
What is cystic fibrosis
Cystic fibrosis is an inherited genetic disease characterized by the buildup of thick, sticky mucus that can damage many of your body’s organs – severely affecting your lungs, your digestive system (pancreas and liver) and sometimes the reproductive system. Cystic fibrosis affects the exocrine glands, which produce body fluids such as saliva, sweat, mucus, tears and enzymes. Mucus is a slippery substance that lubricates and protects the linings of the airways, digestive system, reproductive system, and other organs and tissues. In people with cystic fibrosis, the body produces mucus that is abnormally thick and sticky. This abnormal mucus can clog the airways, leading to severe problems with breathing and bacterial infections in the lungs. These infections cause chronic coughing, wheezing, and inflammation. Over time, mucus buildup and infections result in permanent lung damage, including the formation of scar tissue (fibrosis) and cysts in the lungs.
Most people with cystic fibrosis also have digestive problems. Some affected babies have meconium ileus, a blockage of the intestine that occurs shortly after birth. Other digestive problems result from a buildup of thick, sticky mucus in the pancreas. The pancreas is an organ that produces insulin (a hormone that helps control blood sugar levels). The pancreas also makes enzymes that help digest food. In people with cystic fibrosis, mucus blocks the ducts of the pancreas, reducing the production of insulin and preventing digestive enzymes from reaching the intestines to aid digestion. Problems with digestion can lead to diarrhea, malnutrition, poor growth, and weight loss. In adolescence or adulthood, a shortage of insulin can cause a form of diabetes known as cystic fibrosis-related diabetes mellitus.
A person who has cystic fibrosis often need to eat foods high in calories, fats, sugars and salt. However, the features of cystic fibrosis and their severity varies among affected individuals.
Cystic fibrosis is the most common life threatening genetic disease affecting children. Cystic fibrosis used to be considered a fatal disease of childhood. Through newborn screening using the heel prick test, most babies are diagnosed within the first few weeks of life. Every state in the U.S. now routinely screens newborns for cystic fibrosis. Early diagnosis means treatment can begin immediately. Symptoms usually start in early childhood and vary from child to child, but the condition gets slowly worse over time, with the lungs and digestive system becoming increasingly damaged. Advances in treatment have seen improved quality of life and life expectancy for those with cystic fibrosis, but there is no cure and complications can increase with age.
Cystic fibrosis is a common genetic disease within the white population in the United States. The disease occurs in 1 in 2,500 to 3,500 white newborns. Cystic fibrosis is less common in other ethnic groups, affecting about 1 in 17,000 African Americans and 1 in 31,000 Asian Americans.
Cystic fibrosis is an autosomal recessive condition – that means that a person needs two defective genes (one from each parent) to develop the condition. Approximately 12 million Americans or 1 in every 20 people living in this country, is a cystic fibrosis carrier. The gene that has been thought to cause cystic fibrosis is called cystic fibrosis transmembrane conductance regulator or CFTR. This gene has been located on chromosome 7.
In general, the condition results in defective chloride ion transport across the epithelial cells and increased viscosity of bodily secretions, specifically secretions from the respiratory tract (i.e. lungs, throat) and pancreas. As a result, the patient is predisposed to pancreatic insufficiency and recurrent chest infections. Due to the abnormal transport of chloride ions, this abnormality can be detected in sweat. This abnormality lead to a diagnostic test for cystic fibrosis, named the sweat test.
Adults with cystic fibrosis experience health problems affecting the respiratory, digestive, and reproductive systems. Most men with cystic fibrosis have congenital bilateral absence of the vas deferens, a condition in which the tubes that carry sperm (the vas deferens) are blocked by mucus and do not develop properly. Men with congenital bilateral absence of the vas deferens are unable to father children (infertile) unless they undergo fertility treatment. Women with cystic fibrosis may experience complications in pregnancy.
Although cystic fibrosis requires daily care, people with the condition are usually able to attend school and work, and often have a better quality of life than people with cystic fibrosis had in previous decades.
With improved treatments and better ways to manage the disease, many people with cystic fibrosis now live well into adulthood.
Cystic fibrosis prognosis
The severity and extent of the disease varies greatly amongst cystic fibrosis patients. Cystic fibrosis condition may also lead to bowel problems such as malaborption, bowel obstructions and constipation.
Due to recent developments into the management of cystic fibrosis, the prognosis has greatly improved. Over 90% of children diagnosed with cystic fibrosis will live to their teens. The estimated median survival age of children born in after 1990 will be 40 years. This may be further improved upon in the future by gene therapy techniques.
Cystic fibrosis life expectancy
Improvements in screening and treatments mean people with cystic fibrosis now may live into their mid- to late 30s, on average, and some are living into their 40s and 50s.
Cystic fibrosis causes
What causes cystic fibrosis
In cystic fibrosis, a defect (mutation) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene changes a protein that regulates the movement of salt in and out of cells. The result is thick, sticky mucus in the respiratory, digestive and reproductive systems, as well as increased salt in sweat.
Many different defects can occur in the gene. The type of gene mutation is associated with the severity of the condition.
Children need to inherit one copy of the gene from each parent in order to have the disease. If children inherit only one copy, they won’t develop cystic fibrosis. However, they will be carriers and possibly pass the gene to their own children.
Risk factors for cystic fibrosis
- Family history. Because cystic fibrosis is an inherited disorder, it runs in families.
- Race. Although cystic fibrosis occurs in all races, it is most common in white people of Northern European ancestry.
Cystic fibrosis genetics
The gene responsible for cystic fibrosis, the cystic fibrosis transmembrane conductance regulator (CFTR) gene was discovered in 1989. More than 2,000 mutations (different forms of the gene) have been discovered, but only a few are common. The cystic fibrosis transmembrane conductance regulator (CFTR) gene provides instructions for making a channel that transports negatively charged particles called chloride ions into and out of cells. Chloride is a component of sodium chloride, a common salt found in sweat. Chloride also has important functions in cells; for example, the flow of chloride ions helps control the movement of water in tissues, which is necessary for the production of thin, freely flowing mucus.
Mutations in the CFTR gene disrupt the function of the chloride channels, preventing them from regulating the flow of chloride ions and water across cell membranes. As a result, cells that line the passageways of the lungs, pancreas, and other organs produce mucus that is unusually thick and sticky. This mucus clogs the airways and various ducts, causing the characteristic signs and symptoms of cystic fibrosis.
Other genetic and environmental factors likely influence the severity of the condition. For example, mutations in genes other than CFTR might help explain why some people with cystic fibrosis are more severely affected than others. Most of these genetic changes have not been identified, however.
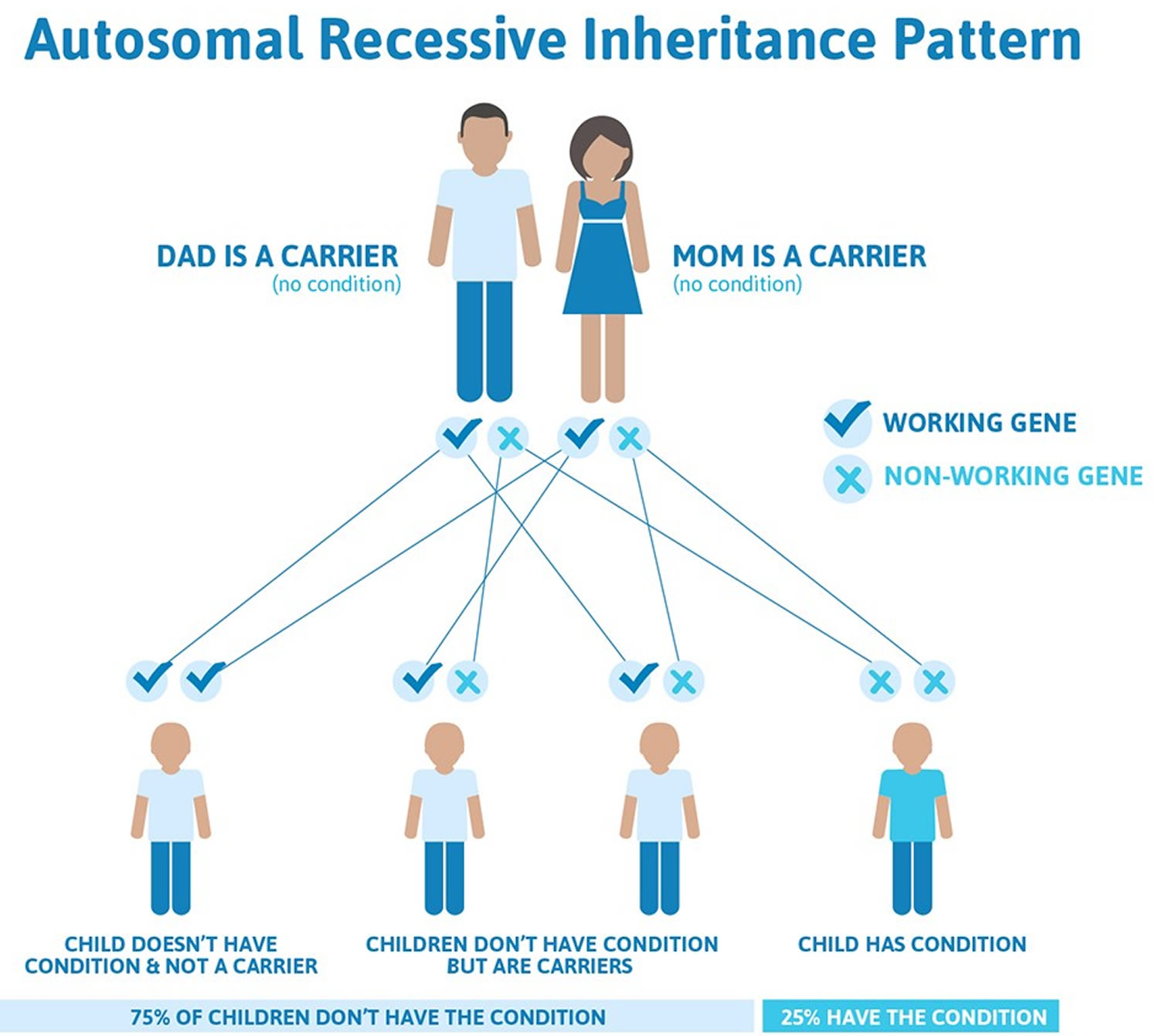
Cystic fibrosis is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
For a baby to be born with cystic fibrosis, both parents must be carriers of the faulty cystic fibrosis gene. The child will have inherited the faulty gene from each parent.
- Approximately 12 million Americans or 1 in every 20 people living in this country, is a cystic fibrosis carrier.
The carrier parents, who are not themselves affected by cystic fibrosis, have one unaffected gene and one faulty cystic fibrosis gene. If both parents are carriers, a child has:
- A one-in-four chance of being born with cystic fibrosis,
- A two-in-four chance of being a carrier, like their parents, but not having the disease, and
- A one-in-four chance of being completely free of the condition – neither having cystic fibrosis nor being a carrier of the faulty cystic fibrosis gene
Note that the odds are the same for each successive pregnancy.
Figure 1. Cystic fibrosis autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Cystic fibrosis carrier
People with only one faulty copy of the cystic fibrosis transmembrane conductance regulator (CFTR) gene that causes cystic fibrosis will not have the condition and are not at risk of developing it. Carriers do not experience any detrimental symptoms as a result of their status, although any children they have with another carrier could be born with cystic fibrosis.
Can I find out if my partner and I are carriers?
It is now possible to test for the CFTR gene using a simple mouthwash or blood test. Check with your local laboratory if mouthwash testing is available. In the case of a mouthwash test, cells lining the cheek are collected by spitting saliva into a special tube; DNA can be extracted from these cells and then analyzed. In relatives of people diagnosed with cystic fibrosis, the exact gene alterations found in their affected family member are established wherever possible, no matter how rare the alteration. A test for the more common gene alterations is performed on the partner who does not have a family history of the condition.
Because the test for common mutations only detects about 90 – 95% of mutations, a negative result does not entirely rule out the possibility of the person being a cystic fibrosis carrier, but the statistical likelihood is reduced to less than 1 in 250. Depending on the number of genetic cystic fibrosis alterations tested for in your health region, this figure may differ slightly. A positive result shows that the person is definitely a carrier, even if there is no family history.
Very occasionally, neither cystic fibrosis gene alteration is identified in the person with cystic fibrosis, because the mutations may be very rare ones that are not detected in the routine test. In these cases, testing for cystic fibrosis carriers in a relative becomes a little more complicated, but is generally still possible by testing the entire cystic fibrosis gene for mutations. Once the cystic fibrosis-carrying gene from each parent has been identified, it is then possible to test for that pattern in the relative. Except in the case of a full brother or sister of someone with cystic fibrosis, a negative result does not confirm with 100% certainty that you are not a carrier, but it does mean that you have a much lower risk. The Table 1 below shows how the level of risk of having a child with cystic fibrosis can be estimated once testing has taken place and suggests possible courses of action.
Table 1. Cystic Fibrosis – Family Genetic Testing
| Results | Risk of having a child with cystic fibrosis | Action |
|---|---|---|
| Both partners are carriers | 1 in 4 | - Genetic counseling. - Options are discussed including tests and support available in current or future pregnancy. - Pre-implantation genetic diagnosis is also explained. |
| Relative carrier, partner negative | Less than 1 in 1,000 | - Reassurance - Low Risk. Tests are not routinely offered in pregnancy - Check offspring for cystic fibrosis |
| Relative negative, partner carrier | Less than 1 in 2,000 Unless the relative is a full brother or sister of the person with cystic fibrosis, when a negative test means non-carrier (risk zero). | - Re-test to ensure there is no sample mix-up - Reassure – low risk - No routine tests in pregnancy - Check offspring for cystic fibrosis if sickly. |
| Both partners negative | Less than 1 in 250,000 | Strong reassurance |
What are the options if my partner and I are both carriers of the cystic fibrosis gene?
All available options will be explained during your genetic counseling session, so that you can decide what is right for you. All decisions are for you to make, and there will be trained staff available in the genetics department to clarify the situation and to discuss the implications of all possible courses of action. Antenatal tests, with the options of ending or continuing the pregnancy, are discussed, including any risks that may be associated with the tests. Pre-implantation genetic diagnosis using IVF (in-vitro fertilization) techniques to ensure that only an embryo free of cystic fibrosis is implanted into the womb are also explained.
The future
Work is being carried out on techniques capable of detecting cystic fibrosis cells in the embryo using a sample of the mother’s blood. This would avoid the need for chorionic biopsy (where a piece of the developing placenta is taken at 10 – 12 weeks) or amniocentesis (where a sample of the fluid surrounding the fetus is taken), the current tests available for antenatal diagnosis.
Also as our ability to detect more cystic fibrosis genes improves, it will be possible to test for the rarer mutations that currently make the carrier status test less than 100% accurate due to missed, rare or previously unidentified mutations. This is called a false-negative result.
Home testing
The Cystic Fibrosis Foundation recommends that genetic testing is undertaken with professional clinical support and that the results of a genetic test are discussed with a genetic counselor, who can explain in full the results you may get and the options available to you as an individual.
Using a home genetic testing kit would mean that you do not have professional support available to you if and when you need it, as well as being less reliable. For these reasons, the Cystic Fibrosis Foundation advises against the use of home genetic testing kits.
Cystic fibrosis prevention
If you or your partner has close relatives with cystic fibrosis, you both may want to undergo genetic testing before having children. The test, which is performed in a lab on a sample of blood, can help determine your risk of having a child with cystic fibrosis.
If you’re already pregnant and the genetic test shows that your baby may be at risk of cystic fibrosis, your doctor can conduct additional tests on your developing child.
Genetic testing isn’t for everyone. Before you decide to be tested, you should talk to a genetic counselor about the psychological impact the test results might carry.
Cystic fibrosis symptoms
Cystic fibrosis signs and symptoms vary, depending on the severity of the disease. Even in the same person, symptoms may worsen or improve as time passes. Some people may not experience symptoms until adolescence or adulthood.
Children presenting with cystic fibrosis may experience recurrent chest infections and failure to thrive (i.e. not growing to normal weight and height compared to children of the same age). The child may also suffer from very smelly, light colored stools. 10% of children that had a history of meconium ileus will have cystic fibrosis.
The build-up of sticky mucus in the lungs can cause breathing problems and increases the risk of lung infections. Over time, the lungs may stop working properly.
Mucus also clogs the pancreas (the organ that helps with digestion), which stops enzymes reaching food in the gut and helping with digestion.
This means most people with cystic fibrosis don’t absorb nutrients from food properly and need to eat more calories to avoid malnutrition.
Symptoms of cystic fibrosis include:
- recurring chest infections
- wheezing, coughing, shortness of breath and damage to the airways (bronchiectasis)
- difficulty putting on weight and growing
- jaundice
- diarrhea, constipation, or large, smelly poo
- a bowel obstruction in newborn babies (meconium ileus) – surgery may be needed
People with the condition can also develop a number of related conditions, including diabetes, thin, weakened bones (osteoporosis), infertility in males, and liver problems.
Respiratory signs and symptoms
The thick and sticky mucus associated with cystic fibrosis clogs the tubes that carry air in and out of your lungs. This can cause signs and symptoms such as:
- A persistent cough that produces thick mucus (sputum)
- Wheezing
- Breathlessness
- Exercise intolerance
- Repeated lung infections
- Inflamed nasal passages or a stuffy nose
Digestive signs and symptoms
The thick mucus can also block tubes that carry digestive enzymes from your pancreas to your small intestine. Without these digestive enzymes, your intestines aren’t able to completely absorb the nutrients in the food you eat. The result is often:
- Foul-smelling, greasy stools
- Poor weight gain and growth
- Intestinal blockage, particularly in newborns (meconium ileus)
- Severe constipation
Frequent straining while passing stool can cause part of the rectum — the end of the large intestine — to protrude outside the anus (rectal prolapse). When this occurs in children, it may be a sign of cystic fibrosis. Parents should consult a physician knowledgeable about cystic fibrosis. Rectal prolapse in children may sometimes require surgery. Rectal prolapse in children with cystic fibrosis is less common than it was in the past, which may be due to earlier testing, diagnosis and treatment of cystic fibrosis.
Cystic fibrosis diagnosis
In United States, all newborn babies are screened for cystic fibrosis as part of the newborn blood spot test (heel prick test) carried out shortly after they’re born.
A heel prick blood test is performed to check for the enzyme IRT (immune reactive trypsin) is measured. If the levels are high, its suggestive of cystic fibrosis and the infants undergoes a sweat test.
Cystic fibrosis testing
If the screening test suggests a child may have cystic fibrosis, they’ll need these additional tests to confirm they have the condition:
- The sweat test. This is the gold-standard diagnostic test. It is based on the pathological feature of cystic fibrosis, which is abnormal chloride ion transport. If the patient has two successive tests showing chloride levels above 60mmol/L (normal is under 15mmol/L), its conclusive that the patient has cystic fibrosis.
- A genetic test (CF genotyping) may be used to confirm the diagnosis. A sample of blood or saliva is checked for the faulty gene that causes cystic fibrosis
- A blood test is also performed on infants.
These tests can also be used to diagnose cystic fibrosis in older children and adults who didn’t have the newborn test.
The genetic test can also be used to see whether someone is a “carrier” of cystic fibrosis in cases where the condition runs in the family.
This test can be important for someone who thinks they may have the faulty gene and wishes to have children.
Cystic fibrosis complications
People with cystic fibrosis also have a higher risk of developing other conditions.
These include:
- weak and brittle bones (osteoporosis). People with cystic fibrosis are at higher risk of developing a dangerous thinning of bones – medicines called bisphosphonates can sometimes help
- electrolyte imbalances and dehydration. Because people with cystic fibrosis have saltier sweat, the balance of minerals in their blood may be upset. Signs and symptoms include increased heart rate, fatigue, weakness and low blood pressure.
- cystic fibrosis-related diabetes – insulin and a special diet may be needed to control blood sugar levels
- nasal polyps and sinus infections – steroids, antihistamines, antibiotics or sinus flushes can help
- liver problems
- fertility problems – it’s possible for women with cystic fibrosis to have children, but men won’t be able to father a child without help from fertility specialists (see a doctor or fertility specialist for more advice)
They’re more likely to pick up infections, and more vulnerable to complications if they do develop an infection, which is why people with cystic fibrosis shouldn’t meet face to face.
Respiratory system complications
- Damaged airways (bronchiectasis). Cystic fibrosis is one of the leading causes of bronchiectasis, a condition that damages the airways. This makes it harder to move air in and out of the lungs and clear mucus from the airways (bronchial tubes).
- Chronic infections. Thick mucus in the lungs and sinuses provides an ideal breeding ground for bacteria and fungi. People with cystic fibrosis may often have sinus infections, bronchitis or pneumonia.
- Growths in the nose (nasal polyps). Because the lining inside the nose is inflamed and swollen, it can develop soft, fleshy growths (polyps).
- Coughing up blood (hemoptysis). Over time, cystic fibrosis can cause thinning of the airway walls. As a result, teenagers and adults with cystic fibrosis may cough up blood.
- Pneumothorax. This condition, in which air collects in the space that separates the lungs from the chest wall, also is more common in older people with cystic fibrosis. Pneumothorax can cause chest pain and breathlessness.
- Respiratory failure. Over time, cystic fibrosis can damage lung tissue so badly that it no longer works. Lung function usually worsens gradually, and it eventually can become life-threatening.
- Acute exacerbations. People with cystic fibrosis may experience worsening of their respiratory symptoms, such as coughing and shortness of breath, for several days to weeks. This is called an acute exacerbation and requires treatment in the hospital.
Digestive system complications
- Nutritional deficiencies. Thick mucus can block the tubes that carry digestive enzymes from your pancreas to your intestines. Without these enzymes, your body can’t absorb protein, fats or fat-soluble vitamins.
- Diabetes. The pancreas produces insulin, which your body needs to use sugar. Cystic fibrosis increases the risk of diabetes. Around 30 percent of people with cystic fibrosis develop diabetes by age 30.
- Blocked bile duct. The tube that carries bile from your liver and gallbladder to your small intestine may become blocked and inflamed, leading to liver problems and sometimes gallstones.
- Intestinal obstruction. Intestinal obstruction can happen to people with cystic fibrosis at all ages. Children and adults with cystic fibrosis are more likely than are infants to develop intussusception, a condition in which a section of the intestines folds in on itself like an accordion.
- Distal intestinal obstruction syndrome (DIOS). DIOS is partial or complete obstruction where the small intestine meets the large intestine.
Reproductive system complications
Almost all men with cystic fibrosis are infertile because the tube that connects the testes and prostate gland (vas deferens) is either blocked with mucus or missing entirely. Certain fertility treatments and surgical procedures sometimes make it possible for men with cystic fibrosis to become biological fathers.
Although women with cystic fibrosis may be less fertile than other women, it’s possible for them to conceive and to have successful pregnancies. Still, pregnancy can worsen the signs and symptoms of cystic fibrosis, so be sure to discuss the possible risks with your doctor.
Cystic fibrosis treatment
There’s no cure for cystic fibrosis, but a range of treatments can help control the symptoms, prevent or reduce complications, and make the condition easier to live with.
Cystic fibrosis is a disease that affects many systems. Therefore, its essential that a multidisciplinary approach is taken in the management of this disease.
The main goals in the management of cystic fibrosis are:
- Promote sufficient growth and nutrition
- Preventing and controlling infections that occur in the lungs
- Removing and loosening mucus from the lungs
- Prevent the progression of further lung disease
- Treating and preventing intestinal blockage
This is achieved with a team of health professionals including pediatricians, physiotherapists, dieticians, counselors and family doctors. Specific treatments for cystic fibrosis are:
- Antibiotics to reduce risk of chest infections
- Pain relievers such as Ibuprofen may reduce lung deterioration
- Mucus-thinning drugs to reduce viscosity of mucus, making it easier to breathe
- Bronchodilators to increase air entry
- Physio exercises to remove mucus in the chest
- Improved nutrition ( high calorie diet, vitamin and mineral rich) to maintain weight and ensure essential nutrients are consumed
- Refer for counseling and support groups as chronic diseases can cause a great amount of stress for the patient and family
- Oral enzymes should be taken to aid in digestion and absorption of food
- In severe cases, a lung transplant may be suggested due to severity of disease and poor quality of life
Medicines for lung problems
People with cystic fibrosis may need to take different medicines to treat and prevent lung problems. These may be swallowed, inhaled or injected.
Medicines for lung problems include:
- antibiotics to prevent and treat chest infections
- medicines to make the mucus in the lungs thinner and easier to cough up – for example, dornase alfa, hypertonic saline and mannitol dry powder
- medicine to help reduce the levels of mucus in the body – for example, ivacaftor taken on its own (Kalydeco) or in combination with lumacaftor (Orkambi, but this is only available on compassionate grounds if people fulfill several criteria set by the manufacturer)
- bronchodilators to widen the airways and make breathing easier
- steroid medicine to treat small growths inside the nose (nasal polyps)
It’s also important that people with cystic fibrosis are up-to-date with all routine vaccinations and have the flu jab each year once they’re old enough.
Exercise
Any kind of physical activity, like running, swimming or football, can help clear mucus from the lungs and improve physical strength and overall health.
A physiotherapist can advise on the right exercises and activities for each individual.
Airway clearance techniques
Airway clearance helps to stop thick mucus from building up and blocking airways in the lungs, reducing infections and preventing lung damage.
There are many different techniques, and your specialist cystic fibrosis physiotherapist will work closely with you to find the technique, or combination of techniques, that works best for you. Everyone’s lungs are different and the amount of airway clearance needed will vary from person to person. No one technique has been shown to be better than another, and this may be because each person’s chest can behave very differently, and people may prefer different techniques at different times in their life. It is important that you are happy with the technique you’re using as, despite it being essential, physiotherapy to clear the chest can be a treatment that people struggle to do regularly. Some techniques require no equipment, and others use a device.
These include:
- Active Cycle of Breathing Techniques – a cycle of deep breathing, huffing, coughing and relaxed breathing to move mucus.
- Autogenic Drainage – a series of gentle controlled breathing techniques that moves mucus from smaller to bigger airways using controlled breathing at different levels. Uses fast flow rates as you sigh out but ensures the airways are kept open.
- Positive Expiratory Pressure – breathing out through a mask or mouthpiece against a resistance to build up pressure behind mucus to move it.
- Oscillating Positive Expiratory Pressure e.g. Flutter®, Acapella® – breathing out against a resistance to create positive expiratory pressure and also vibrate your airways to move mucus.
- High Frequency Chest Wall Oscillation (the vest) – an electric air pulse generator that connects to an inflatable jacket (vest) to vibrate the chest.
The evidence for airway clearance techniques in people with cystic fibrosis has been analyzed in five Cochrane reviews published between 2000 and 2011. There is currently insufficient evidence to suggest that one technique is better than another.
However two recent well-constructed studies in cystic fibrosis show that if using High Frequency Vest Oscillation, (the Vest), the patients may not breathe as well using it or may get more infections and get them more quickly. These studies were done in children. There are at least two studies in adults that show less or just the same amount of secretions cleared with the Vest compared to other techniques.
The Association of Chartered Physiotherapists in Cystic Fibrosis therefore recommends that the Vest is not used as the only airway clearance technique especially in children with cystic fibrosis. The Association of Chartered Physiotherapists in Cystic Fibrosis recognizes that some patients may prefer to use the Vest and feel benefit from using it. We advise people thinking about wanting to use the Vest to discuss the pros and cons with their physiotherapist. The Association of Chartered Physiotherapists in Cystic Fibrosis recommends that for people with cystic fibrosis choosing to use the Vest some other airway clearance technique such as at least huffing and coughing is done with the Vest on.
Physiotherapists should consider patient choice and circumstances, and clinical reasoning of the person with cystic fibrosis’s signs and symptoms when recommending techniques. Techniques may be combined depending on patient needs.
There are other devices not named above. Be sure to discuss any new airway clearance technique that you would like to try with your cystic fibrosis physiotherapist. They will discuss whether there is good research for the device and whether there are any positives and/or negatives to consider.
How much airway clearance do you need?
The length and number of airway clearance sessions will vary from person to person, and may need to increase if you have a chest infection.
For few or no lung secretions, treatment sessions may only need to last 10–15 minutes, but if there are many it could take 45–60 minutes. Most people do two treatments a day, but the number and length of your sessions may need to increase depending on the volume of secretions your lungs are producing. If no mucus is present, exercise may play a greater role in your airway clearance techniques. Your cystic fibrosis physiotherapist will be able to advise what is suitable for you.
When should airway clearance start?
A specialist cystic fibrosis physiotherapist should assess all newly diagnosed people with cystic fibrosis and advise when and how they should start doing airway clearance. This may not be immediately in screened infants with no symptoms, and exercise/activity may play a greater role. At all ages, the advice will be based on an individual assessment.
Who will do my airway clearance?
The families of babies with cystic fibrosis will be taught how to do airway clearance with their child. As you grow older with cystic fibrosis, your physiotherapist will aim to find a technique that you can carry out independently so that you do not have to rely on someone else to help. Breathing exercises can be introduced from the age of two to three in the form of a game. Older children can start doing airway clearance themselves with supervision. Most teenagers and adults become completely independent, and only need help if they have increased mucus.
What about exercise?
Exercise helps with general fitness, health and well-being, and bone health. Improved fitness has been shown to be important in the long term prognosis of cystic fibrosis, and people with cystic fibrosis should aim to maintain an active lifestyle. Physical exercise is also often used as part of an airway clearance regime. Exercise can help loosen mucus in the lungs and make airway clearance techniques quicker and easier. Your cystic fibrosis physiotherapist should be able to answer any questions about the type and intensity of exercise suitable for you.
What about your posture?
As you get older it is easy to adopt poor postures, which can lead to back pain and impact upon lung function. It is therefore important for people with cystic fibrosis to maintain good posture. Your physiotherapist will be able to offer advice and help to assess and treat any joint or back problems you may have.
What about incontinence?
Some people with cystic fibrosis may develop problems with urinary or fecal incontinence (leaking wee or poo) when coughing and/or exerting themselves. If this should happen to you it is important to tell your physiotherapist as soon as possible and not be embarrassed, as they can teach you pelvic floor exercises to help. They may also refer you on to a specialist. For more information see our fact sheet.
Inhalation therapy
Many people with cystic fibrosis will take inhaled medications as part of their treatment. This may be to loosen the mucus, hydrate the airways, open up the airways, reduce inflammation of the airways and/or to deliver antibiotics to treat infections. These medications may come in nebulized (inhaling a mist) or dry powder form, and often the timing of these alongside airway clearance and exercise is important to ensure your treatment is as effective as possible. Please ask your doctor for advice.
Cystic fibrosis diet
Eating well is important for people with cystic fibrosis because the mucus can make it difficult to digest food and absorb nutrients.
The pancreas often doesn’t work properly, making it even harder to digest food.
A dietitian will advise on how to take in extra calories and nutrients to avoid malnutrition.
They may recommend a high-calorie diet, vitamin and mineral supplements, and taking digestive enzyme capsules with food to help with digestion.
Only around 10% of people with cystic fibrosis retain any useful pancreatic function, meaning that digestive juices (enzymes) do not reach the stomach in order to break down food properly so that the body can use it to produce energy.
This means that most people with cystic fibrosis require enzyme capsules with all meals and snacks. The amount of capsules needed depends on the food being eaten, and varies from person to person, but it can be as many as 60 a day.
Being a healthy weight can improve the chances of fighting off chest infections, so a suitable diet is important. It is also necessary to build up reserves in case of weight loss during times of illness.
A healthy diet for someone with cystic fibrosis is:
- High in energy (calories) – the body of someone with cystic fibrosis has to work harder, and has higher energy requirements.
- Rich in fat and protein – to compensate for the amount wasted as food is not fully digested.
As with any diet, the exact amount of these elements varies by age and from person to person.
As a general principle, people with cystic fibrosis often require 20 to 50% more calories each day than people without cystic fibrosis, however some may need considerably more than this.
This high calorie intake is often hard to achieve as many people with cystic fibrosis experience reduced appetite, especially during episodes of infection – the very time when your body’s energy requirements are at their highest. Many people with cystic fibrosis need to take enzymes to digest and absorb fat-containing foods. Forgetting or taking too few enzymes will cause you to lose fat (energy) in your stools and this results in some of the fat you have eaten being wasted. This can also lead to weight loss or difficulty in gaining weight despite eating well. Even if you are taking your enzymes appropriately you will continue to lose some fat in your stools and this contributes to your increased energy requirements.
What is a high energy diet?
This is a diet that contains an increased amount of calories. Calories are a measure of how much energy a food contains. Dietary fat provides the most energy (calories) in the smallest volume and is the reason that your dietician encourage you to use high fat foods and snacks whenever possible, together with the appropriate number of enzymes. This should be in combination with the right balance of foods from other food groups – starchy foods (bread, pasta, rice and cereals), proteins (meat, fish, cheese and meat substitutes), dairy foods (milk, cheese, and yogurts) and fruit and vegetables.
Although a diet high in energy can be hard to achieve, there are simple guidelines that you can follow to help increase your calorie intake:
Fatty foods
- Fat is the richest source of energy (calories). One gram of fat contains nine calories, which is more than twice the amount found in protein or carbohydrate. Fat is also a good source of essential fatty acids and fat soluble vitamins
- Where possible you should choose unsaturated fats and oils as they give the same amount of calories but in a healthier form. Monounsaturated fats include olive oil and rapeseed oil. Polyunsaturated fats include sunflower, soya, sesame and corn oil. Soft spreads made from olive oil, rapeseed oil etc can be used to replace butter and lard
- The essential omega 3 fats are also important and are found mainly in oily fish. You should try to include these in your diet
- Try to eat plenty of fat, making sure you take enough enzymes with your food, as fatty foods require more enzymes
- Fry your food in olive oil or rapeseed oil, add olive oil, butter or margarine to vegetables and potatoes and spread butter or margarine generously on bread
- Olive oil can be drizzled on food to increase the flavor and energy content
- Toss pasta in olive oil before serving or drizzle olive oil over pasta dishes
- Add mayonnaise, olive oil, dips or dressings to sandwiches and salads
- Choose higher fat foods such as pastries, crisps, nuts, seeds, chocolate, cakes, biscuits and chips
- Single, double or whipped cream can be added to puddings, sauces and soups
- Add extra olive oil/butter/margarine to vegetables, potatoes, pasta, bread and toast
- Try to have more fried foods
- When grilling or roasting foods, drizzle them with olive oil or vegetable oil and baste with oil regularly
- Add olive oil/mayonnaise/salad cream to sandwiches and salads
- Always add a dressing (oil or mayonnaise) to your salads
- Avoid using low fat spread or eating reduced fat foods
Starchy (carbohydrate) foods
- Starchy foods are another good energy provider and should be a part of every meal
- Starchy foods include breakfast cereals, pasta, potatoes, rice and bread
- Make them higher in calories by brushing with olive oil, garnishing with a drizzle of oil, frying them or adding lots of milk, butter, olive oil, mayonnaise etc
- Mash potatoes with butter, olive oil spread or margarine and mix in some cream, crème fraiche or cheese
- Toss pasta in olive oil before serving or drizzle olive oil over pasta dishes
- Add chopped nuts or dried fruit to cereal, yogurt, milk pudding, fruit and ice cream
Sugary foods
- Sugary foods such as jam, honey, marmalade, syrup, fizzy drinks, tinned fruit in syrup, cakes, biscuits, sweets and chocolates are also energy rich
- Put plenty of sugar into hot drinks, on cereals and in desserts
- Spread jam, honey, marmalade and chocolate spread thickly on bread and toast
- Add sugar, honey, jam or syrup to cereal, yogurt, milk pudding, fruit and ice cream
- Avoid foods which are labelled as being ‘low sugar’ or sweetened with artificial sweetener, such as diet yogurts, diet squashes and diet fizzy drinks
Your teeth are very important to your health. Remember to clean your teeth after eating sugary food or taking sweetened drinks and to visit your dentist regularly.
Milk and dairy products
- Avoid low fat dairy products such as low fat yogurts, semi-skimmed and skimmed milk and low fat cheeses such as cottage cheese. If these are the only types of dairy products you like they are still a good source of calcium
- Milk and dairy products are an important source of energy and calcium
- Use full cream or Channel Island milk and try to have between one and two pints daily in drinks, desserts such as custard, instant whips or milk pudding, savoury sauces such as cheese or parsley sauce and on cereals
- Add double cream to ordinary milk when making any milk-based savory and sweet dishes or milky drinks
- Use milk instead of water when making up condensed or packet soup.
- Stir in double cream or olive oil just before serving
- Always add milk and olive oil/butter/margarine to mashed potato
- Cheese is an ideal snack; serve as cheese on toast, cheese and crackers etc. In addition cheese can be used to add extra calories to your food, for example it
- can be added to mashed potatoes, baked beans or pasta dishes
- Sprinkle grated cheese on top of vegetables, potatoes, soups and sauces
- Yogurts and ice cream make a quick dessert or snack. Try to choose whole milk, thick and creamy, Greek- or custard- style yoghurts and real dairy ice creams
Protein foods
- Protein foods come in two varieties:
- Animal proteins such as meat, fish, eggs, milk and dairy products
- Vegetable proteins such as beans, peas, lentils and nuts
- Try to keep your protein intake high by having a good helping at each meal, including breakfast. It will also help if your snacks include some protein
- Fatty or oil-rich fish are good source of protein as well as a good source of the omega 3 fatty acids. Try eating oil-rich fish such as salmon, herring, sardines,
- mackerel, pilchards, trout, kippers and fresh tuna regularly
- If you are vegetarian make sure you replace meat or fish with a variety of protein sources such as soya mince or other meat substitutes, beans, seeds, lentils, tofu, nuts, cheese or eggs
- Avoid eating raw eggs and do not add them to high energy drinks. They do not add many calories and may be a Salmonella risk
Fruit and vegetables
- Fruit and vegetables are generally low in energy, but do provide vitamins and some minerals, so they are an important part of a balanced diet
- Try to eat fruit and vegetables everyday – fresh, frozen or tinned – but if your appetite is poor do not fill yourself up with fruit and vegetables at the expense of higher calorie foods
- Some fruit and vegetables e.g. bananas, beans, pulses and root vegetables such as potato, turnip and parsnip are higher in calories
- Try to have a glass of fruit juice or squash with added vitamin C daily
- Use fruit in pies and crumbles or serve with double cream or dairy ice cream
- Add double cream, ice cream or custard to fruit
- Add sauces, olive oil or butter to vegetables or fry them
Calcium
- Calcium is essential to the body for maintaining strong and healthy bones. Therefore it is important that you have plenty of calcium rich foods each day
- The best sources of calcium are dairy products such as milky drinks, cheese, yogurts and dairy ice cream
- If you don’t like these types of foods, non-dairy calcium sources include baked beans, tinned fish with bones (salmon, sardines and pilchards) and white bread
- Also look out for foods that may have calcium added to them such as soya milk, drinks and cereal bars
- Generally non-dairy foods contain less calcium and therefore you will need to eat more of them to ensure an adequate calcium intake. If you only like low fat varieties of milk and milk products continue to have these as they contain similar amounts of calcium as full cream versions
- If you are concerned about your calcium intake talk to your cystic fibrosis dietitian
Is this really a healthy diet?
For most people a high fat and high calorie diet would not be considered a healthy diet. However for many people with cystic fibrosis this is the most appropriate diet to help achieve and maintain a good healthy weight. This in turn helps to fight infections and stay healthy for longer. It is almost the opposite of the diet recommended for most other adults which is low in fat, sugar and salt with an increased intake of dietary fiber. This type of diet is not suitable for most people with cystic fibrosis because it is bulky and filling and is unlikely to provide enough energy.
Not everyone is the same and some people with cystic fibrosis may gain too much weight. These patients may need to reduce their fat and energy intake and should be assessed and advised on an individual basis by a specialist cystic fibrosis dietitian.
To give your high fat diet a more healthy emphasis where possible you should try to use monounsaturated cooking oils or spreads such as olive oil, rapeseed oil, olive oil spread instead of lard or butter and to eat fruit and vegetables daily. These can be incorporated as high fiber, high energy snacks including dried fruit, fruit pies and crumbles served with cream or ice cream. Additional fat e.g. olive oil, butter, cream, cheese can be added to dishes after the rest of the family has been served to ensure the whole family get the type of diet they need.
Pancreatic enzymes
Approximately 85% or more of adults with cystic fibrosis are pancreatic insufficient. This means the pancreas is unable to produce or release enough digestive enzymes into the small intestine and so food is not digested and can not be absorbed in the gut. To help to compensate for the lack of digestive enzymes most people with cystic fibrosis have to take replacement pancreatic enzymes to digest their food properly. The enzyme dose needed varies from one adult to another. Your cystic fibrosis team will advise on appropriate enzyme doses and it is important to discuss any changes to your enzyme intake with the team before changing our dose. There is a wide range of strengths of pancreatic enzymes available. When taking pancreatic enzymes it is also important to drink plenty of fluids.
Making your enzymes work for you
To get the most benefit from your enzymes, follow these guidelines:
- Take enzymes with every meal, fat-containing snack or milky drink
- Spread your enzymes throughout the meal
- High fat meals will need more enzymes than low fat meals
- Most snacks will require fewer enzymes than a meal but be cautious as some snacks are very fatty and can require as many or even more enzymes than a meal
- Be flexible about the dosage and timing of enzymes
- Enzymes do not need to be taken with fruit, jelly, sorbet, boiled, chewy or jelly sweets, squash, fizzy drinks or fruit juices
Remember there is no standard dose of enzyme. You should take the amount required to control your bowels. Sometimes people with cystic fibrosis produce too much stomach acid and this can make pancreatic enzymes less effective. If you are experiencing problems with heartburn, reflux or abdominal cramps then please discuss with your cystic fibrosis team and they can prescribe appropriate medications to help.
Distal intestinal obstruction syndrome, fluid and fiber
Distal intestinal obstruction syndrome (a blockage in the bowel) – usually referred to as DIOS – may be caused by not taking enough enzymes and if you are dehydrated this can make things worse. Sometimes it can occur for no known reason. You should make sure you drink enough fluid and aim to have 30 ml per kg body weight of fluid each day. For example if you weigh 50 kg this is 1500 ml per day, the equivalent of about six to eight glasses/mugs of liquid each day.
- Some drinks such as sweet, fizzy caffeine-containing drinks or strong coffee may dehydrate you further. Water or diluted squash are best
- When the weather is hot or you are in a hot room you will need more fluid
- If you are doing a lot of exercise the fluid you lose by sweating will need to be replaced by drinking more
- Your body will lose fluid if your blood sugars are too high. Make sure that if you have diabetes it is well controlled
- Taking more fiber in your diet may help prevent distal intestinal obstruction syndrome. Try including a high fiber cereal at breakfast, some wholemeal bread, and increasing the amount of fruit and vegetables you eat. If you increase your fiber intake you will need to increase the amount of fluid you drink.
Vitamin supplements
Malabsorption of the fat-soluble vitamins A, D, E and K is likely in most people with cystic fibrosis, especially those who are pancreatic insufficient. Without supplements, blood levels of these vitamins may become low and occasionally deficiency symptoms can occur. Fat-soluble vitamins should be taken at a mealtime when enzymes will also be taken.
Recommended daily supplements (starting doses):
- Vitamin A – 4000-8000 iu (International Units)
- Vitamin D – 400-800 iu
- Vitamin E – 50-200 iu
Diabetes is common in adults and adolescents with cystic fibrosis. It occurs in approximately 30% of people with cystic fibrosis by the age of 25 years. Cystic fibrosis-related diabetes is less common in children.
Cystic fibrosis-related diabetes is different from the two main types of diabetes (Type 1 and Type 2 diabetes) and has features of both.
In addition, people with cystic fibrosis may develop high blood sugar levels
during periods of lung infection or while taking oral steroids. This may be temporary and may resolve when the acute infection is treated or when the steroids are reduced or stopped.
Some people may be concerned that the sugary diet that they have eaten may contribute to them developing cystic fibrosis-related diabetes. There are many other risk factors that cause or contribute to people with cystic fibrosis developing cystic fibrosis-related diabetes. Any concerns should be discussed with your doctor or dietitian.
While some people can control their blood sugar levels by taking tablets, in cystic fibrosis-related diabetes most people are best treated with injections of insulin. Insulin cannot be taken by mouth because it is destroyed in the stomach. It is usually given as an injection two to four times a day.
Maintaining a healthy body weight is one of the most important steps you can take to ensure good health. People with cystic fibrosis-related diabetes still need to eat their usual high calorie, high protein and high fat diet to help achieve and maintain a healthy body weight. This is the opposite of the usual advice for diabetics and it can become confusing.
In cystic fibrosis, more energy (calories) is needed in the diet and the dose of insulin can usually be tailored to individual requirements and people can be taught to adjust their insulin dose to their dietary intake. Keeping your blood sugars at a near normal level will also help to maintain/improve your weight.
Surgical and other procedures
- Nasal polyp removal. Your doctor may recommend surgery to remove nasal polyps that obstruct breathing.
- Oxygen therapy. If your blood oxygen level declines, your doctor may recommend that you breathe pure oxygen to prevent high blood pressure in the lungs (pulmonary hypertension).
- Endoscopy and lavage. Mucus may be suctioned from obstructed airways through an endoscope.
- Feeding tube. Cystic fibrosis interferes with digestion, so you can’t absorb nutrients from food very well. Your doctor may suggest temporarily using a feeding tube to deliver extra nutrition while you sleep. This tube may be inserted in your nose and guided to your stomach, or it may be surgically implanted into the abdomen.
- Bowel surgery. If a blockage develops in your bowel, you may need surgery to remove it. Intussusception, where a section of bowel has folded in on itself, also may require surgical repair.
Lung transplants
In severe cases of cystic fibrosis, when the lungs stop working properly and all medical treatments have failed to help, a lung transplant may be recommended.
If you have severe breathing problems, life-threatening lung complications or increasing resistance to antibiotics used to treat lung infections, lung transplantation may be an option. Because bacteria line the airways in diseases that cause permanent widening of the large airways (bronchiectasis), such as cystic fibrosis, both lungs need to be replaced.
A lung transplant is a serious operation that carries risks, but it can greatly improve the length and quality of life for people with severe cystic fibrosis.
Cystic fibrosis does not recur in transplanted lungs. However, other complications associated with cystic fibrosis — such as sinus infections, diabetes, pancreas problems and osteoporosis — can still occur after a lung transplant.
Lifestyle and home remedies
You can manage your condition and minimize complications in several ways. Always talk to your doctor before starting home remedies.
Pay attention to nutrition and fluid intake
Cystic fibrosis can cause malnourishment because the enzymes needed for digestion can’t reach your small intestine, preventing food from being absorbed. People with cystic fibrosis may need a significantly higher number of calories daily than do people without the condition.
A healthy diet is important to maintain good lung function. It’s also important to drink lots of fluids, which can help thin the mucus in your lungs. You may work with a dietitian to develop a nutrition plan.
Most people with cystic fibrosis need to take pancreatic enzyme capsules with every meal and snack. In addition, your doctor may recommend:
- Antacids
- Supplemental high-calorie nutrition
- Special fat-soluble vitamins
- Extra fiber to prevent intestinal blockage
- Extra salt, especially during hot weather or before exercising
- Adequate water during hot weather
Keep immunizations up to date
In addition to other usual childhood vaccines, people with cystic fibrosis should have the annual flu vaccine and any other vaccines their doctor recommends. Cystic fibrosis doesn’t affect the immune system, but children with cystic fibrosis are more likely to develop complications when they become sick.
Exercise
Regular exercise helps loosen mucus in your airways, and strengthens your heart. For many people with cystic fibrosis, participating in sports can improve confidence and self-esteem. Anything that gets you moving, including walking and biking, can help.
Eliminate smoke
Don’t smoke in your home or car, and don’t allow other people to smoke around you or your child. Secondhand smoke is harmful for everyone, but especially for people with cystic fibrosis.
Encourage hand-washing
Teach all the members of your family to wash their hands thoroughly before eating, after using the bathroom, when coming home from work or school, and after being around a person who is sick. Hand-washing is the best way to protect against infection.
Attend medical appointments
You’ll have ongoing care from your doctor and other medical professionals. Make sure to attend your regular follow-up appointments. Take your medications as prescribed and follow therapies as instructed. Contact your doctor if you experience any signs or symptoms such as severe constipation, more mucus than usual, blood in your mucus or reduced energy.
Coping and support
If you or someone you love has cystic fibrosis, you may experience strong emotions such as anger or fear. These issues are especially common in teens. Talking openly about how you feel can help. It also may help to talk with others who are dealing with the same issues.
That might mean joining a support group for parents of children with cystic fibrosis. Older children with the disorder may want to join a cystic fibrosis group to meet and talk with others who have the disease.
If you or your child is depressed or anxious, it may help to meet with a psychologist. He or she may suggest medications or other treatments as well.
Spend time with friends and family. Having their support can help you manage stress and reduce anxiety. Ask your friends or family for help if you need it.
Take time to learn about your or your child’s condition. If your child has cystic fibrosis, encourage him or her to learn about the condition. Find out how medical care is managed for children with cystic fibrosis as they grow older into adulthood. Ask your child’s doctor if you have questions about your child’s care as he or she becomes older.
References



{kind=link}