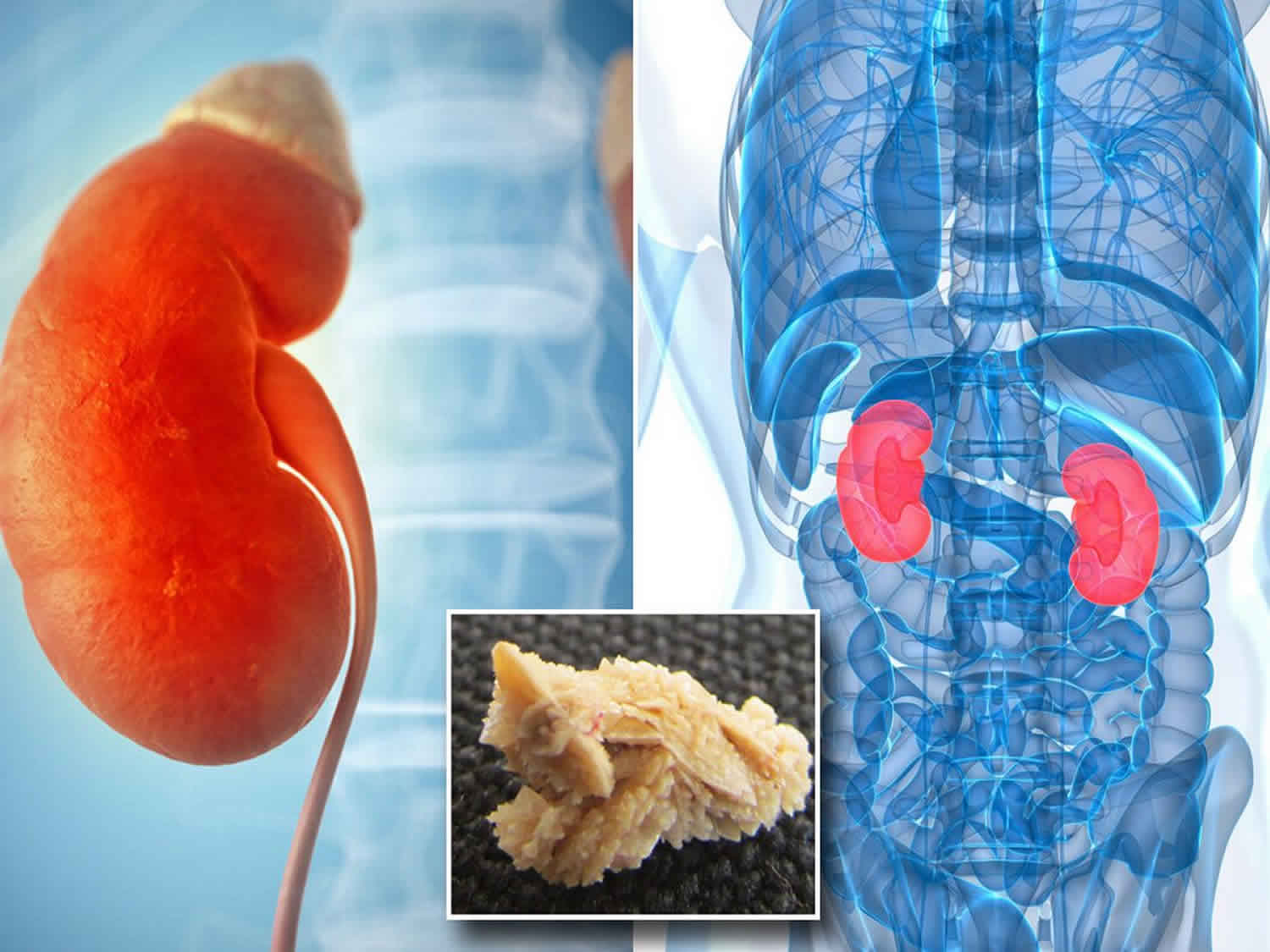
Hyperoxaluria
Hyperoxaluria is a condition where you have too much oxalate in your urine. Oxalate is a natural chemical in your body, and it’s also found in certain types of food. There is no known need for oxalate by the human body, it is normally eliminated as waste through the kidneys. But too much oxalate in your urine can cause serious problems when the excess oxalate binds with calcium in the urine to form calcium oxalate, a hard compound that is the main component of kidney and bladder stones. Deposits of calcium oxalate can damage the kidneys and other organs and lead to blood in the urine (hematuria), urinary tract infections, kidney damage, end stage renal disease (ESRD), and injury to other organs. Over time, kidney function decreases such that the kidneys can no longer excrete as much oxalate as they receive. As a result oxalate levels in the blood rise, and the substance gets deposited in tissues throughout the body (systemic oxalosis), particularly in bones and the walls of blood vessels. Oxalosis in bones can cause fractures. Hyperoxaluria can be caused by eating too many oxalate-rich foods, an intestinal disease or an inherited (genetic) disorder (e.g., primary hyperoxaluria). In some persons the cause of hyperoxaluria is not known, but may result from changes in the way kidneys handle normal amounts of body oxalate. Hyperoxaluria is uncommon, though can be found in about 20 percent of individuals with kidney stones. Quick diagnosis and treatment of hyperoxaluria is important to the long-term health of your kidneys.
Primary hyperoxaluria is a rare inherited condition characterized by recurrent kidney stones and bladder stones 1. In primary hyperoxaluria, the liver doesn’t create enough of a certain protein (enzyme) that prevents overproduction of oxalate, or the enzyme doesn’t work properly. Very large amounts of oxalate are produced when there is not enough of these enzymes in the liver. Excess oxalate is eliminated through your kidneys, in your urine. The extra oxalate can combine with calcium to create kidney stones and crystals, which can damage the kidneys and cause them to stop working (renal failure). Moreover, unlike dietary or enteric hyperoxaluria, the amount of oxalate in the urine is not greatly affected by changes in dietary oxalate.
There are three types of primary hyperoxaluria that differ in their severity and genetic cause.
- In primary hyperoxaluria type 1, kidney stones typically begin to appear anytime from childhood to early adulthood, and end stage renal disease can develop at any age.
- Primary hyperoxaluria type 2 is similar to type 1, but end stage renal disease develops later in life.
- In primary hyperoxaluria type 3, affected individuals often develop kidney stones in early childhood, but few cases of this type have been described so additional signs and symptoms of this type are unclear.
The enzymes involved in the three types of primary hyperoxaluria are as follows:
- Primary hyperoxaluria type 1: alanine-glyoxylate aminotransferase, or AGT
- Primary hyperoxaluria type 2: glyoxylate/hydroxypyruvate reductase, or GR/HPR
- Primary hyperoxaluria type 3: 4-hydroxy-2-oxoglutarate aldolase, or HOGA
Primary hyperoxaluria is estimated to affect 1 in 58,000 individuals worldwide 1. Type 1 is the most common form, accounting for approximately 80 percent of cases. Types 2 and 3 each account for about 10 percent of cases.
It is possible that other types of primary hyperoxaluria will be discovered. Some patients have high urine oxalates and kidney stones but do not have a genetic mutation that causes one of the three known types of primary hyperoxaluria. Future research may help us to better understand the cause of the high urine oxalate in these patients.
Primary hyperoxaluria type 1
Primary hyperoxaluria type 1 is caused by a deficiency of the liver peroxisomal enzyme alanine-glyoxylate-aminotransferase (AGT), which catalyzes the conversion of glyoxylate to glycine 2. When alanine-glyoxylate-aminotransferase activity is absent, glyoxylate is converted to oxalate, which forms insoluble calcium oxalate crystals that accumulate in the kidney and other organs. Individuals with primary hyperoxaluria type 1 are at risk for recurrent nephrolithiasis (deposition of calcium oxalate in the renal pelvis / urinary tract), nephrocalcinosis (deposition of calcium oxalate in the renal parenchyma), or end-stage renal disease (ESRD). Age at onset of symptoms ranges from infancy to the sixth decade. Approximately 10% of affected individuals present in infancy or early childhood with nephrocalcinosis, with or without nephrolithiasis, and failure to thrive related to renal failure. The majority of individuals with primary hyperoxaluria type 1 present in childhood or early adolescence, usually with symptomatic nephrolithiasis and normal or reduced kidney function. The remainder of affected individuals present in adulthood with recurrent renal stones and a mild-to-moderate reduction in kidney function. The natural history of untreated primary hyperoxaluria type 1 is one of progressive decline in renal function as a result of calcium oxalate deposits in kidney tissue and complications of nephrolithiasis (e.g., obstruction and infection) with eventual progression to oxalosis (widespread tissue deposition of calcium oxalate) and death from end-stage renal disease and/or complications of oxalosis.
Primary hyperoxaluria type 1 diagnosis
The diagnosis of primary hyperoxaluria type 1 is established in a proband with hyperoxaluria or hyperoxalemia by identification biallelic pathogenic variants in AGXT on molecular genetic testing. Failure to identify at least one AGXT pathogenic variant should prompt examination for other types of primary hyperoxaluria and in occasional circumstances may require consideration of liver biopsy to assay the activity of the enzyme alanine-glyoxylate-aminotransferase (AGT). However, given the wide availability of genetic testing, liver biopsy to obtain tissue for enzymatic activity is now rarely employed.
Primary hyperoxaluria type 1 treatment
Treatment of manifestations
Reduction of calcium oxalate supersaturation in the urine to minimize stone formation; reduction of calcium oxalate supersaturation in the plasma in individuals with end-stage renal disease (ESRD) to minimize systemic deposition of calcium oxalate crystals; treatment of kidney stones; reduction of oxalate biosynthesis; dialysis; organ transplantation as either liver transplantation or combined liver/kidney transplantation.
Prevention of primary manifestations
To reduce calcium oxalate supersaturation in the urine: maintenance of high fluid intake; inhibition of calcium oxalate crystal formation with potassium or sodium citrate, pyrophosphate-containing solutions. To reduce oxalate biosynthesis: pyridoxine supplements for those who are pyridoxine responsive.
Surveillance
At regular intervals, obtain serum creatinine to estimate glomerular filtration rate (GFR), renal ultrasound examinations or other kidney imaging, urinalysis; and periodic fundoscopic eye examinations. Additionally:
- In those with reduced GFR (<60 mL/min/1.73 m²): regular measurement of plasma oxalate and more frequent monitoring of kidney function.
- In individuals with greatly reduced GFR (<30 mL/min/1.73 m²) or rapid deterioration in function: frequent assessment of serum creatinine and plasma oxalate. At regular intervals obtain x-ray examination of the long bones, electrocardiogram for conduction abnormalities, echocardiogram for evidence of oxalate cardiomyopathy, hemoglobin, thyroid function testing, and frequent clinical evaluation for additional complications of oxalosis.
Agents/circumstances to avoid
Dehydration from any cause can lead to irreversible kidney failure and should be strictly avoided. Individuals with primary hyperoxaluria type 1 should avoid intake of vitamin C that exceeds the recommended daily allowance, loop diuretics, high doses of nonsteroidal anti-inflammatory medications (NSAIDs) or other medications that can compromise kidney function; and large intake of foods high in oxalate (e.g., chocolate, rhubarb, starfruit).
Evaluation of relatives at risk
Early diagnosis of at-risk relatives enables early institution of treatment and preventive measures.
Primary hyperoxaluria type 2
Primary hyperoxaluria type 2 is caused by deficiency of the enzyme glyoxylate reductase/hydroxypyruvate reductase, is characterized by recurrent nephrolithiasis (deposition of calcium oxalate in the renal pelvis/urinary tract), nephrocalcinosis (deposition of calcium oxalate in the renal parenchyma), and end-stage renal disease (ESRD) 3. After end-stage renal disease, oxalosis (widespread tissue deposition of calcium oxalate) usually develops. Symptom onset is typically in childhood.
Primary hyperoxaluria type 2 diagnosis
The diagnosis of primary hyperoxaluria type 2 is established in a proband by identification of biallelic pathogenic variants in GRHPR (glyoxylate reductase/hydroxypyruvate reductase) by molecular genetic testing. If no pathogenic variants or only one pathogenic variant is identified by molecular genetic testing, identification of reduced glyoxylate reductase enzyme activity on liver biopsy can establish the diagnosis of primary hyperoxaluria type 2.
Primary hyperoxaluria type 2 treatment
Treatment of manifestations
Reduction of urinary calcium oxalate supersaturation through adequate daily fluid intake and treatment with inhibitors of calcium oxalate crystallization (orthophosphate, potassium citrate, and magnesium); temporary intensive dialysis for end-stage renal disease (ESRD), followed by transplantation.
Surveillance
Biannual assessment of renal function, urinalysis with measurements of urine oxalate excretion (using 24-hour collection if easy to facilitate or spot urine oxalate to creatinine ratio), and calcium oxalate saturation if available, blood pressure, and full blood count including hematocrit; assessment of renal stone burden every six to 12 months by urinary tract imaging (renal ultrasound or CT); assessment of cardiac, skin, bone, joint, eye, thyroid, and hematologic involvement annually after progression to end-stage renal disease (ESRD).
Agents/circumstances to avoid
Dehydration from any cause can lead to irreversible kidney failure and should be strictly avoided. Ascorbate (vitamin C) ingestion and foods rich in oxalate (chocolate, rhubarb, and star fruit) may cause additional minimal increase in urinary oxalate levels in select individuals; excess should be discouraged; high salt (sodium) diet should be discouraged; excessive stone interventions with extracorporal shock wave lithotripsy.
Evaluation of relatives at risk
For asymptomatic at-risk relatives offer urine analysis and, if indicated by the results of urine analysis, molecular genetic testing (if the pathogenic variants in the family are known) so that early diagnosis can inform treatment.
Primary hyperoxaluria type 3
Primary hyperoxaluria type 3 is characterized by recurring calcium oxalate stones beginning in childhood or adolescence and on occasion, nephrocalcinosis or reduced kidney function 4. In 50%-65% of individuals with primary hyperoxaluria type 3 stone formation begins prior to age five years. Although the frequency and severity of stone activity often seem to abate by adolescence and adulthood, stone formation can occur throughout life and some adults have many stones. To date systemic oxalosis has not been reported in primary hyperoxaluria type 3.
Primary hyperoxaluria type 3 diagnosis
The diagnosis of primary hyperoxaluria type 3 is established in a proband with calcium oxalate kidney stone(s) and/or nephrocalcinosis, urine oxalate >0.7 mmol/1.73 m2/24 hours (in those with preserved kidney function), increased concentration of plasma oxalate (particularly in those with reduced kidney function), and biallelic (homozygous or compound heterozygous) pathogenic variants in HOGA1.
Primary hyperoxaluria type 3 treatment
Treatment of manifestations
Reduction of urine supersaturation of calcium oxalate by maintaining high oral fluid intake at all times and use of an inhibitor of calcium oxalate crystallization such as potassium or sodium citrate; prevention of stone complications by prompt relief of urinary tract obstruction and treatment of urinary tract infections; and avoidance of marked dietary oxalate excess.
Surveillance
For those who are stable, annual clinical assessment of: stone-related symptoms (pain), frequency of passage of urinary stones and/or gravel, urinary tract infection; adherence to high fluid intake and medication schedule; annual: assessment of kidney function by serum creatinine, measurement of plasma oxalate concentration in those with impaired renal function, 24-hour urine oxalate and supersaturation study, and renal ultrasound examination or other imaging to monitor for stone formation.
Agents/circumstances to avoid
Intravascular volume contraction, delays in treatment of acute stone episodes, marked dietary oxalate excess, high-dose ascorbic acid, and nephrotoxic agents.
Evaluation of relatives at risk
Presymptomatic diagnosis and treatment is warranted in relatives at risk to reduce stone formation and the risk of nephrocalcinosis and chronic kidney disease.
Pregnancy management
Adequate fluid intake throughout the pregnancy.
Oxalosis
Oxalosis is a rare metabolic disorder that occurs when the kidneys stop eliminating calcium oxalate crystals from the body through the urine. Because the kidneys stop working, oxalate crystals are deposited elsewhere in the body. Oxalosis in its late stages can cause a variety of complications outside the kidney, including bone disease, anemia, skin ulcers, heart and eye problems, and in children, a failure to develop and grow normally.
Hyperoxaluria causes
Primary hyperoxaluria
Primary hyperoxaluria is a rare, inherited (genetic) disorder of liver metabolism that often results in life-threatening damage to the kidneys. In this type, the liver doesn’t create enough of a certain protein (enzyme) that prevents overproduction of oxalate, or the enzyme doesn’t work properly. Unlike dietary or enteric hyperoxaluria, the amount of oxalate in the urine is not greatly affected by changes in dietary oxalate.
Mutations in the AGXT, GRHPR, and HOGA1 genes cause primary hyperoxaluria types 1, 2, and 3, respectively. These genes provide instructions for making enzymes that are involved in the breakdown and processing of protein building blocks (amino acids) and other compounds. The enzyme produced from the HOGA1 gene is involved in the breakdown of an amino acid, which results in the formation of a compound called glyoxylate. This compound is further broken down by the enzymes produced from the AGXT and GRHPR genes.
Mutations in the AGXT, GRHPR, or HOGA1 gene lead to a decrease in production or activity of the respective proteins, which prevents the normal breakdown of glyoxylate. AGXT and GRHPR gene mutations result in an accumulation of glyoxylate, which is then converted to oxalate for removal from the body as a waste product. HOGA1 gene mutations also result in excess oxalate, although researchers are unsure as to how this occurs. Oxalate that is not excreted from the body combines with calcium to form calcium oxalate deposits, which can damage the kidneys and other organs.
Primary hyperoxaluria inheritance pattern
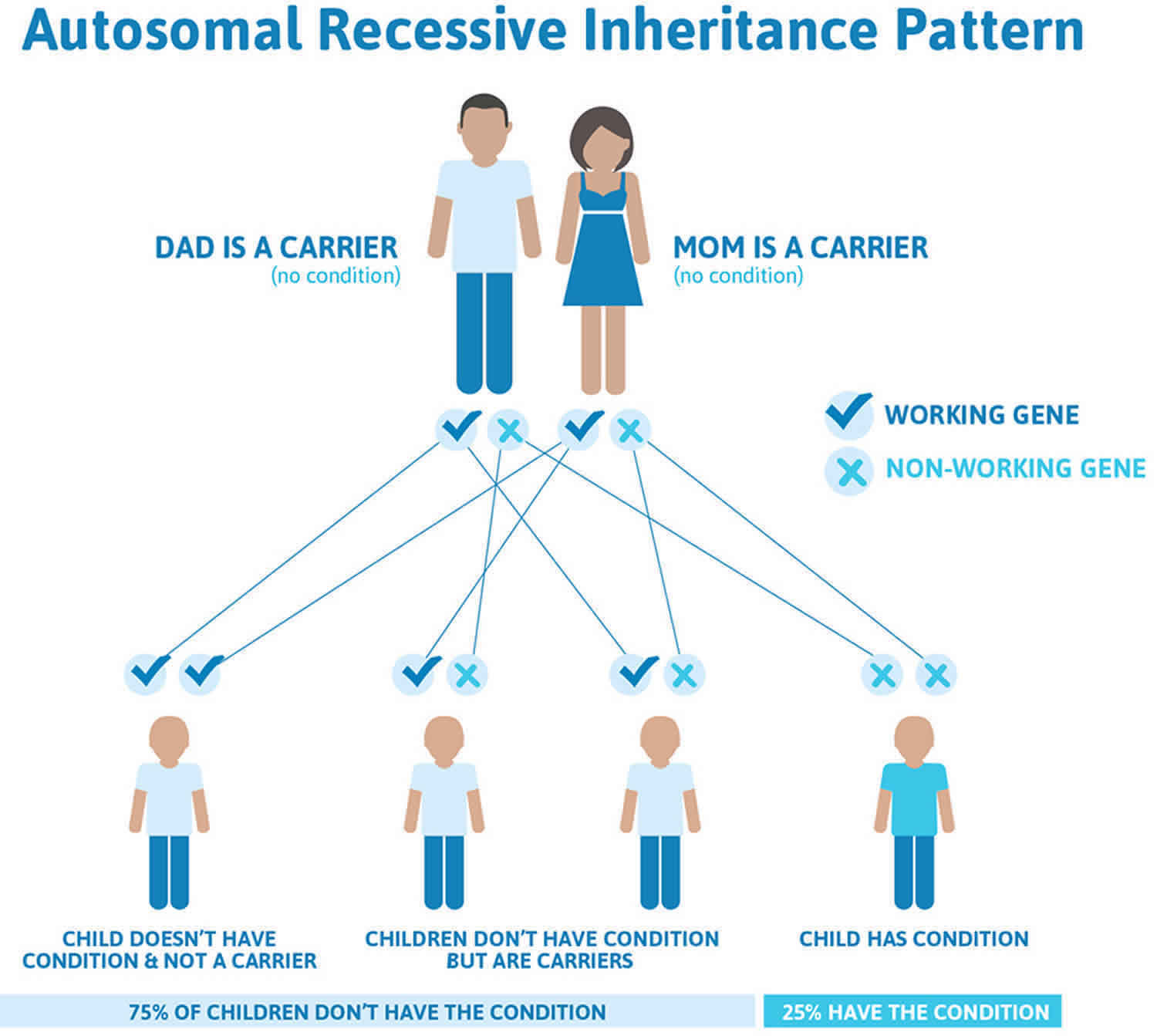
Primary hyperoxaluria is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Primary hyperoxaluria autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Secondary hyperoxaluria
Secondary hyperoxaluria is caused by excess absorption of oxalate from the gastrointestinal tract due to underlying gastrointestinal conditions such as gastric bypass, Crohn’s Disease, short bowel syndrome or other malabsorption disorders causing enteric hyperoxaluria. Avoiding, foods high in oxalate in particularly important.
Hyperoxaluria symptoms
Commonly, kidney stones are the first sign of hyperoxaluria. Kidney stones are uncommon in childhood. Kidney stones that form in children and teenagers are likely to be caused by an underlying condition, such as hyperoxaluria. For this reason, all young people with kidney stones should have a thorough evaluation, including measurement of oxalate in the urine. Adults with recurrent kidney stones also should be evaluated for oxalate in the urine.
Symptoms of a kidney stone can include the following:
- Severe or sudden abdominal or flank pain
- Blood in the urine
- Frequent urge to urinate
- Pain when urinating
- Fever and chills
- Primary hyperoxaluria that goes untreated can eventually damage your kidneys. Over time your kidneys may stop working. For some people, this is the first sign of the disease.
Symptoms of kidney failure can include the following:
- Decrease in urine output or no urine output at all
- Feeling generally ill, tired or heavy fatigue
- Loss of appetite, nausea and vomiting
- Pale skin color related to anemia
Oxalosis in its late stages can cause a variety of complications outside the kidney, including bone disease, anemia, skin ulcers, heart and eye problems, and in children, a failure to develop and grow normally.
Hyperoxaluria diagnosis
Schedule an appointment with your doctor to conduct a thorough physical exam including a medical history and dietary consult. Tests to diagnose hyperoxaluria may include:
- Urine tests to measure oxalate, glycolate and glycerate.
- Blood tests to check oxalate levels in the blood and kidney function.
- Kidney X-ray, ultrasound, or computerized tomography (CT scan) which can show kidney stones, calcium oxalate deposits and provide an internal view of the kidneys and urinary tract.
Stone analysis to determine the type of kidney stone once it passed or surgically removed.
If these tests indicate that you might have primary hyperoxaluria (primary hyperoxaluria) then additional testing will be necessary. These tests include:
- Genetic testing to look for inherited (genetic) mutations for primary hyperoxaluria, testing is currently available for three types — primary hyperoxaluria type 1, primary hyperoxaluria type 2 and primary hyperoxaluria type 3.
- Kidney biopsy to check for oxalate deposits.
- Echocardiogram to check for oxalate deposits in the heart.
- Eye exam to check for oxalate deposits in the eyes.
- Bone marrow biopsy to check for oxalate deposit in the bones.
- Liver biopsy to look for enzyme deficiencies in the liver — only needed in rare cases where genetic testing does not reveal primary hyperoxaluria.
Genetic testing
Genetic testing uses laboratory methods to look at your genes, which are the DNA instructions you inherit from your mother and your father. Genetic tests may be used to identify whether or not you have primary hyperoxaluria and help your doctor assess your response to treatments. You may decide to undergo a genetic test if you:
- have signs of primary hyperoxaluria.
- have an increased risk of having primary hyperoxaluria.
- think you might pass primary hyperoxaluria on to your children.
- are pregnant and want your fetus to be tested for a disease.
Genes come in pairs. primary hyperoxaluria occurs in individuals who have received two copies of an autosomal recessive gene, one copy from each parent. Recessive inheritance means both genes in a pair must be abnormal to cause primary hyperoxaluria. People with only one defective gene in the pair are considered carriers. However, a parent can pass the abnormal gene to their children. If primary hyperoxaluria is suspected or confirmed, brothers and sisters of a patient should be tested.
Medical genetics counselors experienced in hyperoxaluria can help guide your decisions and testing. Tests such as blood levels of oxalate, urine or blood levels of glycolate or glycerate, DNA testing and liver biopsy enzyme analysis are not widely available and can only be arranged by a few selected laboratories.
Hyperoxaluria treatment
It is important to seek medical care that takes into account the type, symptoms and severity of hyperoxaluria. Treatment will depend on the individual needs and how well the patient responds to treatment.
Reducing oxalate
The primary goal is to prevent or reduce the accumulation of calcium oxalate crystal formation in your kidneys.
- Medications. Prescription doses of vitamin B-6 (pyridoxine) can be effective in reducing oxalate in some people with hyperoxaluria. Oral preparation of neutral phosphates and citrate also can be effective to help prevent the formation of calcium oxalate crystals. Other medications like thiazide diuretics may also be considered, depending on the abnormalities present in your urine.
- High fluid intake. If you have normal kidney function your doctor will likely tell you to drink more water or other fluids. The extra fluid flushes the kidneys, prevents oxalate crystal buildup and helps prevent kidney stones from forming. Most important — drinking enough fluid will help reduce your chances of a kidney stone.
- Dietary modifications. For patients with enteric or dietary hyperoxaluriaz changes in diet may include restricting foods high in oxalate, limiting salt, decreasing sugar and reducing animal protein such as meat, eggs and fish. Restricting foods rich in oxalates such as rhubarb, beets, okra, spinach, Swiss chard, sweet potatoes, nuts, tea, chocolate and soy products. Also, it is important to ensure that you are getting enough calcium from food. Dietary restrictions may not be as important for people with primary hyperoxaluria. Follow the advice of your doctor or registered dietician.
Table 1. High oxalate foods by content of oxalate
| FOOD | SERVING | OXALATE milligram (mg)/SERVING |
| Spinach, cooked | 1/2 cup | 755 |
| Spinach, raw | 1 cup | 656 |
| Rhubarb | 1/2 cup | 541 |
| Rice Bran | 1 cup | 281 |
| Buckwheat Groats | 1 cup cooked | 133 |
| Almonds | 1 oz or 22 kernels | 122 |
| Miso Soup | 1 cup | 111 |
| Wheat Berries | 1 cup cooked | 98 |
| Corn Grits | 1 cup | 97 |
| Corn Grits | 1 cup | 97 |
| Baked Potato with Skin | 1 medium | 97 |
| Soy Flour | 1 cup | 94 |
| Bulgur, cooked | 1 cup | 86 |
| Navy Beans | 1/2 cup | 76 |
| Beets | 1/2 cup | 76 |
| Cocoa Powder | 4 tsp | 67 |
| Hot Chocolate (homemade) | 1 cup | 65 |
| Brown Rice Flour | 1 cup | 65 |
| Brown Rice Flour | 1 cup | 65 |
| Cornmeal | 1 cup | 64 |
| Cornmeal | 1 cup | 64 |
| Millet, cooked | 1 cup | 62 |
| Okra | 1/2 cup | 57 |
| Bran Flakes with Raisins, Single Brand | 1 cup | 57 |
| Post Original Shredded Wheat & Bran | 1 1/4 cup | 53 |
| French Fries | 4 oz | 51 |
| French Fries (homemade or fast food) | 4 oz or 1/2 cup | 51 |
| Cashews | 1 oz or 18 kernels | 49 |
| Raspberries | 1 cup | 48 |
| Soy Beans | 1/2 Cup | 48 |
| Nabisco Honey Nut Shredded Wheat Bite Size | 1 cup | 47 |
| Kellog Raisin Bran | 1 cup | 46 |
| Spoonsize Shredded Wheat | 1 cup | 45 |
| Nabisco Shredded Wheat | 2 biscuits | 42 |
| Stevia – Plant only, processed is 0 | 1 tsp | 42 |
| Post Fruit & Fiber Dates, Raisins & Walnuts | 1 cup | 41 |
| Raisin Squares Mini-Wheats | 3/4 cup | 41 |
| Barley Flour | 1/2 Cup | 41 |
| Miso | 1 cup | 40 |
| Miso | 1 cup | 40 |
| Yams | 1/2 cup, cubed | 40 |
| Bagel NY Style | 1 Bagel | 40 |
| Lentil Soup | 1 cup | 39 |
| Mixed Nuts (with Peanuts) | 1 oz | 39 |
| Chocolate Syrup | 2 Tbs | 38 |
| Chocolate Syrup | 2 Tbs | 38 |
| Candies with Nuts (ex Snickers) | 2 oz | 38 |
| Candies with Nuts (ex Snickers) | 2 oz | 38 |
| Pancakes (dry mix) | 4 pancakes | 37 |
| General Mills Multi-Bran Chex | 1 cup | 36 |
| Post 40% Bran | 3/4 cup | 36 |
| Stuffing | 1 cup | 36 |
| Kellog Special K Low Carb | 1/2 Cup | 35 |
| Post Cranberry Almond Crunch (Morning Traditions) | 1 cup | 35 |
| Bamboo Shoots | 1 cup | 35 |
| Kellog Complete Wheat Bran | 3/4 cup | 34 |
| General Mills Total Raisin Bran | 1 cup | 31 |
| Rutabaga | 1/2 cup mashed | 31 |
| Walnuts | 1 cup or 7 nuts | 31 |
| Brownies | 1 oz or 1/2 brownie | 31 |
| Dried Pineapples | 1/2 cup | 30 |
| Turnip | 1/2 cup mashed | 30 |
| Mashed Potatoes | 1 cup | 29 |
| Orange | 1 fruit | 29 |
| Wheat Flour, Whole Grain | 1 cup | 29 |
| Wheat Flour, Whole Grain | 1 cup | 29 |
| Fudge Sauce | 2 Tbs | 28 |
| Fudge Sauce | 2 Tbs | 28 |
| Kellog Frosted Mini-Wheats | 1 cup | 28 |
| Sweet Potatoes | 1 cup | 28 |
| Kellog Raisin Bran Crunch | 1 cup | 27 |
| Peanuts | 1 oz | 27 |
| Soy Protein Isolate | 1 oz | 27 |
| Carrot Juice | 1 cup | 27 |
| Kellog All-Bran Original | 1/2 cup | 26 |
| Post 100% Bran | 1/3 cup | 25 |
| Post Banana Nut Crunch | 1 cup | 25 |
| Chili with Beans | 1 cup | 24 |
| Dates | 1 date | 24 |
| Dried Figs | 5 pieces/fruits | 24 |
| General Mills Oatmeal Crisp with Almonds | 1 cup | 24 |
| General Mills Raisin Nut Bran | 1 cup | 24 |
| Veggie Burger | 1 pattie | 24 |
| Brown Rice, cooked | 1 cup | 24 |
| General Mills Honey Nut Clusters | 1 cup | 23 |
| Lasagna | 1 serving | 23 |
| Lasagna with meat | 1 serving | 23 |
| Pancakes (Homemade) | 4 pancakes | 22 |
| Potato Chips | 1 oz | 21 |
| Potato Chips | 1 oz | 21 |
| Fava Beans | 1/2 cup | 20 |
| Kellog All-Bran Buds | 1/2 cup | 20 |
| Kellog Mueslix Apple & Almond Crunch | 2/3 cup | 20 |
| Soy Milk | 1 Cup | 20 |
| Celery Raw | 1/2 Cup | 19 |
| Avocados | 1 fruit | 19 |
| Cream of Wheat | 1 cup | 18 |
| Olives | approx 10 olives | 18 |
| Post Great Grains Crunch Pecan | 2/3 cup | 18 |
| V8 Juice | 1 cup | 18 |
| General Mills Basic 4 | 1 cup | 17 |
| Mueslix | 2/3 cup | 17 |
| Post Great Grains Raisin, Dates & Pecans | 2/3 cup | 17 |
| Potato Salad | 1/3 cup | 17 |
| Pumpkin Seeds | 1 cup, cooked | 17 |
| Tomato Sauce | 1/2 cup | 17 |
| All-Purpose Flour | 1 cup | 17 |
| All-Purpose Flour | 1 cup | 17 |
| Brussel Sprouts Raw | 1/2 Cup | 17 |
| Burritos with beans | 1 burrito | 17 |
| Farina Cereal | 1 cup | 16 |
| Kellog Low Fat Granola with Raisins | 2/3 cup | 16 |
| Kiwi | 1 fruit | 16 |
| Peanut Butter Reduced Fat | 1 Tbs | 16 |
| Refried Beans | 1/2 cup | 16 |
| Tahini | 1 Tbs | 16 |
| Burritos with beans & meat | 1 burrito | 16 |
| Cake (homemade) | 1 piece | 16 |
| Couscous | 1 cup | 15 |
| Kellog Cracklin’ Oat Bran | 3/4 cup | 15 |
| Kellog Smart Start | 1 cup | 15 |
| Lemonade (frozen from concentrate) | 8 oz | 15 |
| Quaker Low Fat 100% Natural Granola with Raisins | 3/4 cup | 15 |
| Parsnip | 1/2 cup | 15 |
| Red Kidney Beans | 1/2 cup | 15 |
| Trail Mix | 1 oz | 15 |
| Cake (store brand) | 1 piece | 15 |
| Carrots, raw | 1/2 Cup | 15 |
| Danish Pastry Homemade | 1 pastry | 14 |
| Kellog Kashi Go Lean | 3/4 cup | 14 |
| Pistachios | 1 oz or 48 kernels | 14 |
| Post Grape Nuts | 1/2 cup | 14 |
| Tea, Brewed | 1 cup | 14 |
| Tomato Juice | 1 cup | 14 |
| Cheeseburger with bun | 1 burger & bun | 13 |
| Clam Chowder | 1 cup | 13 |
| Enchilada with Cheese & beef | 1 enchilada | 13 |
| Enchilada with Chicken | 1 enchilada | 13 |
| French Toast | 2 slices | 13 |
| French Toast | 2 slices | 13 |
| General Mills Fiber One | 1/2 cup | 13 |
| General Mills Nature Valley Cinnimon & Raisins Granola | 3/4 cup | 13 |
| General Mills Oatmeal Raisin Crisp | 1 cup | 13 |
| Kellog Just Right Fruit & Nut | 1 cup | 13 |
| Kellog Puffed Kashi | 1 cup | 13 |
| Nachos with Cheese | 6-8 chips | 13 |
| Peanut Butter | 1 Tbs | 13 |
| Pizza with Cheese | 2 slices | 13 |
| Red River Cereal | 1/4 cup | 13 |
| Rice Dream | 1 cup | 13 |
| Sweet Rolls Low Fat | 1 pastry | 13 |
| Tofu | 3.5oz | 13 |
| Quaker 100% Natural Granola Oats & Honey | 1/2 cup | 13 |
| English Muffin Whole Wheat | 1 muffin | 12 |
| English Muffins Whole Wheat ONLY | 1 muffin | 12 |
| Grapefruit | 1/2 fruit | 12 |
| Grilled Cheese Sandwich | 1 sandwich | 12 |
| Sunflower Seeds | 1 cup | 12 |
| Tacos | 1 small taco | 12 |
| General Mills Harmony | 1 1/4 cup | 11 |
| General Mills Wheaties Raisin Bran | 1 cup | 11 |
| Kellog All-Bran with Extra Fiber | 1/2 cup | 11 |
| Kellog Cocoa Krispies | 3/4 cup | 11 |
| Pancakes (Homemade) | 4 cakes | 11 |
| Spaghetti | 1 cup cooked | 11 |
| Uncle Sam | 1 cup | 11 |
| White Rice Flour | 1 cup | 11 |
| White Rice Flour | 1 cup | 11 |
| Cake (Low Fat Only) | 1 piece | 11 |
| Celery, Cooked | 1 cup | 10 |
| Chocolate Chip Cookies (store brand) | 1 cookie | 10 |
| Collards | 1 cup | 10 |
| Kellog Kashi Good Friends | 3/4 cup | 10 |
| Quaker Oat Bran | 1 1/4 cup | 10 |
| Pancakes (mix) | 4 cakes | 10 |
| Pecans | 1 oz or 15 halves | 10 |
| Blueberry Muffins | 1 muffin | 9 |
Kidney stone management
Calcium oxalate kidney stones are common in people with hyperoxaluria, but they don’t always need to be treated. Depending on your diagnosis, you may need nothing more than to take pain medication and drink lots of water to pass a kidney stone.
Small stones with minimal symptoms
Most small kidney stones won’t require invasive treatment. You may be able to pass a small stone by:
- Drinking water. Drinking as much as 2 to 3 quarts (1.9 to 2.8 liters) a day may help flush out your urinary system. Unless your doctor tells you otherwise, drink enough fluid — mostly water — to produce clear or nearly clear urine.
- Pain relievers. Passing a small stone can cause some discomfort. To relieve mild pain, your doctor may recommend pain relievers such as ibuprofen (Advil, Motrin IB, others), acetaminophen (Tylenol, others) or naproxen sodium (Aleve).
- Medical therapy. Your doctor may give you a medication to help pass your kidney stone. This type of medication, known as an alpha blocker, relaxes the muscles in your ureter, helping you pass the kidney stone more quickly and with less pain.
Large stones and those that cause symptoms
A person with a larger stone or one that blocks urine flow and causes pain, may need a team of urologists who specialize in stone disease to perform a more specialized treatment, such as:
- Extracorporeal shock wave lithotripsy (ESWL). Extracorporeal shock wave lithotripsy uses sound waves to create strong vibrations (shock waves) that break the stones into tiny pieces that can be passed in your urine. The procedure lasts about 45 to 60 minutes and can cause moderate pain, so you may be under sedation or light anesthesia to make you comfortable. Extracorporeal shock wave lithotripsy can cause blood in the urine, bruising on the back or abdomen, bleeding around the kidney and other adjacent organs, and discomfort as the stone fragments pass through the urinary tract.
- Ureteroscopy. To remove a smaller stone in your ureter or kidney, your doctor may pass a thin lighted tube (ureteroscope) equipped with a camera through your urethra and bladder to your ureter. Once the stone is located, special tools can snare the stone or break it into pieces that will pass in your urine. Your doctor may then place a small tube (stent) in the ureter to relieve swelling and promote healing. You may need general or local anesthesia during this procedure.
- Percutaneous nephrolithotomy. In this procedure, a wire-thin viewing instrument called a nephroscope is used to locate and remove the stone through a small incision in the persons back. The procedure is performed by a urologist in a hospital with general anesthesia. The person may have to stay in the hospital for one to two days after the procedure to allow for any residual stone fragments to drain from the kidney into a urine collection bag. Your doctor may recommend this surgery if extracorporeal shock wave lithotripsy (ESWL) was unsuccessful.
Dialysis
Depending on the severity of your hyperoxaluria, your kidneys may eventually stop working. Dialysis is a treatment used to replace some of the kidney’s function of cleaning the blood of toxins and removing extra fluids. There are two types of dialysis
Hemodialysis cleans blood by removing it from the body and passing it through a dialyzer, or artificial kidney. The dialyzer is a filter with two parts: one for blood and another for a sterile dialysis cleansing fluid, called dialysate. The filter between these two parts has very small pores, allowing some tiny particles to pass through. The particles that are filtered include the toxins that need to be removed from the body such as urea, creatinine and potassium, while larger blood cells and protein the body needs cannot pass through. The filtered blood is then returned to the body. The process of removing blood from the body, filtering it and returning it takes time. Hemodialysis treatment usually takes three to five hours and is repeated three or more times a week.
Peritoneal dialysis cleans your blood inside your body. Dialysate flows through a catheter into your abdomen (belly). Waste, chemicals and extra fluid in your blood pass from tiny blood vessels (capillaries) in the lining of your abdominal cavity (peritoneum) into the sterile cleansing solution —dialysate. The solution contains a sugar that draws wastes and extra fluid through the capillaries in your peritoneum into your abdomen. The solution, along with the waste products from your blood and any excess fluid, drain into a sterile collection bag. With peritoneal dialysis, treatments can be done at home (while you are sleeping), at work or while traveling.
Kidney dialysis is only temporary since dialysis typically does not remove the amount of oxalate formed or absorbed in the body. Dialysis continues until a new kidney can be transplanted. While on dialysis, most patients with primary hyperoxaluria, will continue to build up oxalate in body tissues, thus developing oxalosis.
Kidney transplants
For patients with kidney failure, a kidney transplant may be necessary. Depending on the the type and severity of primary hyperoxaluria, a kidney transplant or kidney and liver transplant may be needed. The type of transplant is based on each patient’s disease characteristics and needs.
References- Primary hyperoxaluria. https://ghr.nlm.nih.gov/condition/primary-hyperoxaluria
- Milliner DS, Harris PC, Cogal AG, et al. Primary Hyperoxaluria Type 1. 2002 Jun 19 [Updated 2017 Nov 30]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1283
- Rumsby G, Hulton SA. Primary Hyperoxaluria Type 2. 2008 Dec 2 [Updated 2017 Dec 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK2692
- Milliner DS, Harris PC, Lieske JC. Primary Hyperoxaluria Type 3. 2015 Sep 24. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK316514
- How To Eat A Low Oxalate Diet. https://kidneystones.uchicago.edu/how-to-eat-a-low-oxalate-diet





{kind=link}