
Leigh syndrome
Leigh syndrome also called Leigh’s disease, is a rare, inherited neurodegenerative condition that usually becomes apparent in the first year of life. Rarely, it begins in the teenage or adult years. Leigh syndrome is characterized by progressive loss of mental and movement abilities (psychomotor regression) and typically results in death within two to three years, usually due to respiratory failure. A small number of individuals do not develop symptoms until adulthood or have symptoms that worsen more slowly.
Early symptoms of Leigh syndrome seen in infancy may include poor sucking ability, loss of head control and motor skills, loss of appetite,; vomiting, diarrhea, seizures and difficulty swallowing (dysphagia), which disrupts eating 1. These problems often result in an inability to grow and gain weight at the expected rate (failure to thrive). As the condition progresses, symptoms may include weakness and lack of muscle tone (hypotonia), spasticity, involuntary muscle contractions (dystonia), movement disorders and cerebellar ataxia. Loss of sensation and weakness in the limbs (peripheral neuropathy), common in people with Leigh syndrome, may also make movement difficult. Complications can lead to impairment of respiratory, heart and kidney function 2. The term “Leigh-like syndrome” is often used for people with features that are strongly suggestive of Leigh syndrome but who do not meet the diagnostic criteria 3.
Several other features may occur in people with Leigh syndrome. Many individuals with this condition develop weakness or paralysis of the muscles that move the eyes (ophthalmoparesis); rapid, involuntary eye movements (nystagmus); or degeneration of the nerves that carry information from the eyes to the brain (optic atrophy). Severe breathing problems are common, and these problems can worsen until they cause acute respiratory failure. Some affected individuals develop hypertrophic cardiomyopathy, which is a thickening of the heart muscle that forces the heart to work harder to pump blood. In addition, a substance called lactate can build up in the body, and excessive amounts are often found in the blood, urine, or the fluid that surrounds and protects the brain and spinal cord (cerebrospinal fluid) of people with Leigh syndrome.
The signs and symptoms of Leigh syndrome are caused in part by patches of damaged tissue (lesions) that develop in the brains of people with this condition. A medical procedure called magnetic resonance imaging (MRI) reveals characteristic lesions in certain regions of the brain. These regions include the basal ganglia, which help control movement; the cerebellum, which controls the ability to balance and coordinates movement; and the brainstem, which connects the brain to the spinal cord and controls functions such as swallowing and breathing. The brain lesions are often accompanied by loss of the myelin coating around nerves (demyelination), which reduces the ability of the nerves to activate muscles used for movement or relay sensory information from the rest of the body back to the brain.
The classical form of Leigh syndrome develops during infancy (infantile necrotizing encephalopathy) and usually begins between the ages of 3 months and 2 years. This form of the disease affects males and females in equal numbers.
In cases of Leigh syndrome that are inherited as an X-linked recessive trait, the symptoms typically develop during infancy. Almost twice as many males as females are affected by this form of the disease.
In some rare cases, Leigh syndrome may begin during late adolescence or early adulthood (adult-onset subacute necrotizing encephalomyelopathy). In these cases, which affect twice as many males as females, the progression of the disease is slower than the classical form of the disease.
Researchers once believed that the classical form of Leigh syndrome accounted for approximately 80 percent of cases. Leigh syndrome in general is rare and is estimated to affect about 1 in 30,000 to 1 in 40,000 people at birth 2. Mitochondrial DNA-associated Leigh syndrome, which is more rare than nuclear gene-encoded Leigh syndrome, is likely to occur in about 1 in 100,000 to 1 in 140,000 births 3. Leigh syndrome is more common in certain populations. For example, Leigh’s disease occurs in approximately 1 in 2,000 newborns in the Saguenay Lac-Saint-Jean region of Quebec, Canada and in approximately 1 in 1,700 individuals on the Faroe Islands (between Norway and Iceland) 2.
The inheritance of Leigh syndrome depends on where the responsible gene is located in each case. This is because it can be due to mutations in either mitochondrial DNA or nuclear DNA 4:
- Mitochondrial DNA-associated Leigh syndrome follows a mitochondrial inheritance pattern (also called maternal inheritance).
- Nuclear gene-encoded Leigh syndrome may be inherited in an autosomal recessive or X-linked manner.
Treatment is based on the symptoms present and depends on the type of Leigh syndrome a person has 2. While life expectancy depends on the cause of Leigh syndrome in each person, most do not survive past mid-childhood or adolescence 1.
What is known about Leigh syndrome diagnosed in adolescence or adulthood?
A very small number of individuals with Leigh syndrome live beyond 10 years of age. Leigh syndrome in adolescents or adults may occur in people who have Leigh syndrome who survive into adulthood, in people with a late onset of symptoms, or in people who experience spontaneous recovery. Adults with Leigh syndrome may have no neurological abnormalities. Rarely, they may have typical features of Leigh syndrome or more generalized mitochondrial disorders 5.
If a family has a child with Leigh syndrome, what is the risk of future children being affected? Is this condition inherited from the mother or father?
Leigh syndrome can be inherited as either an autosomal recessive trait, an X-linked recessive trait, or as a mutation found within the mitochondrial DNA 6.
In autosomal recessive inheritance, a person inherits mutations in two genes, one from the father and one from the mother. If an individual receives one normal gene and one gene for the disease, the person will be a carrier and usually does not show symptoms. The risk for two carrier parents to both pass on the gene mutation and, therefore, have an affected child is 25% with each pregnancy. There is also a 50% risk in each pregnancy of having a child who is also a carrier; and a 25% of having a child that is neither affected nor a carrier 6.
In X-linked recessive disorders, a person can also inherit the condition from either parent. Females have two X chromosomes, but males have an X and a Y chromosome. In females, disease traits on the X chromosome can be masked by the normal gene on the other X chromosome. Since males only have one X chromosome, they will show features of the disease if they inherit a gene mutation on the X chromosome. Men with X-linked disorders transmit the gene to all their daughters, who are carriers, but never to their sons. Women who are carriers of an X-linked disorder have a 50% risk of passing on the carrier condition to their daughters, and a 50% chance of transmitting the disease to their sons 6.
In some cases, Leigh syndrome may be inherited from the mother as a mutation found within the mitochondrial DNA (mtDNA). All human mtDNA comes from the mother. Thus, an affected mother will pass the traits to all of her children, but only the daughters will pass the mutation(s) onto the next generation 6.
The genetic mutations that are present in the mtDNA may outnumber the normal copies of the genes. Symptoms may not occur until mutations are present in a significant percentage of the mitochondria. The uneven distribution of normal and mutant mtDNA in different tissues of the body can affect different organ systems in individuals from the same family and can result in a variety of symptoms in affected family members 6.
A genetics professional can help determine how Leigh syndrome is being transmitted in a particular family.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Am I at risk to be a carrier of Leigh syndrome if my maternal half sister has an affected son?
If your maternal half sister has a son with Leigh syndrome, there is a chance you carry a mutation responsible for Leigh syndrome. However, the chance depends on the genetic status of your sister and the inheritance pattern of Leigh syndrome in your sister’s family. (Detailed information about how Leigh syndrome may be inherited is provided below). For example:
- If your nephew has mitochondrial DNA-associated Leigh syndrome and your sister has an mtDNA mutation, you and your mother are also at risk 7. A child always shares the same mtDNA with his/her maternal siblings and mother 8.
- If your nephew has nuclear gene-encoded Leigh syndrome and it is inherited in an autosomal recessive manner, there is a chance you are a carrier if your sister inherited her mutation from your mother. If so, your chance to be a carrier of autosomal recessive Leigh syndrome would be 50%. If your sister inherited her mutation from her father, it is assumed you are not at risk to be a carrier (since you have different fathers).
- If your nephew has nuclear gene-encoded Leigh syndrome and it is inherited in an X-linked recessive manner, there is a 50% chance you are a carrier for this type of Leigh syndrome. This is assuming that your sister’s X chromosome with the mutation was inherited from your mother. If it was inherited from her father, he would have likely been affected with Leigh syndrome.
The genetics of Leigh syndrome, particularly mitochondrial DNA-associated Leigh syndrome, is very complex. Identifying the mode of inheritance in each family is necessary to learn about who is at risk to be a carrier and who is at risk to be affected.
Genetic testing of affected family members can confirm the mode of inheritance in a particular family. Carrier testing for at-risk relatives is then possible if the disease-causing mutation in a person or family has been identified 3. If a specific mutation causing Leigh syndrome cannot be identified and more than one family member is affected, a genetics professional may be able to take an “educated guess” about the inheritance mode based on careful evaluation of the family history 8.
People with personal questions and concerns about the inheritance of Leigh syndrome and risks to family members are strongly encouraged to speak with a genetic counselor or other genetics professional.
Leigh syndrome causes
Leigh syndrome can be caused by mutations in one of more than 75 different genes. In humans, most genes are found in DNA in the cell’s nucleus, called nuclear DNA (nDNA). However, some genes are found in DNA in specialized structures in the cell called mitochondria. This type of DNA is known as mitochondrial DNA (mtDNA). While most people with Leigh syndrome have a mutation in nuclear DNA (nDNA), about 20 percent have a mutation in mtDNA. Nuclear DNA (nDNA) is contained in the nucleus of a cell and is inherited from both biological parents. Mitochondrial DNA (mtDNA) is contained in the mitochondria of cells and is inherited exclusively from the child’s mother.
Most genes associated with Leigh syndrome are involved in the process of energy production in mitochondria. Mitochondria use oxygen to convert the energy from food into a form cells can use through a process called oxidative phosphorylation. Five protein complexes, made up of several proteins each, are involved in this process. The complexes are named complex I, complex II, complex III, complex IV, and complex V. During oxidative phosphorylation, the protein complexes drive the production of adenosine triphosphate (ATP), the cell’s main energy source, through a step-by-step transfer of negatively charged particles called electrons. Many of the gene mutations associated with Leigh syndrome affect proteins in these complexes or disrupt their assembly. These mutations reduce or eliminate the activity of one or more of these complexes, which can lead to Leigh syndrome.
Disruption of complex I, also called NADH:ubiquinone oxidoreductase, is the most common cause of Leigh syndrome, accounting for nearly one third of cases of the condition. At least 25 genes involved in the formation of complex I, found in either nuclear or mitochondrial DNA, have been associated with Leigh syndrome.
Disruption of complex IV, also called cytochrome c oxidase or COX, is also a common cause of Leigh syndrome, underlying approximately 15 percent of cases. One of the most frequently mutated genes in Leigh syndrome is SURF1. This gene, which is found in nuclear DNA, provides instructions for making a protein that helps assemble the COX protein complex (complex IV). This complex, which is involved in the last step of electron transfer in oxidative phosphorylation, provides the energy that will be used in the next step of the process to generate ATP. Mutations in the SURF1 gene typically lead to an abnormally short SURF1 protein that is broken down in cells, resulting in the absence of functional SURF1 protein. The loss of this protein reduces the formation of normal COX complexes, which impairs mitochondrial energy production.
The most common mtDNA mutation in Leigh syndrome affects the MT-ATP6 gene, which provides instructions for making a piece of complex V, also known as the ATP synthase protein complex. Using the energy provided by the other protein complexes, the ATP synthase complex generates ATP. MT-ATP6 gene mutations, found in approximately 10 percent of people with Leigh syndrome, block the generation of ATP. Other mtDNA mutations associated with Leigh syndrome decrease the activity of other oxidative phosphorylation protein complexes or lead to reduced formation of mitochondrial proteins, all of which impair mitochondrial energy production.
Other gene mutations associated with Leigh syndrome decrease the activity of one or more oxidative phosphorylation protein complexes or affect additional steps related to energy production. For example, Leigh syndrome can be caused by mutations in genes that form the pyruvate dehydrogenase complex or coenzyme Q10, both of which are involved in mitochondrial energy production. Mutations in genes that direct the replication of mtDNA or the production of mitochondrial proteins can also disrupt mitochondrial energy production.
Although the exact mechanism is unclear, researchers believe that impaired oxidative phosphorylation can lead to cell death because of decreased energy available in the cell. Certain tissues that require large amounts of energy, such as the brain, muscles, and heart, seem especially sensitive to decreases in cellular energy. Cell death in the brain likely causes the characteristic lesions seen in Leigh syndrome, which contribute to the signs and symptoms of the condition. Cell death in other sensitive tissues may also contribute to the features of Leigh syndrome.
Leigh syndrome inheritance pattern
Leigh syndrome can have different inheritance patterns. It is most commonly inherited in an autosomal recessive pattern, which means that to be affected, a person must have a mutation in both copies of the responsible gene in each cell. Affected people inherit one mutated copy of the gene from each parent, who is referred to as a carrier. Carriers of an autosomal recessive condition typically do not have any signs or symptoms (they are unaffected). When two carriers of an autosomal recessive condition have children, each child has a (see Figure 1):
- 25% chance to be affected
- 50% chance to be an unaffected carrier like each parent
- 25% chance to be unaffected and not a carrier
Autosomal recessive inheritance applies to most of the Leigh syndrome-associated genes contained in nuclear DNA, including SURF1. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
In approximately 20 percent of people with Leigh syndrome, the condition is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA, including MT-ATP6. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA mutations only from their mother. These disorders can appear in every generation of a family and affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children. Occasionally, mutations in mtDNA occur spontaneously, and there is no history of Leigh syndrome in the family.
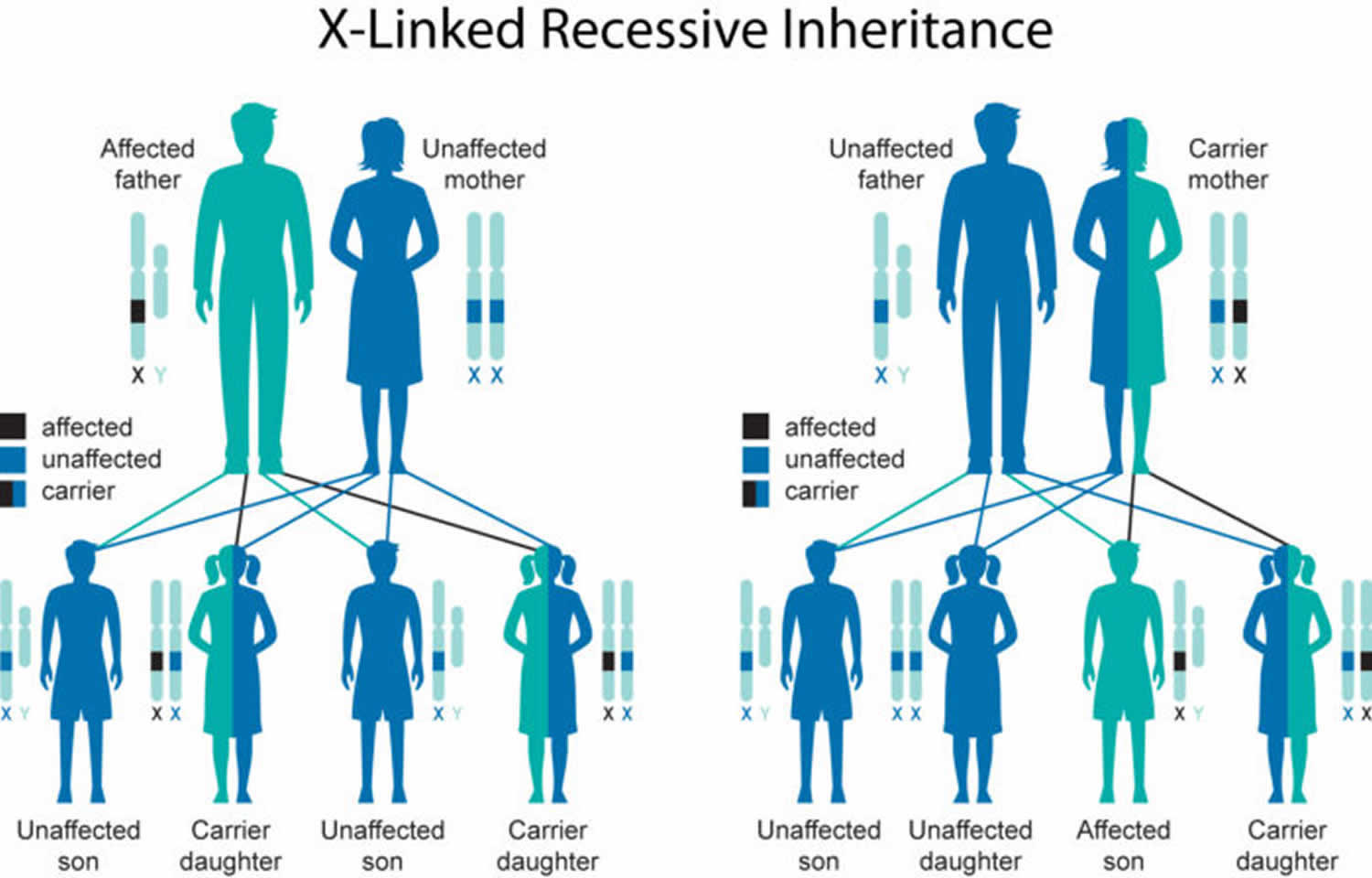
In a small number of affected individuals with mutations in nuclear DNA, Leigh syndrome is inherited in an X-linked recessive pattern. The condition has this pattern of inheritance when the mutated gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
If a mother is a carrier of an X-linked recessive condition and the father is not, the risk to children depends on each child’s sex (see Figure 2).
- Each son has a 50% chance to be unaffected, and a 50% chance to be affected
- Each daughter has a 50% chance to be unaffected, and a 50% chance to be an unaffected carrier
If a father has the condition and the mother is not a carrier, all sons will be unaffected, and all daughters will be unaffected carriers.
Figure 1. Leigh syndrome autosomal recessive inheritance pattern

Figure 2. Leigh syndrome X-linked inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Leigh syndrome symptoms
The symptoms of Leigh syndrome vary greatly from person to person. Very rarely, affected people with near-normal neurologic findings have been reported. Most people with Leigh syndrome have central nervous system and peripheral nervous system abnormalities, without involvement of other body systems 5.
Central nervous system abnormalities may include:
- Developmental delay or regression
- Nystagmus
- Ophthalmoparesis (weakness in the muscles that control eye movement)
- Optic atrophy
- Ataxia
- Dysphagia
- Retinitis pigmentosa
- Deafness
Peripheral nervous system abnormalities may include polyneuropathy and myopathy 5.
Although most people with Leigh syndrome only have neurological abnormalities, some people also have non-neurologic abnormalities. These may include 5:
- Distinct physical features
- Hormone abnormalities resulting in short stature or hypertrichosis
- Heart abnormalities (hypertrophic or dilated cardiomyopathy)
- Gastrointestinal symptoms such as diarrhea
The symptoms of classical Leigh syndrome (infantile necrotizing encephalopathy), a rapidly progressive neurological disorder, usually begin between the ages of 3 months and 2 years. In most children, the first noticeable sign is the loss of previously acquired motor skills. When there is early onset (i.e., 3 months), loss of head control and poor sucking ability may be the first noticeable symptoms. This may be accompanied by a profound loss of appetite, recurrent vomiting, irritability, continuous crying and possible seizure activity. Delays in reaching developmental milestones may also occur. Affected infants may fail to grow and gain weight at the expected rate (failure to thrive).
If the onset of Leigh syndrome is later in childhood (e.g., 24 months), a child may experience difficulty articulating words (dysarthria) and coordinating voluntary movements such as walking or running (ataxia). Previously acquired intellectual skills may diminish and intellectual disability may also occur.
Progressive neurological deterioration associated with Leigh syndrome is marked by a variety of symptoms including generalized weakness, lack of muscle tone (hypotonia), clumsiness, tremors, muscle spasms (spasticity) that result in slow, stiff movements of the legs, and/or the absence of tendon reflexes. Further neurological development is delayed.
Episodes of lactic acidosis may occur and are characterized by abnormally high levels of lactic acid in the blood, brain and other tissues of the body. Periodically, levels of carbon dioxide in the blood may also be abnormally elevated (hypercapnia). Lactic acidosis and hypercapnia can lead to psychomotor regression and respiratory, heart, or kidney impairment.
Children with Leigh syndrome usually develop respiratory problems including the temporary cessation of spontaneous breathing (apnea), difficulty breathing (dyspnea), abnormally rapid breathing (hyperventilation), and/or abnormal breathing patterns (Cheyne-Stokes). Some infants may also experience difficulty swallowing (dysphagia). Visual problems may include abnormally rapid eye movements (nystagmus), sluggish pupils, crossed eyes (strabismus), paralysis of certain eye muscles (ophthalmoplegia), deterioration of the nerves of the eyes (optic atrophy), and/or visual impairment leading to blindness.
Leigh syndrome may also affect the heart. Some children with this disorder may have abnormal enlargement of the heart (hypertrophic cardiomyopathy) and overgrowth of the fibrous membrane that divides the various chambers of the heart (asymmetric septal hypertrophy). Disease affecting the nerves outside of the central nervous system (peripheral neuropathy) may eventually occur, causing progressive weakness of the arms and legs.
The symptoms of the X-linked infantile form of Leigh syndrome are similar to those of classical Leigh syndrome. The symptoms of the adult-onset form of Leigh syndrome (subacute necrotizing encephalomyelopathy), a very rare form of the disorder, generally begin during adolescence or early adulthood. Initial symptoms are generally related to vision and may include such abnormalities as blurred “filmy” central visual fields (central scotoma), colorblindness, and/or progressive visual loss due to degeneration of the optic nerve (bilateral optic atrophy). The neurological problems associated with the disease progress slowly in this form of the disorder. At about 50 years of age, affected individuals may find it progressively difficult to coordinate voluntary movements (ataxia). Additional late symptoms may include partial paralysis and involuntary muscle movements (spastic paresis), sudden muscle spasms (clonic jerks), grand mal seizures, and/or varying degrees of dementia.
Leigh syndrome diagnosis
Leigh syndrome may be diagnosed by using the following criteria, defined by Rahman et al. in 1996 4:
- Progressive neurologic disease with motor and intellectual developmental delay
- Signs and symptoms of brainstem and/or basal ganglia disease
- Raised lactate concentration in blood and/or cerebrospinal fluid (CSF)
- The presence of one or more of the following:
- Characteristic features on brain imaging (CT scan or MRI)
- Typical nervous system tissue changes
- Typical nervous system tissue changes in a similarly affected sibling
After these criteria are met and a diagnosis of Leigh syndrome is made, molecular genetic testing can then differentiate between mtDNA-associated Leigh syndrome (caused by mutations in mtDNA) and nuclear gene-encoded Leigh syndrome (caused by mutations in nuclear DNA) 4. A diagnosis of nuclear gene-encoded Leigh syndrome can be made either by identifying a mutation in nuclear DNA, or by excluding the presence of a mutation in mtDNA 4.
Because not all patients have increased lactate levels, recent studies proposed new diagnostic criteria excluding the raised lactate levels as a prerequisite. The remaining criteria are similar, but add mitochondrial dysfunction as a criterion 4.
A diagnosis of Leigh-like syndrome may be considered in individuals who do not meet the strict diagnostic criteria but have features resembling Leigh syndrome 4.
What tests are considered when diagnosing Leigh syndrome?
Tests that may be useful in diagnosing Leigh syndrome include, measuring lactic acid concentration in body fluids (i.e., blood, urine, and/or cerebrospinal fluid, the fluid that surrounds the brain and spinal cord), brain imaging, muscle biopsy, respiratory chain enzyme studies, and genetic testing.
Is there genetic testing available for the mitochondrial and nuclear genes known to cause Leigh syndrome?
Although genetic testing is not available for all genes associated with Leigh syndrome, there is genetic testing available for both mitochondrial genes and nuclear genes associated with the condition.
Leigh syndrome treatment
Treatment of Leigh syndrome is directed toward the specific symptoms present in each person 6. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, cardiologists, neurologists, specialists who assess and treat hearing problems (audiologists), eye specialists, and other health care professionals may need to systematically and comprehensively plan an effective child’s treatment.
The most common treatment for Leigh’s disease is thiamine (Vitamin B1) or thiamine derivatives 1. Some people with Leigh syndrome may experience a temporary symptomatic improvement and a slight slowing of the progression of the disease. In those patients with Leigh syndrome who also have a deficiency of pyruvate dehydrogenase enzyme complex, a high fat, low carbohydrate diet may be recommended.
Oral sodium bicarbonate or sodium citrate may also be prescribed to manage lactic acidosis. Researchers are currently testing dichloroacetate to establish its effectiveness in treating lactic acidosis. In individuals who have the X-linked form of Leigh’s disease, a high-fat, low-carbohydrate diet may be recommended.
Supportive care for Leigh syndrome includes treatment of acidosis, seizures, dystonia, and cardiomyopathy, and attention to nutritional status 4.
- Seizures. Appropriate antiepileptic drugs tailored to the type of seizure should be administered under the supervision of a neurologist. Sodium valproate and barbiturates should be avoided because of their inhibitory effects on the mitochondrial respiratory chain 9.
- Dystonia.
- Benzhexol, baclofen, tetrabenezine, and gabapentin may be useful, alone or in various combinations; an initial low dose should be started and gradually increased until symptom control is achieved or intolerable side effects occur.
- Botulinum toxin injection has also been used in individuals with Leigh syndrome and severe intractable dystonia.
- Cardiomyopathy. Medical therapy may be required and should be supervised by a cardiologist.
- Nutrition. Regular assessment of daily caloric intake and adequacy of dietary structure including micronutrients and feeding management is indicated.
- Psychological support for the affected individual and family is essential.
Because anesthesia can potentially aggravate respiratory symptoms and bring on respiratory failure, careful consideration should be given to its use and close monitoring prior to, during, and after its use 10.
Progression and new symptoms should be monitored regularly (typically every 6-12 months). Evaluations with a neurologist, ophthalmologist, audiologist, and cardiologist are recommended 4.
Specific treatment is possible for the three nuclear gene-encoded Leigh-like syndromes (milder conditions with similar features). These include biotin-thiamine-responsive basal ganglia disease (BTBGD), biotinidase deficiency, and coenzyme Q10 deficiency caused by mutation of PDSS2 4.
Leigh syndrome prognosis
The prognosis for individuals with Leigh’s disease is poor. Individuals who lack mitochondrial complex IV activity and those with pyruvate dehydrogenase deficiency tend to have the worst prognosis and die within a few years. Those with partial deficiencies have a better prognosis, and may live to be 6 or 7 years of age. Some have survived to their mid-teenage years.
Leigh syndrome life expectancy
Leigh’s disease life expectancy depends on the cause of Leigh syndrome in each person, most do not survive past mid-childhood or adolescence 1.
References- Leigh’s Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Leighs-Disease-Information-Page
- Leigh syndrome. https://ghr.nlm.nih.gov/condition/leigh-syndrome
- Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. 2003 Oct 30 [Updated 2017 Sep 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1173
- Rahman S, Thorburn D. Nuclear Gene-Encoded Leigh Syndrome Overview. 2015 Oct 1. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK320989
- Finsterer J. Leigh and Leigh-Like Syndrome in Children and Adults. Pediatr Neurol. 2008; http://www.ncbi.nlm.nih.gov/pubmed/18805359
- Leigh syndrome. https://rarediseases.org/rare-diseases/leigh-syndrome
- Chinnery PF. Mitochondrial Disorders Overview. 2000 Jun 8 [Updated 2014 Aug 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1224
- Inheritance and Genetics. https://www.umdf.org/what-is-mitochondrial-disease/inheritance-and-genetics/
- Anderson CM, Norquist BA, Vesce S, Nicholls DG, Soine WH, Duan S, Swanson RA. Barbiturates induce mitochondrial depolarization and potentiate excitotoxic neuronal death. J Neurosci. 2002;22:9203–9.
- Niezgoda J, Morgan PG. Anesthetic considerations in patients with mitochondrial defects. Paediatr Anaesth. 2013;23:785–93.





{kind=link}