individuals who are receiving supplemental copper under medical supervision.
Table 3. Tolerable Upper Intake Levels (ULs) for Copper
| Age | Male | Female | Pregnancy | Lactation |
|---|---|---|---|---|
| Birth to 6 months | None established* | None established* | ||
| 7–12 months | None established* | None established* | ||
| 1–3 years | 1,000 mcg | 1,000 mcg | ||
| 4–8 years | 3,000 mcg | 3,000 mcg | ||
| 9–13 years | 5,000 mcg | 5,000 mcg | ||
| 14–18 years | 8,000 mcg | 8,000 mcg | 8,000 mcg | 8,000 mcg |
| 19+ years | 10,000 mcg | 10,000 mcg | 10,000 mcg | 10,000 mcg |
Footnote: * Breast milk, formula, and food should be the only sources of copper for infants.
[Source 30 ]Copper deficiency disease
Menkes disease
Menkes disease or Menkes syndrome (Menkes kinky hair syndrome) or copper transport disease, is an inherited disorder in which the body has a problem absorbing copper resulting in copper deficiency. Mutations in the ATP7A gene located on the X chromosome cause Menkes syndrome (Menkes kinky hair syndrome). The mutation leads to uneven distribution of copper in the body. It may build up in tissues of the intestines and kidneys, for example, but may be deficient in areas such as the brain.
Menkes disease affects development, both mental and physical. It is characterized by sparse, kinky hair; failure to gain weight and grow at the expected rate (failure to thrive); and developmental delay, nervous system deterioration. Additional signs and symptoms include weak muscle tone (hypotonia), sagging facial features, seizures, developmental delay, and intellectual disability. Children with Menkes disease (Menkes syndrome) typically begin to develop symptoms during infancy and often do not live past age 3. Early treatment with copper may improve the prognosis in some affected individuals. In rare cases, symptoms begin later in childhood.
Occipital horn syndrome (sometimes called X-linked cutis laxa) is a less severe form of Menkes disease that begins in early to middle childhood. It is characterized by wedge-shaped calcium deposits in a bone at the base of the skull (the occipital bone), coarse hair, and loose skin and joints.
The incidence of Menkes disease and occipital horn syndrome is estimated to be 1 in 100,000 newborns.
Outlook (Prognosis)
Most children with Menkes disease die within the first few years of life.
Menkes disease causes
Mutations in the ATP7A gene located on the X chromosome cause Menkes disease (Menkes kinky hair syndrome). The ATP7A gene provides instructions for making a protein that is important for regulating copper levels in the body. Copper is necessary for many cellular functions, but it is toxic when present in excessive amounts. Mutations in the ATP7A gene result in poor distribution of copper to the body’s cells. Copper accumulates in some tissues, such as the small intestine and kidneys, while the brain and other tissues have unusually low levels of copper. The decreased supply of copper can reduce the activity of numerous copper-containing enzymes that are necessary for the structure and function of bone, skin, hair, blood vessels, and the nervous system. The signs and symptoms of Menkes syndrome and occipital horn syndrome are caused by the reduced activity of these copper-containing enzymes.
Menkes disease inheritance pattern
Menkes disease is inherited in an X-linked recessive pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause Menkes disease (Menkes kinky hair syndrome). In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause Menkes disease (Menkes kinky hair syndrome). Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons because males receive the Y chromosome from their father.
In about one-third of cases, Menkes disease is caused by new mutations in the ATP7A gene. People with a new mutation do not have a history of the disorder in their family.
Menkes disease tests
Once Menkes disease is suspected, tests that may be done include:
- Ceruloplasmin blood test (substance that transports copper in the blood)
- Copper blood test
- Skin cell culture
- X-ray of the skeleton or x-ray of the skull
- Gene testing to check for defect of the ATP7A gene
Menkes disease treatment
Treatment usually only helps when started very early in the course of the disease. Injections of copper into a vein or under the skin have been used with mixed results and depends on whether the ATP7A gene still has some activity.
Zinc-induced myeloneuropathy
Copper and zinc are known to compete for absorption 100. Study has confirmed a decline in serum zinc levels as copper supplementation continued and that monitoring of serum zinc levels is necessary when copper supplementation is performed. Zinc-induced myeloneuropathy is also caused by a deficit in copper 101. Zinc-induced myeloneuropathy is the human equivalent to the copper deficiency myelopathy occurring in ruminants (“enzootic ataxia”) 102. Zinc-induced myeloneuropathy occurs mainly after sustained zinc exposure 103. Regular use of zinc-containing dental fixatives has been identified as a triggering factor 104. Patients undergoing gastrojejunal bypass surgery are also at risk 105. The first disturbances are sensory symptoms in the feet. After a few years, zinc-induced myeloneuropathy mimics the subacute combined degeneration of the spinal cord, a disorder due to a deficit in vitamin B12. MRI of the spinal cord demonstrates signal changes in the dorsal columns. Chronic zinc ingestion may also cause bone marrow suppression with anemia, thrombocytopenia and neutropenia. By contrast to Wilson’s disease, urinary copper levels are typically decreased 106. Although copper replacement may revert cytopenias, neurological deficits are irreversible in 40% of cases, highlighting the importance of detecting the hypocupremic state as soon as possible.
Copper deficiency anemia
It was found in the 1930s that copper is vital for effective erythropoiesis 107. The hematological manifestations of deficiency are typically anemia or neutropenia, or more rarely pancytopenia 108. The anemia is usually normocytic or macrocytic, but may rarely be microcytic. A retrospective review of 40 patients with copper deficiency defined the typical bone marrow findings of hypocellularity, vacuolisation of granulocyte and erythroid precursors, iron-containing plasma cells and ring sideroblasts 109. Owing to the similarity of the haematological findings, copper deficiency may be mistaken for a myelodysplastic syndrome. Some haematology textbooks do not list copper deficiency as a differential of myelodysplastic syndrome 110, and in cases with concomitant myelopathy the neurologist may be the first to consider copper deficiency as the unifying diagnosis.
The mechanisms underlying the hematological effects of copper deficiency have not been fully elucidated. The anemia appears to be partly attributable to impaired iron metabolism. Copper is an essential component of both ceruloplasmin and hephaestin, which are necessary for normal iron homeostasis. Both have enzymatic activity as ferroxidases and are involved in iron export from non-intestinal 111 and intestinal cells 112, respectively. In copper deficiency, iron is seen to accumulate in the liver and can be released by intravenous caeruloplasmin. Although the literature suggests that copper deficiency typically causes a microcytic anaemia akin to iron deficiency, this pattern was reported in only one of the cases reviewed herein. The remaining cases where mean corpuscular volume (MCV) was reported were either macrocytic (9 cases) or normocytic (3 cases). The difference between copper deficiency and iron deficiency anaemia may be due to iron utilisation being impaired only at certain steps or the contribution of other mechanisms. For example, copper deficiency seems to impair the production of heme from ferric iron and protoporphyrin, possibly because decreased energy production (cytochrome-c oxidase) in erythrocytes impinges on the synthetic pathway 113. In addition, alterations in erythrocyte membrane fluidity and oxidative damage cause reduced red cell lifespan in copper-deficient rats 114.
Suggestions for the mechanism leading to neutropenia include destruction of myeloid precursors and impaired maturation within the bone marrow, impaired release of mature neutrophils from the bone marrow and increased serum clearance of neutrophils 115. Indeed, the bone marrow of copper-deficient patients contains a decreased number of myeloid precursors which show vacuolisation 109.
Familial benign copper deficiency
Familial benign copper deficiency is a rare disorder of mineral absorption and transport characterized by low blood copper level (hypocupremia) that manifests as failure to thrive, mild anemia, repeated seizures, hypotonia, and seborrheic skin 116. Spurring of the femur and tibia are also noted on radiographic imaging. Symptoms are reversible or improve with supplements of oral copper. There have been no further descriptions in the literature since 1988 117.
Mehes and Petrovicz 118 found hypocupremia with normal ceruloplasmin levels in a 21-month-old boy admitted to hospital because of repeated seizures and failure to thrive. He had blond curly hair, spurring of the femurs and tibias, and mild anemia, but his mental development, electroencephalogram, and hair structure on microscopic examination were normal. His condition improved with supplements of oral copper, but as soon as these were reduced or stopped, hypocupremia and seizures recurred. Photographs showed curly hair and an appearance of the nose and lips reminiscent of that in the infantile hypercalcemia syndrome. The father and 2 brothers were physically and biochemically normal. The mother, aged 28, was notably thin, had always been pale and vulnerable to infections but had no seizures. Her face was seborrheic and her hair so thin that the top of the head was almost bald. Serum copper was low. The mother’s brother was also thin and had been frequently ill as a child but had never had seizures. He had had blond, ‘extremely curly’ hair, but had been bald since age 24 years. His skin was seborrheic and serum copper was low. His 2 sons were healthy with brown, slightly curly hair and normal serum copper levels. Deficient dietary intake of copper and excessive renal loss were excluded. The authors suggested either X-linked or autosomal dominant inheritance and a defect in intestinal absorption of copper. Mehes and Petrovicz 117 provided a 7-year follow-up. Low serum copper levels with marginally low/normal ceruloplasmin values persisted. It was thought that seborrhea improved with copper supplementation.
Copper deficiency causes
Upper gastrointestinal surgery
Certain types of upper gastrointestinal surgery can reduce the effective absorption area for particular micronutrients. Deficiencies of iron and vitamin B12 are especially common 119, with a lack of intrinsic factor contributing to the latter. Whilst neurological complications of gastrectomy for peptic ulcer disease have long been recognized, the underlying aetiology often remained obscure 120. The association between a remote history of upper gastrointestinal surgery, copper deficiency and myelopathy was first described in 2001 121, and has since been increasingly reported following both non-bariatric 122 and bariatric intervention 123.
Partial gastrectomy has rarely been used to treat peptic ulcer disease since the advent of effective pharmacological therapy. The number of cases of gastrectomy-related copper deficiency is small relative to the number of past procedures. However, it is likely that many further cases will be diagnosed because undiagnosed hypocupraemia is common in patients with a history of gastrectomy 124, copper deficiency may manifest over 40 years after the operation 125 and awareness amongst physicians continues to increase.
The annual number of bariatric operations continues to grow, with the commonest procedure being a Roux-en-Y gastric bypass. Post-operatively, food bypasses most of the stomach and the entire duodenum. Copper deficiency tends to develop sooner after bariatric than after non-bariatric operations, probably due to the greater reduction in the effective absorption area for copper. Case numbers of copper deficiency will rise with the growing use of bariatric surgery and with the ageing of the present cohort.
Malabsorption
In half the cases of copper deficiency attributed to malabsorption, celiac disease was diagnosed; in the other half, no cause of malabsorption was established. In three cases of idiopathic copper malabsorption, no detailed investigation findings were reported 120. In the fourth case, malabsorption was not specific to copper: the patient was cachectic and exhibited concomitant deficiencies of iron, vitamin D and vitamin E. A D-xylose absorption test was compatible with proximal intestinal malabsorption, but upper gastrointestinal endoscopy, duodenal biopsies and faecal microbiology failed to identify an underlying cause 126.
Drug-induced copper deficiency
Rosa 127 reviewed a series of cases of infants born to women who received d-penicillamine during pregnancy for a variety of conditions, including connective-tissue abnormalities and rheumatoid arthritis. Abnormalities observed in the infants included lax skin, hyperflexibility of the joints, fragility of the veins, and numerous soft tissue abnormalities. It was suggested that the malformations were in part due to a drug-induced copper deficiency during embryonic or fetal development. Similar abnormalities were produced with d-penicillamine in rodent models 128, and the teratogenicity of the drug can be modulated by maternal dietary copper intake 129. The above suggest that in humans maternal copper status should be monitored when d-penicillamine is taken during pregnancy.
Little is known about the influence of other copper chelating drugs such as captopril, disulfiram, dimercaptosuccinic acid, and triethylenetetramine on human prenatal development. Given the significant teratogenic effects that can be associated with similar drugs in experimental animals, it is reasonable to suggest that these drugs can pose a substantial risk to the conceptus if maternal dietary copper intake is low.
A condition of severe copper deficiency can be rapidly induced in experimental animals through the use of a number of chelating drugs, including disulfiram, D-penicillamine, triethylenetetramine, and dimercaptosuccinic acid 130. Each of those drugs is known to be teratogenic. The abnormalities produced are reminiscent of those induced by dietary copper deficiency. Although the teratogenicity of D-penicillamine, triethylenetetramine, and dimercaptosuccinic acid can be modulated by the amount of copper in the mother’s diet (Cohen et al. 1983; Mark-Savage et al. 1983), it is important to note that drugs that bind copper typically also bind zinc. Thus, the teratogenic effects of those drugs might be due to a combination of copper and zinc deficiencies.
In contrast to the effects of zinc, drugs and chemicals that induce an acute-phase response in the mother do not necessarily influence copper uptake by the embryo or fetus, and in most cases, fetal copper concentrations are unaffected by chemicals that induce transitory acute-phase responses 131. That is not surprising, because maternal plasma copper concentrations are increased during an acute-phase response (due to the hepatic production and release of the cuproenzyme ceruloplasmin), and zinc concentrations are decreased. Ceruloplasmin has been postulated to be involved directly in copper transport to the embryo and fetus 132. It must be noted that the mechanisms underlying copper transport into the embryo and fetus is an area of active research.
Disease-induced copper deficiency
Copper deficiency can occur secondarily to such diseases as chronic diarrhea, diabetes, alcoholism, and hypertension 133. In the case of maternal diabetes and alcoholism, disease-induced deficiencies of copper in the embryo or fetus have been postulated to contribute to the teratogenesis 134. Mothers with diabetes are reported to have low concentrations of copper in erythrocytes 135. In addition, maternal hypocupremia is often noted in cases of spontaneous abortion or rupture of the fetal and placental membranes 136.
Copper-diet interactions
Zinc
Zinc intakes, well in excess of the amount normally found in the diet, can decrease copper absorption in adults 137. In one case report, an infant who was given 16 to 24 mg/day of zinc developed copper deficiency 138. Very high doses of zinc have been used to treat patients with Wilson’s disease, an inborn error of copper metabolism resulting in copper toxicity 139. This zinc-induced inhibition of copper absorption could be the result of competition for a common, apically oriented transporter or the induction of metallothionein in intestinal cells by zinc. Because this protein has a higher binding affinity for copper than for zinc, copper is retained within enterocytes and its absorption is reduced. This response has been used as a therapy to diminish copper absorption in patients with Wilson’s disease 140. The interaction could also be responsible for reducing copper absorption during consumption of zinc supplements. When zinc-to-copper ratios of 2:1, 5:1, and 15:1 were fed to humans, there were limited effects on copper absorption 141.
Iron
High iron intakes may interfere with copper absorption in infants. Infants fed a formula containing low concentrations of iron absorbed more copper than infants consuming the same formula with a higher iron concentration 142. Such an interaction has been reported to produce reduced copper status in infants 143.
Fructose
Studies in rats demonstrated that diets very high in fructose were associated with increased severity of copper deficiency in rats 144, but a similar effect was not observed in pigs 145, which have cardiovascular systems and gastrointestinal tracts more similar to those of humans. The effects were inconsistent in humans 146 but did not result in copper depletion, and the extremely high levels of fructose fed (20 percent of energy intake) suggest the effect would not be relevant to normal diets.
Gene-induced copper deficiency
There are at least two genetic defects that are expressed as copper-deficiency syndromes (Menkes disease and occipital horn syndrome). Both disorders are due to defects in a copper-transporting P-type ATPase. Infants with Menkes disease are characterized by progressive degeneration of the brain and spinal cord, hypothermia, connective-tissue abnormalities, and failure to thrive. Menkes disease has been recognized as a disorder of copper metabolism for over 20 years. The prognosis for infants with the disorder is poor and death typically occurs before 3 years of age 133. Similar to the blotchy mouse, the developmental abnormalities associated with Menkes disease are thought to be the consequence of low activity of numerous cuproenzymes during embryonic and fetal development. Those cuproenzymes include dopamine-B-monoxygenase, peptidylglycine a-amidating monooxygenase, cytochrome c oxidase, lysyl oxidase, and CuZnSOD 147. The aberrant pattern of the plasma and cerebrospinal fluid of Menkes patients has been attributed to low activity of dopamine-B-monoxygenase 148. Moreover, individuals with Menkes disease are characterized by low activity of ceruloplasmin and peptidylglycine a-amidating monooxygenase 149. Thus, low activity of numerous enzymes that rely on the amidation of peptides for their activity might occur as a secondary effect of low peptidylglycine a-amidating monooxygenase activity.
Groups at Risk of Copper Inadequacy
The following groups are most likely to have inadequate copper status.
People with Celiac disease
In a study of 200 adults and children with celiac disease, of which 69.9% claimed to maintain a gluten-free diet, 15% had copper deficiency (less than 70 mcg/dL in serum in men and girls younger than 12 years and less than 80 mcg/dL in women older than 12 years and/or ceruloplasmin (CP) less than 170 mg/L) as a result of intestinal malabsorption resulting from the intestinal lining alterations associated with celiac disease 150. In its 2009 clinical guidelines for celiac disease, the American College of Gastroenterology notes that people with celiac disease appear to have an increased risk of copper deficiency and that copper levels normalize within a month of adequate copper supplementation while eating a gluten-free diet 151.
People with Menkes disease
Menkes disease is a rare, X-linked, recessive disorder of copper homeostasis caused by ATP7A gene mutations, which encode a copper-transporting ATPase 12. In these individuals, intestinal absorption of dietary copper drops sharply, leading to signs of copper deficiency, including low serum copper and ceruloplasmin (CP) levels 12, 152. The typical manifestations of Menkes disease include failure to thrive, impaired cognitive development, aortic aneurysms, seizures, and unusually kinky hair 153. Most individuals with Menkes disease die by age 3 years if untreated, but subcutaneous injections of copper starting in the first few weeks after birth can reduce mortality risk and improve development 154.
People taking high doses of Zinc supplements
High dietary intakes of zinc can interfere with copper absorption, and excessive use of zinc supplements can lead to copper deficiency. Reductions in red blood cell copper-zinc superoxide dismutase, a marker of copper status, have been reported with even moderately high zinc intakes of approximately 60 mg/day for up to 10 weeks 40. People who regularly consume high doses of zinc from supplements or use excessive amounts of zinc-containing denture creams can develop copper deficiency because zinc can inhibit copper absorption. This is one reason the Food and Nutrition Board established the Tolerable Upper Intake Level (maximum daily intake unlikely to cause adverse health effects) for zinc at 40 mg/day for adults 40.
Copper deficiency signs and symptoms
Copper plays a role as a cofactor for various enzymes in the human body, and is indispensable for myeloid and neurological structure and function 155. Copper deficiency leads to functional disorders of hematopoiesis such as anemia and neutropenia, as well as neurological disorders such as myelopathy 156. Symptoms accompanying the copper deficiency included normocytic, hypochromic anemia, leukopenia, and neutropenia. Osteoporosis was observed in copper-deficient infants and growing children 157.
Acquired copper deficiency has long been recognised as a cause of anemia and, less commonly, other cytopenias 158. It was only in 2001 that an association with myelopathy was reported 121. The neurologic manifestations of copper deficiency may be similar to the myeloneuropathy observed with vitamin B12 deficiency 159. The clinical and radiological picture is usually indistinguishable from subacute combined degeneration due to vitamin B12 (cobalamin) deficiency 160 and the syndrome is thought to represent the human counterpart of swayback or enzootic ataxia, a copper deficiency myelopathy occurring in ruminants 161. Other, less frequently reported and less clearly causally related neurological associations of acquired copper deficiency include isolated peripheral neuropathy 110, motor neuron disease 162, myopathy 163, cerebral demyelination 164, cognitive dysfunction 165 and optic neuropathy 166.
Though copper deficiency is thought to be rare in developed countries, the neurologic symptoms can be profound and are frequently irreversible, making awareness and early diagnosis essential 167.
Copper deficiency test
Copper testing may be ordered when a person has signs and symptoms that may be due to a copper deficiency, such as:
- Abnormally low numbers of neutrophils, a type of white blood cell (neutropenia)
- Osteoporosis
- Anemia
- Less commonly, neurologic symptoms and delayed growth in children
One or more of the copper tests may be ordered periodically when monitoring is recommended.
A hepatic (liver) copper test may be ordered to further investigate copper storage when copper and ceruloplasmin results are abnormal or equivocal.
Copper testing is primarily used to help diagnose Wilson disease, a rare inherited disorder that can lead to excess storage of copper in the liver, brain, and other organs. Less commonly, a copper test may be used to detect copper excess due to another condition, to detect a copper deficiency, or to monitor treatment for one of these conditions.
Rarely, a copper test may be used to help diagnose Menkes kinky hair syndrome (Menkes disease), a rare inherited disorder of copper transport dysfunction.
Copper is an essential mineral but in excess, it can be toxic. In the blood from normal, healthy humans, more than 95% of the copper is incorporated into the enzyme ceruloplasmin and only a small amount is in a “free” or unbound state (loosely bound to albumin).
Typically, a total blood copper test is ordered along with a ceruloplasmin level. If the results from these tests are abnormal or unclear, then they may be followed by a 24-hour urine copper test to measure copper elimination and/or a copper test performed on a liver biopsy to evaluate copper storage in the liver.
Sometimes a free (unbound) blood copper test is also ordered. If Wilson disease is suspected, genetic testing may be performed to detect mutations in the ATP7B gene. However, these tests have limited availability and are usually performed in special reference or research laboratories.
Normal copper reference values
- 0-2 months: 0.40-1.40 mcg/mL
- 3-6 months: 0.40-1.60 mcg/mL
- 7-9 months: 0.40-1.70 mcg/mL
- 10-12 months: 0.80-1.70 mcg/mL
- 13 months-10 years: 0.80-1.80 mcg/mL
- > or =11 years: 0.75-1.45 mcg/mL
When is copper test ordered?
One or more copper tests are ordered along with ceruloplasmin when someone has signs and symptoms that a health practitioner suspects may be due to Wilson disease, excess copper storage, or copper poisoning. These signs and symptoms may include:
- Anemia
- Nausea, abdominal pain
- Jaundice
- Fatigue
- Behavioral changes
- Tremors
- Difficulty walking and/or swallowing
- Dystonia
Copper tests may also be ordered to aid in the diagnosis of:
- Primary biliary cirrhosis
- Primary sclerosing cholangitis
What does abnormal copper test result mean?
Copper test results must be evaluated in context and are usually compared to ceruloplasmin levels. Abnormal copper results are not diagnostic of a specific condition; they indicate the need for further investigation. Interpretation can be complicated by the fact that ceruloplasmin is an acute phase reactant – ceruloplasmin may be elevated whenever inflammation or severe infections are present. Both ceruloplasmin and copper are also increased during pregnancy and with estrogen and oral contraceptive use.
- Serum copper below the normal range is associated with Wilson disease, as well as a variety of other clinical situations (see below). Excess use of denture cream containing zinc can cause hypocupremia.
- Low blood copper concentrations along with increased urine copper levels, low ceruloplasmin levels, and increased hepatic copper are typically seen with Wilson disease.
- Serum concentrations above the normal range are seen in primary biliary cirrhosis and primary sclerosing cholangitis, as well as a variety of other clinical situations (see below).
- Increased blood and urine copper concentrations and normal or increased ceruloplasmin levels may indicate exposure to excess copper or may be associated with conditions that decrease copper excretion, such as chronic liver disease, or that release copper from tissues, such as acute hepatitis.
- Increased hepatic copper may be present with chronic conditions.
- Decreased blood and urine copper concentrations and decreased ceruloplasmin may indicate a copper deficiency.
- A normal hepatic copper test may indicate that copper metabolism is functioning properly or that the distribution of copper in the liver is uneven and the sample is not representative of the person’s condition.
- If a person is being treated for Wilson disease or copper toxicity with drugs that bind copper (chelators), then that person’s 24-hour urine copper levels may be high until body copper stores decrease. Eventually, blood copper and 24-hour urine copper concentrations should normalize.
- If someone is being treated for a condition related to copper deficiency and the person’s ceruloplasmin and total copper concentrations begin to rise, then the condition is likely responding to the treatment.
- Medications such as carbamazepine and phenobarbital can increase blood copper levels. They may also be elevated with rheumatoid arthritis and with some cancers and decreased with a variety of conditions associated with malabsorption, such as cystic fibrosis.
- Total serum copper concentrations are normally low at birth, rise over the next few years, peak, and then decline slightly to a relatively stable level.
Low serum copper, most often due to excess iron or zinc ingestion and infrequently due to dietary copper deficit, results in severe derangement in growth and impaired erythropoiesis. Low serum copper is also observed in hepatolenticular degeneration (Wilson disease) due to a decrease in the synthesis of ceruloplasmin and allelic variances in cellular metal ion transporters. In Wilson disease, the albumin-bound copper may actually be increased, but ceruloplasmin copper is low, resulting in low serum copper. However, during the acute phase of Wilson disease (fulminant hepatic failure), ceruloplasmin and copper may be normal; in this circumstance, hepatic inflammation causes increased release of ceruloplasmin. It is useful to relate the degree of liver inflammation to the ceruloplasmin and copper-see discussion on hypercupremia below. Significant hepatic inflammation with normal ceruloplasmin and copper suggest acute Wilson disease.
Other disorders associated with decreased serum copper concentrations include malnutrition, hypoproteinemia, malabsorption, nephrotic syndrome, Menkes disease, copper toxicity, and megadosing of zinc-containing vitamins (zinc interferes with normal copper absorption from the gastrointestinal tract).
Hypercupremia is found in primary biliary cirrhosis, primary sclerosing cholangitis, hemochromatosis, malignant diseases (including leukemia), thyrotoxicosis, and various infections. Serum copper concentrations are also elevated in patients taking contraceptives or estrogens and during pregnancy.
Since the gastrointestinal (GI) tract effectively excludes excess copper, it is the gastrointestinal tract that is most affected by copper ingestion. Increased serum concentration does not, by itself, indicate copper toxicity.
Table 4. Copper test results interpretation
| Test | Wilson Disease | Copper Toxicity | Menkes Disease (Kinky Hair Syndrome) | Copper Deficiency |
|---|---|---|---|---|
| Copper, blood | Low but may be normal | High | Low | Low |
| Copper, serum free | High | High | Low | Low |
| Ceruloplasmin | Low but may be normal | High | Low | Low |
| Copper, urine | Very high | High | Low | Low |
| Copper, liver/hepatic* | Positive but, depending on the site sampled, may be negative | High or normal | Low | Low |
Footnote: *Excess copper in the liver is often unevenly distributed and may not be detected in a sample. Care must be taken, especially with a 24-hour urine sample, not to contaminate the sample with an external source of copper. Talk to the health practitioner and/or the laboratory that will perform the test about necessary precautions. If a urine or blood copper test result is higher than expected, the health practitioner may have the test repeated with a new sample to confirm the findings.
Should everyone’s copper metabolism be evaluated?
General screening for copper concentrations is not recommended or necessary. Many people with conditions not associated with copper, such as people with infections or inflammation, may have temporarily increased concentrations.
Can I choose either a blood (total or free) or urine copper test?
These tests provide complementary information and your healthcare provider will determine which tests are necessary to evaluate your condition.
Copper deficiency treatment
Treatment of copper deficiency consists of parenteral (IV) and oral copper replacement until normal copper levels in blood are achieved. The response to copper replacement is variable and likely depends on the degree and duration of copper depletion. The hematologic abnormalities have been reported to correct within four to six weeks; however, improvements in neurologic symptoms are modest and often difficult to demonstrate objectively. Paresthesias may resolve, but significant gait abnormalities may remain, consistent with clinical observations in reported cases 168. Despite malabsorption being the likely mechanism underlying some copper deficiency after roux-en-Y gastric bypass surgery, oral supplementation has been found to correct copper deficiency, suggesting that super-saturation of intestinal copper transport systems is a reliable way to maintain adequate serum levels 169.
Copper deficiency treatment dose
Typical daily doses were equivalent to 2–4 mg of elemental copper 170. In one case attributed to non-bariatric gastrointestinal surgery, administration of 2 mg of oral copper per day normalized blood parameters and improved neurological symptoms. However, a subsequent symptomatic and biochemical relapse necessitated that the dose be incremented to 4 mg and then 6 mg, which achieved sustained remission 171. Another patient with idiopathic zinc overload was poorly compliant with oral copper supplementation at 2 mg/day. His serum copper levels remained low and the neurological deficit progressed, though serum ceruloplasmin increased to a value at the low end of the normal range and the anaemia resolved. The copper dose was gradually escalated to 8 mg/day but compliance remained poor and he continued to deteriorate neurologically. Subsequently, the patient reported better compliance and his neurological status improved. However, his highest recorded serum copper level remained below the normal range, and it was unclear whether this reflected insufficient dosage, poor compliance or other factors. Copper doses of up to 9 mg per day have been reported 122.
References- Gabreyes AA, Abbasi HN, Forbes KP, McQuaker G, Duncan A, Morrison I. Hypocupremia associated cytopenia and myelopathy: a national retrospective review. Eur J Haematol. 2013 Jan;90(1):1-9. doi: 10.1111/ejh.12020
- Halfdanarson TR, Kumar N, Li CY, Phyliky RL, Hogan WJ. Hematological manifestations of copper deficiency: a retrospective review. Eur J Haematol. 2008 Jun;80(6):523-31. doi: 10.1111/j.1600-0609.2008.01050.x
- Prohaska JR. Copper. In: Erdman JW, Macdonald IA, Zeisel SH, eds. Present Knowledge in Nutrition. 10th ed. Washington, DC: Wiley-Blackwell; 2012:540-53.
- Prodan CI, Holland NR, Wisdom PJ, Burstein SA, Bottomley SS. CNS demyelination associated with copper deficiency and hyperzincemia. Neurology. 2002 Nov 12;59(9):1453-6. doi: 10.1212/01.wnl.0000032497.30439.f6
- Angotti LB, Post GR, Robinson NS, Lewis JA, Hudspeth MP, Lazarchick J. Pancytopenia with myelodysplasia due to copper deficiency. Pediatr Blood Cancer. 2008 Nov;51(5):693-5. doi: 10.1002/pbc.21661
- Forbes GM, Forbes A. Micronutrient status in patients receiving home parenteral nutrition. Nutrition. 1997 Nov-Dec;13(11-12):941-4. doi: 10.1016/s0899-9007(97)00334-1
- Chen CC, Takeshima F, Miyazaki T, Murase K, Ohtani H, Isomoto H, Shikuwa S, Omagari K, Mizuta Y, Ozono Y, Kohno S. Clinicopathological analysis of hematological disorders in tube-fed patients with copper deficiency. Intern Med. 2007;46(12):839-44. doi: 10.2169/internalmedicine.46.6264
- Jayakumar S, Micallef-Eynaud PD, Lyon TD, Cramb R, Jilaihawi AN, Prakash D. Acquired copper deficiency following prolonged jejunostomy feeds. Ann Clin Biochem. 2005 May;42(Pt 3):227-31. doi: 10.1258/0004563053857879
- Griffith DP, Liff DA, Ziegler TR, Esper GJ, Winton EF. Acquired copper deficiency: a potentially serious and preventable complication following gastric bypass surgery. Obesity (Silver Spring). 2009 Apr;17(4):827-31. doi: 10.1038/oby.2008.614
- Tümer Z, Møller LB. Menkes disease. Eur J Hum Genet. 2010 May;18(5):511-8. doi: 10.1038/ejhg.2009.187
- Menkes disease. https://medlineplus.gov/ency/article/001160.htm
- Collins JF. Copper. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2014:206-16.
- Fairweather-Tait SJ, Harvey LJ, Collings R. Risk-benefit analysis of mineral intakes: case studies on copper and iron. Proc Nutr Soc. 2011 Feb;70(1):1-9. doi: 10.1017/S0029665110003873
- Prohaska JR. Impact of copper deficiency in humans. Ann N Y Acad Sci. 2014 May;1314:1-5. doi: 10.1111/nyas.12354
- Uchino K, Quang LV, Enomoto M, Nakano Y, Yamada S, Matsumura S, Kanasugi J, Takasugi S, Nakamura A, Horio T, Murakami S, Goto M, Mizuno S, Yamamoto H, Watarai M, Hanamura I, Takami A. Cytopenia associated with copper deficiency. EJHaem. 2021 Aug 26;2(4):729-737. doi: 10.1002/jha2.278
- Copper. https://lpi.oregonstate.edu/mic/minerals/copper
- Harris ZL, Klomp LW, Gitlin JD. Aceruloplasminemia: an inherited neurodegenerative disease with impairment of iron homeostasis. Am J Clin Nutr. 1998 May;67(5 Suppl):972S-977S. doi: 10.1093/ajcn/67.5.972S
- Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439-58. doi: 10.1146/annurev.nutr.22.012502.114457
- Lahey ME, Gubler CJ, Chase MS, Cartwright GE, Wintrobe MM. Studies on copper metabolism. II. Hematologic manifestations of copper deficiency in swine. Blood. 1952;7(11):1053-1074. https://doi.org/10.1182/blood.V7.11.1053.1053
- Wintrobe MM, Cartwright GE, Lahey ME, Gubler CJ. The role of copper in hemopoiesis. Trans Assoc Am Physicians. 1951;64:310-315.
- Cartwright GE, Gubler CJ, Bush JA, Wintrobe MM. Studies of copper metabolism. XVII. Further observations on the anemia of copper deficiency in swine. Blood. 1956;11(2):143-153. https://doi.org/10.1182/blood.V11.2.143.143
- Bustos RI, Jensen EL, Ruiz LM, Rivera S, Ruiz S, Simon F, Riedel C, Ferrick D, Elorza AA. Copper deficiency alters cell bioenergetics and induces mitochondrial fusion through up-regulation of MFN2 and OPA1 in erythropoietic cells. Biochem Biophys Res Commun. 2013 Aug 2;437(3):426-32. doi: 10.1016/j.bbrc.2013.06.095
- Peled T, Landau E, Prus E, Treves AJ, Nagler A, Fibach E. Cellular copper content modulates differentiation and self-renewal in cultures of cord blood-derived CD34+ cells. Br J Haematol. 2002 Mar;116(3):655-61. doi: 10.1046/j.0007-1048.2001.03316.x. Erratum in: Br J Haematol 2002 May;117(2):485.
- Prohaska JR. Impact of copper limitation on expression and function of multicopper oxidases (ferroxidases). Adv Nutr. 2011 Mar;2(2):89-95. doi: 10.3945/an.110.000208
- Turnlund JR. Copper. In: Shils ME, Shike M, Ross A, Caballero B, Cousins RA, eds. Modern Nutrition in Health and Disease. 10th ed. Baltimore: Lipincott Williams & Wilkins; 2006:289-299.
- Uauy R, Olivares M, Gonzalez M. Essentiality of copper in humans. Am J Clin Nutr. 1998 May;67(5 Suppl):952S-959S. doi: 10.1093/ajcn/67.5.952S
- Eid C, Hémadi M, Ha-Duong NT, El Hage Chahine JM. Iron uptake and transfer from ceruloplasmin to transferrin. Biochim Biophys Acta. 2014 Jun;1840(6):1771-81. doi: 10.1016/j.bbagen.2014.01.011
- Squitti, R.; Polimanti, R. Copper phenotype in Alzheimer’s disease: Dissecting the pathway. Am. J. Neurodegener. Dis. 2013, 2, 46–56
- Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol. 2011 Nov 8;21(21):R877-83. doi: 10.1016/j.cub.2011.09.040
- Copper. https://ods.od.nih.gov/factsheets/Copper-HealthProfessional
- Owen CAJ. Biochemical Aspects of Copper: Copper Proteins, Ceruloplasmin, and Copper Protein Binding. Park Ridge, NJ: Noyes Publications; 1982.
- Allen KG, Klevay LM. Copper: an antioxidant nutrient for cardiovascular health. Curr Opin Lipidol. 1994 Feb;5(1):22-8. doi: 10.1097/00041433-199402000-00005
- Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration. Toxics 2014, 2(2), 327-345; doi:10.3390/toxics2020327 https://www.mdpi.com/2305-6304/2/2/327/htm
- Wilson Disease. https://medlineplus.gov/wilsondisease.html
- Wilson Disease. https://rarediseases.org/rare-diseases/wilson-disease
- U.S. Department of Agriculture, Agricultural Research Service. What We Eat in America, 2013-2014. https://www.ars.usda.gov/northeast-area/beltsville-md-bhnrc/beltsville-human-nutrition-research-center/food-surveys-research-group/docs/wweia-data-tables
- Klevay LM. Copper. In: Coates PM, Betz JM, Blackman MR, et al., eds. Encyclopedia of Dietary Supplements. 2nd ed. London and New York: Informa Healthcare; 2010:604-11.
- Klevay LM. Is the Western diet adequate in copper? J Trace Elem Med Biol. 2011 Dec;25(4):204-12. doi: 10.1016/j.jtemb.2011.08.146
- Tapiero H, Townsend DM, Tew KD. Trace elements in human physiology and pathology. Copper. Biomed Pharmacother. 2003 Nov;57(9):386-98. doi: 10.1016/s0753-3322(03)00012-x
- Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington (DC): National Academies Press (US); 2001. Available from: https://www.ncbi.nlm.nih.gov/books/NBK222310
- Chambers A, Krewski D, Birkett N, Plunkett L, Hertzberg R, Danzeisen R, Aggett PJ, Starr TB, Baker S, Dourson M, Jones P, Keen CL, Meek B, Schoeny R, Slob W. An exposure-response curve for copper excess and deficiency. J Toxicol Environ Health B Crit Rev. 2010 Oct;13(7-8):546-78. doi: 10.1080/10937404.2010.538657
- Nishiwaki S, Iwashita M, Goto N, Hayashi M, Takada J, Asano T, Tagami A, Hatakeyama H, Hayashi T, Maeda T, Saito K. Predominant copper deficiency during prolonged enteral nutrition through a jejunostomy tube compared to that through a gastrostomy tube. Clin Nutr. 2011 Oct;30(5):585-9. doi: 10.1016/j.clnu.2011.04.008
- Kelley DS, Daudu PA, Taylor PC, Mackey BE, Turnlund JR. Effects of low-copper diets on human immune response. Am J Clin Nutr. 1995 Aug;62(2):412-6. doi: 10.1093/ajcn/62.2.412
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington (DC): National Academies Press (US); 1998. Available from: https://www.ncbi.nlm.nih.gov/books/NBK114310
- Lecumberri E, Goya L, Mateos R, Alía M, Ramos S, Izquierdo-Pulido M, Bravo L. A diet rich in dietary fiber from cocoa improves lipid profile and reduces malondialdehyde in hypercholesterolemic rats. Nutrition. 2007 Apr;23(4):332-41. doi: 10.1016/j.nut.2007.01.013
- Menkes Disease. https://rarediseases.org/rare-diseases/menkes-disease
- Eid, C.; Hémadi, M.; Ha-Duong, N.T.; El Hage Chahine, J.M. Iron uptake and transfer from ceruloplasmin to transferrin. Biochim. Biophys. Acta 2014, 1840, 1771–1781.
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116C, 33–57.
- Hordyjewska, A.; Popiołek, L.; Kocot, J. The many “faces” of copper in medicine and treatment. Biometals 2014.
- Squitti, R.; Polimanti, R. Copper phenotype in Alzheimer’s disease: Dissecting the pathway. Am. J. Neurodegener. Dis. 2013, 2, 46–56.
- Thiele, D.J. Integrating trace element metabolism from the cell to the whole organism. J. Nutr. 2003, 133, 1579S–1580S.
- Linder, M.C. Biochemistry of Copper; Plenum Press: New York, NY, USA, 1991.
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.; Reyes, S.; Halliday, G.M.; Mercer, J.F.; et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol. Aging 2014, 35, 858–866.
- Hopt, A.; Korte, S.; Fink, H.; Panne, U.; Niessner, R.; Jahn, R.; Kretzschmar, H.; Herms, J. Methods for studying synaptosomal copper release. J. Neurosci. Methods 2003, 128, 159–172.
- Huffman, D.L.; O’Halloran, T.V. Function, structure, and mechanism of intracellular copper trafficking proteins. Annu. Rev. Biochem. 2001, 70, 677–701.
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808.
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458.
- Vashchenko, G.; MacGillivray, R.T. Multi-copper oxidases and human iron metabolism. Nutrients 2013, 5, 2289–2313.
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151.
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34.
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19.
- Schlief, M.L.; Craig, A.M.; Gitlin, J.D. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 2005, 25, 239–246.
- Vlachová, V.; Zemková, H.; Vyklický, L., Jr. Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. Eur. J. Neurosci. 1996, 8, 2257–2264.
- Schlief, M.L.; Gitlin, J.D. Copper homeostasis in the CNS: A novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol. Neurobiol. 2006, 33, 81–90.
- Weiser, T.; Wienrich, M. The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Res. 1996, 742, 211–218.
- Kardos, J.; Kovács, I.; Hajós, F.; Kálmán, M.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144.
- McGee, T.P.; Houston, C.M.; Brickley, S.G. Copper block of extrasynaptic GABAA receptors in the mature cerebellum and striatum. J. Neurosci. 2013, 33, 13431–13435.
- Collins JF. Copper nutrition and biochemistry and human (patho)physiology. Adv Food Nutr Res. 2021;96:311-364. doi: 10.1016/bs.afnr.2021.01.005
- Collins JF. Copper. In: Ross AC, Caballero B, Cousins RJ, Tucker KL, Ziegler TR, eds. Modern Nutrition in Health and Disease. 11th ed. Philadelphia: Wolter Kluwer; Lippincott Williams & Wilkins; 2014:206-216.
- Collins JF. Copper. In: Marriott BP, Birt DF, Stalling VA, Yates AA, eds. Present Knowledge in Nutrition. 11th ed: Academic Press; 2020:409-427.
- Vashchenko G, MacGillivray RT. Multi-copper oxidases and human iron metabolism. Nutrients. 2013 Jun 27;5(7):2289-313. doi: 10.3390/nu5072289
- Vasilyev VB. Looking for a partner: ceruloplasmin in protein-protein interactions. Biometals. 2019 Apr;32(2):195-210. doi: 10.1007/s10534-019-00189-1
- Meyer LA, Durley AP, Prohaska JR, Harris ZL. Copper transport and metabolism are normal in aceruloplasminemic mice. J Biol Chem. 2001 Sep 28;276(39):36857-61. doi: 10.1074/jbc.M105361200
- Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999 Sep 14;96(19):10812-7. doi: 10.1073/pnas.96.19.10812
- Kono S. Aceruloplasminemia. Curr Drug Targets. 2012 Aug;13(9):1190-9. doi: 10.2174/138945012802002320
- Thackeray EW, Sanderson SO, Fox JC, Kumar N. Hepatic iron overload or cirrhosis may occur in acquired copper deficiency and is likely mediated by hypoceruloplasminemia. J Clin Gastroenterol. 2011 Feb;45(2):153-8. doi: 10.1097/MCG.0b013e3181dc25f7
- Harris E. Copper. In: O’Dell B, Sunde R, eds. Handbook of Nutritionally Essential Minerals. New York: Marcel Dekker, Inc.; 1997:231-273.
- Johnson MA, Fischer JG, Kays SE. Is copper an antioxidant nutrient? Crit Rev Food Sci Nutr. 1992;32(1):1-31. doi: 10.1080/10408399209527583
- van den Berghe PV, Klomp LW. Posttranslational regulation of copper transporters. J Biol Inorg Chem. 2010 Jan;15(1):37-46. doi: 10.1007/s00775-009-0592-7
- Armendariz AD, Gonzalez M, Loguinov AV, Vulpe CD. Gene expression profiling in chronic copper overload reveals upregulation of Prnp and App. Physiol Genomics. 2004 Dec 15;20(1):45-54. doi: 10.1152/physiolgenomics.00196.2003
- Armendáriz AD, Olivares F, Pulgar R, Loguinov A, Cambiazo V, Vulpe CD, González M. Gene expression profiling in wild-type and metallothionein mutant fibroblast cell lines. Biol Res. 2006;39(1):125-42. doi: 10.4067/s0716-97602006000100015
- González M, Reyes-Jara A, Suazo M, Jo WJ, Vulpe C. Expression of copper-related genes in response to copper load. Am J Clin Nutr. 2008 Sep;88(3):830S-4S. doi: 10.1093/ajcn/88.3.830S
- Mattie MD, McElwee MK, Freedman JH. Mechanism of copper-activated transcription: activation of AP-1, and the JNK/SAPK and p38 signal transduction pathways. J Mol Biol. 2008 Nov 28;383(5):1008-18. doi: 10.1016/j.jmb.2008.08.080
- Gombart AF, Pierre A, Maggini S. A Review of Micronutrients and the Immune System-Working in Harmony to Reduce the Risk of Infection. Nutrients. 2020 Jan 16;12(1):236. doi: 10.3390/nu12010236
- Failla ML, Hopkins RG. Is low copper status immunosuppressive? Nutr Rev. 1998 Jan;56(1 Pt 2):S59-64. doi: 10.1111/j.1753-4887.1998.tb01646.x
- Percival SS. Copper and immunity. Am J Clin Nutr. 1998 May;67(5 Suppl):1064S-1068S. doi: 10.1093/ajcn/67.5.1064S
- Heresi G, Castillo-Duran C, Munoz C, Arevalo M, Schlesinger L. Phagocytosis and immunoglobulin levels in hypocupremic children. Nutr Res. 1985;5:1327-1334.
- Hodgkinson V, Petris MJ. Copper homeostasis at the host-pathogen interface. J Biol Chem. 2012 Apr 20;287(17):13549-55. doi: 10.1074/jbc.R111.316406
- Govind V, Bharadwaj S, Sai Ganesh MR, Vishnu J, Shankar KV, Shankar B, Rajesh R. Antiviral properties of copper and its alloys to inactivate covid-19 virus: a review. Biometals. 2021 Dec;34(6):1217-1235. doi: 10.1007/s10534-021-00339-4
- US Department of Agriculture, Agricultural Research Service. 2022. Nutrient Intakes from Food and Beverages: Mean Amounts Consumed per Individual, by Gender and Age, What We Eat in America, NHANES 2017-March 2020 Prepandemic. https://www.ars.usda.gov/ARSUserFiles/80400530/pdf/1720/Table_25_SNK_GEN_1720.pdf
- Copper. https://ods.od.nih.gov/factsheets/Copper-Consumer
- Food Labeling: Revision of the Nutrition and Supplement Facts Labels. https://www.federalregister.gov/documents/2016/05/27/2016-11867/food-labeling-revision-of-the-nutrition-and-supplement-facts-labels
- Rosado JL. Zinc and copper: proposed fortification levels and recommended zinc compounds. J Nutr. 2003 Sep;133(9):2985S-9S. doi: 10.1093/jn/133.9.2985S
- Pratt WB, Omdahl JL, Sorenson JR. Lack of effects of copper gluconate supplementation. Am J Clin Nutr. 1985 Oct;42(4):681-2. doi: 10.1093/ajcn/42.4.681
- Blumberg JB, Frei B, Fulgoni VL, Weaver CM, Zeisel SH. Contribution of Dietary Supplements to Nutritional Adequacy in Various Adult Age Groups. Nutrients. 2017 Dec 6;9(12):1325. doi: 10.3390/nu9121325
- Trocello JM, Broussolle E, Girardot-Tinant N, Pelosse M, Lachaux A, Lloyd C, Woimant F. Wilson’s disease, 100 years later…. Rev Neurol (Paris). 2013 Dec;169(12):936-43. doi: 10.1016/j.neurol.2013.05.002
- National Research Council Committee on Copper in Drinking Water. Copper in Drinking Water. Washington, DC: National Academies Press; 2000.
- Environmental Protection Agency. Electronic Code of Federal Regulations, Title 40, Part 141. https://www.ecfr.gov/current/title-40/chapter-I/subchapter-D/part-141/subpart-I
- Fitzgerald DJ. Safety guidelines for copper in water. Am J Clin Nutr. 1998 May;67(5 Suppl):1098S-1102S. doi: 10.1093/ajcn/67.5.1098S
- Patterson WP, Winkelmann M, Perry MC. Zinc-induced copper deficiency: megamineral sideroblastic anemia. Ann Intern Med. 1985 Sep;103(3):385-6. doi: 10.7326/0003-4819-103-3-385
- Schleper, B.; Stuerenburg, H.J. Copper deficiency-associated myelopathy in a 46-year-old woman. J. Neurol. 2001, 248, 705–706.
- Bennetts, H.W.; Chapman, F.E. Copper deficiency in sheep in Western Australia: A preliminary account of the aetiology of enzootic ataxia of lambs and anaemia of ewes. Aust. Vet. J. 1937, 13, 138–149.
- Lanska, D.J.; Remler, B. Myelopathy among zinc-smelter workers in Upper Silesia during the late 19th century. Neurology 2014, 82, 1175–1179.
- Gabreyes, A.A.; Abbasi, H.N.; Forbes, K.P.; McQuaker, G.; Duncan, A.; Morrison, I. Hypocupremia associated cytopenia and myelopathy: A national retrospective review. Eur. J. Haematol. 2013, 90, 1–9.
- Choi, E.H.; Strum, W. Hypocupremia-related myeloneuropathy following gastrojejunal bypass surgery. Ann. Nutr. MeTable 2010, 57, 190–192.
- Jaiser, S.R.; Winston, G.P. Copper deficiency myelopathy. J. Neurol. 2010, 257, 869–881.
- Mills ES. The treatment of idiopathic (hypochromic) anemia with iron and copper. Can Med Assoc J. 1930;22:175–178.
- Williams DM. Copper deficiency in humans. Semin Hematol. 1983;20:118–128.
- Halfdanarson TR, Kumar N, Li CY, Phyliky RL, Hogan WJ. Hematological manifestations of copper deficiency: a retrospective review. Eur J Haematol. 2008;80:523–531. doi: 10.1111/j.1600-0609.2008.01050.x
- Gregg XT, Reddy V, Prchal JT. Copper deficiency masquerading as myelodysplastic syndrome. Blood. 2002;100:1493–1495. doi: 10.1182/blood-2002-01-0256
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/S0092-8674(04)00343-5
- Petrak J, Vyoral D. Hephaestin—a ferroxidase of cellular iron export. Int J Biochem Cell Biol. 2005;37:1173–1178. doi: 10.1016/j.biocel.2004.12.007
- Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. Am J Clin Nutr. 1996;63:797S–811S
- Rock E, Gueux E, Mazur A, Motta C, Rayssiguier Y. Anemia in copper-deficient rats: role of alterations in erythrocyte membrane fluidity and oxidative damage. Am J Physiol. 1995;269:C1245–C1249.
- Percival SS. Neutropenia caused by copper deficiency: possible mechanisms of action. Nutr Rev. 1995;53:59–66. doi: 10.1111/j.1753-4887.1995.tb01503.x
- Familial benign copper deficiency. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=1551
- Familial benign copper deficiency: an old case re-examined. Acta Paediatr Hung. 1988-1989;29(3-4):313-5. https://www.ncbi.nlm.nih.gov/pubmed/2978614
- Méhes K, Petrovicz E. Familial benign copper deficiency. Arch Dis Child. 1982;57(9):716-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1627787/pdf/archdisch00754-0076.pdf
- Fujioka K. Follow-up of nutritional and metabolic problems after bariatric surgery. Diabetes Care. 2005;28:481–484. doi: 10.2337/diacare.28.2.481
- Kumar N. Copper deficiency myelopathy (human swayback) Mayo Clin Proc. 2006;81:1371–1384. doi: 10.4065/81.10.1371
- Schleper B, Stuerenburg HJ. Copper deficiency-associated myelopathy in a 46-year-old woman. J Neurol. 2001;248:705–706. doi: 10.1007/s004150170118
- Bartner R, Will M, Conrad J, Engelhardt A, Schwarz-Eywill M. Pancytopenia, arthralgia and myeloneuropathy due to copper deficiency. Med Klin (Munich) 2005;100:497–501. doi: 10.1007/s00063-005-1072-7
- Goldberg ME, Laczek J, Napierkowski JJ. Copper deficiency: a rare cause of ataxia following gastric bypass surgery. Am J Gastroenterol. 2008;103:1318–1319. doi: 10.1111/j.1572-0241.2007.01782_12.x
- Prodan CI, Bottomley SS, Vincent AS, Cowan LD, Greenwood-Van Meerveld B, Holland NR, Lind SE. Copper deficiency after gastric surgery: a reason for caution. Am J Med Sci. 2009;337:256–258. doi: 10.1097/MAJ.0b013e31818ad0ff
- Jaiser SR, Duddy M. Copper deficiency masquerading as subacute combined degeneration of the cord and myelodysplastic syndrome. Adv Clin Neurosci Rehabil. 2007;7:20–21.
- Jung A, Marziniak M. Copper deficiency as a treatable cause of myelopathy. Nervenarzt. 2008;79:421–425. doi: 10.1007/s00115-007-2397-7
- Rosa, F.W. 1986. Teratogen update: Penacillamine. Teratology 33(1):127–31.
- Keen, C.L., P. Mark-Savage, B. Lonnerdal, L.S. Hurley. 1983. b. Teratogenic effects of D-penicillamine in rats: relation to copper deficiency. Drug Nutr. Interact. 2(1):17–34.
- Mark-Savage, P., C.L. Keen, and L.S. Hurley. 1983. Reduction by copper supplementation of teratogenic effects of D-penicillamine. J. Nutr. 113(3):501–510.
- Jasim, S., B.R. Danielsson, H. Tjalve, and L. Dencker. 1985. Distribution of Cu in foetal and adult tissues in mice: influence of sodium diethyldithiocarbamate. Acta Pharmacol. Toxicol. (Copenhagen) 57(4): 262–270.
- Keen, C.L. 1996. Teratogenic effects of essential trace metals: deficiencies and excesses. Pp. 977–1001 in Toxicology of Metals, L.W. Chang, editor; , L. Magos, editor; , and T. Suzuki, editor. , eds. New York: CRC Press.
- Lee S.H., R. Lancey, A. Montaser, N. Madani, M.C. Linder. 1993. Ceruloplasmin and copper transport during the latter part of gestation in the rat. Proc. Soc. Exp. Biol. Med. 203(4):428–39.
- Turnlund, J.P. 1994. Copper. Pp. 231–241 in Modern Nutrition in Health and Disease, Vol. I., 8th Ed., M.E. Shils, editor; , J.A. Olson, editor; , and M. Shike, editor. , eds. Philadelphia: Lea and Febiger.
- Dubick, M.A., C.L. Keen, R.A. DiSilvestro, C.D. Eskelson, J. Ireton, G.C. Hunter. 1999. Antioxidant enzyme activity in human abdominal aortic aneurysmal and occlusive disease. Proc. Soc. Exp. Biol. Med. 220(1): 39–45.
- Speich, M., A. Murat, J.L. Auget, B. Bousquet, and P. Arnaud. 1992. Magnesium, total calcium, phosphorous, copper and Zn in plasma and erythrocytes of venous cord blood from infants of diabetic mothers: Comparison with a reference group by logistic discriminant analysis. Clin Chem. 38(10):2002–2007.
- Kiilholma, P., M. Gronroos, R. Erkkola, P. Pakarinen, and V. Nanto. 1984. The role of calcium, copper, iron and zinc in preterm delivery and premature rupture of fetal membranes. Gynecol. Obstet. Invest. 17(4):194–201.
- Turnlund JR. 1999. Copper. In: Shils ME, editor; , Olson JA, editor; , Shike M, editor; , Ross AC, editor. , eds. Modern Nutrition in Health and Disease , 9th ed. Baltimore: Williams & Wilkins. Pp.241–252.
- Botash AS, Nasca J, Dubowy R, Weinberger HL, Oliphant M. 1992. Zinc-induced copper deficiency in an infant. Am J Dis Child 146:709–711
- Brewer GJ, Hill GM, Prasad AS, Cossack ZT, Rabbani P. 1983. Oral zinc therapy for Wilson’s disease. Ann Intern Med 99:314–319.
- Yuzbasiyan-Gurkan V, Grider A, Nostrant T, Cousins RJ, Brewer GJ. 1992. Treatment of Wilson’s disease with zinc: X. Intestinal metallothionein induction. J Lab Clin Med 120:380–386.
- August D, Janghorbani M, Young VR. 1989. Determination of zinc and copper absorption at three dietary Zn-Cu ratios by using stable isotopic methods in young adult and elderly subjects. Am J Clin Nutr 50:1457–1463.
- Haschke F, Ziegler EE, Edwards BB, Foman SJ. 1986. Effect of iron fortification of infant formula on trace mineral absorption. J Pediatr Gastroenterol Nutr 5:768–773.
- Lonnerdal B, Hernell O. 1994. Iron, zinc, copper and selenium status of breast-fed infants and infants fed trace element fortified milk-based infant formula. Acta Paediatr 83:367–373.
- Fields M, Ferretti RJ, Smith JC, Reiser S. 1984. The interaction of type of dietary carbohydrates with copper deficiency. Am J Clin Nutr 39:289–295.
- Schoenemann HM, Failla ML, Steele NC. 1990. Consequences of severe copper deficiency are independent of dietary carbohydrate in young pigs. Am J Clin Nutr 52:147–154.
- Reiser S, Smith JC, Mertz W, Holbrook JT, Scholfield DJ, Powell AS, Canfield WK, Canary JJ. 1985. Indices of copper status in humans consuming a typical American diet containing either fructose or starch. Am J Clin Nutr 42:242–251.
- Mercer, J.F. 1998. Menkes syndrome and animal models. Am. J. Clin. Nutr. 67(5 Suppl.):1022S–1028S.
- Kaler, S.G. 1998. Diagnosis and therapy of Menkes syndrome, a genetic form of copper deficiency. Am. J. Clin. Nutr. 67(5 Suppl.):1029S–1034S
- Prohaska, J.R., T. Tamura, A.K. Percy, and J.R. Turnlund. 1997. In vitro copper stimulation of plasma peptidylglycine a-amidating monooxygenase in Menkes disease variant with occipital horns. Pediatr. Res. 42(6):862–865.
- Botero-López JE, Araya M, Parada A, Méndez MA, Pizarro F, Espinosa N, Canales P, Alarcón T. Micronutrient deficiencies in patients with typical and atypical celiac disease. J Pediatr Gastroenterol Nutr. 2011 Sep;53(3):265-70. doi: 10.1097/MPG.0b013e3181f988fc
- Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA; American College of Gastroenterology. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol. 2013 May;108(5):656-76; quiz 677. doi: 10.1038/ajg.2013.7
- Costa LS, Pegler SP, Lellis RF, Krebs VL, Robertson S, Morgan T, Honjo RS, Bertola DR, Kim CA. Menkes disease: importance of diagnosis with molecular analysis in the neonatal period. Rev Assoc Med Bras (1992). 2015 Sep-Oct;61(5):407-10. doi: 10.1590/1806-9282.61.05.407
- Hordyjewska A, Popiołek Ł, Kocot J. The many “faces” of copper in medicine and treatment. Biometals. 2014 Aug;27(4):611-21. doi: 10.1007/s10534-014-9736-5
- Kaler SG. Neurodevelopment and brain growth in classic Menkes disease is influenced by age and symptomatology at initiation of copper treatment. J Trace Elem Med Biol. 2014 Oct;28(4):427-30. doi: 10.1016/j.jtemb.2014.08.008
- Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008 Mar;4(3):176-85. doi: 10.1038/nchembio.72
- Jaiser SR, Winston GP. Copper deficiency myelopathy. J Neurol. 2010 Jun;257(6):869-81. doi: 10.1007/s00415-010-5511-x
- Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington (DC): National Academies Press (US); 2001. 7, Copper. Available from: https://www.ncbi.nlm.nih.gov/books/NBK222312
- Wasa M, Satani M, Tanano H, Nezu R, Takagi Y, Okada A. Copper deficiency with pancytopenia during total parenteral nutrition. JPEN J Parenter Enteral Nutr. 1994;18:190–192. doi: 10.1177/0148607194018002190
- Angotti LB, Post GR, Robinson NS, Lewis JA, Hudspeth MP, Lazarchick J. Pancytopenia with myelodysplasia due to copper deficiency. Pediatr Blood Cancer. 2008 Jul 11
- Kumar N, Ahlskog JE, Klein CJ, Port JD. Imaging features of copper deficiency myelopathy: a study of 25 cases. Neuroradiology. 2006;48:78–83. doi: 10.1007/s00234-005-0016-5
- Bennetts HW, Chapman FE. Copper deficiency in sheep in Western Australia: a preliminary account of the aetiology of enzootic ataxia of lambs and anaemia of ewes. Aust Vet J. 1937;13:138–149. doi: 10.1111/j.1751-0813.1937.tb04108.x
- Weihl CC, Lopate G. Motor neuron disease associated with copper deficiency. Muscle Nerve. 2006;34:789–793. doi: 10.1002/mus.20631
- Kumar N, Low PA. Myeloneuropathy and anemia due to copper malabsorption. J Neurol. 2004;251:747–749. doi: 10.1007/s00415-004-0428-x
- Prodan CI, Holland NR, Wisdom PJ, Burstein SA, Bottomley SS. CNS demyelination associated with copper deficiency and hyperzincemia. Neurology. 2002;59:1453–1456. doi: 10.1001/archneur.59.9.1453
- Nations SP, Boyer PJ, Love LA, Burritt MF, Butz JA, Wolfe GI, Hynan LS, Reisch J, Trivedi JR. Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology. 2008;71:639–643. doi: 10.1212/01.wnl.0000312375.79881.94
- Spinazzi M, De Lazzari F, Tavolato B, Angelini C, Manara R, Armani M. Myelo-optico-neuropathy in copper deficiency occurring after partial gastrectomy. Do small bowel bacterial overgrowth syndrome and occult zinc ingestion tip the balance? J Neurol. 2007;254:1012–1017. doi: 10.1007/s00415-006-0479-2
- Prodan CI, Holland NR, Wisdom PJ, Burstein SA, et al. CNS demyelination associated with copper deficiency and hyperzincemia. Neurology. 2002;59:1453–1456
- Prodan CI, Holland NR, Wisdom PJ, Burstein SA, et al. CNS demyelination associated with copper deficiency and hyperzincemia. Neurology. 2002;59:1453–1456.
- Bertinato J, Abbe MRL. Maintaining copper homeostasis: regulation of copper-traficking proteins in response to copper deficiency or overload. J Nutri Biochem. 2004;15:316–322.
- Jaiser SR, Winston GP. Copper deficiency myelopathy. J Neurol. 2010;257(6):869-81. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3691478/
- Prodan CI, Bottomley SS, Holland NR, Lind SE. Relapsing hypocupraemic myelopathy requiring high-dose oral copper replacement. J Neurol Neurosurg Psychiatry. 2006;77:1092–1093. doi: 10.1136/jnnp.2006.096883





{kind=link}