Kennedy’s disease
Kennedy’s disease also known as X-linked spinal and bulbar muscular atrophy, is a rare X-linked recessive, slowly progressive neuromuscular disorder that affects males in which degeneration of lower motor neurons results in weakness, atrophy, and fasciculations (fleeting muscle twitches visible under the skin) of the appendicular and bulbar muscle 1. Kennedy’s disease is an X-linked recessive disease, which means the patient’s mother carries the defective gene on one of her X chromosomes. Daughters of patients with Kennedy’s disease are also carriers and have a 1 in 2 chance of having a son affected with the disease. Parents with concerns about their children may wish to talk to a genetic counselor.
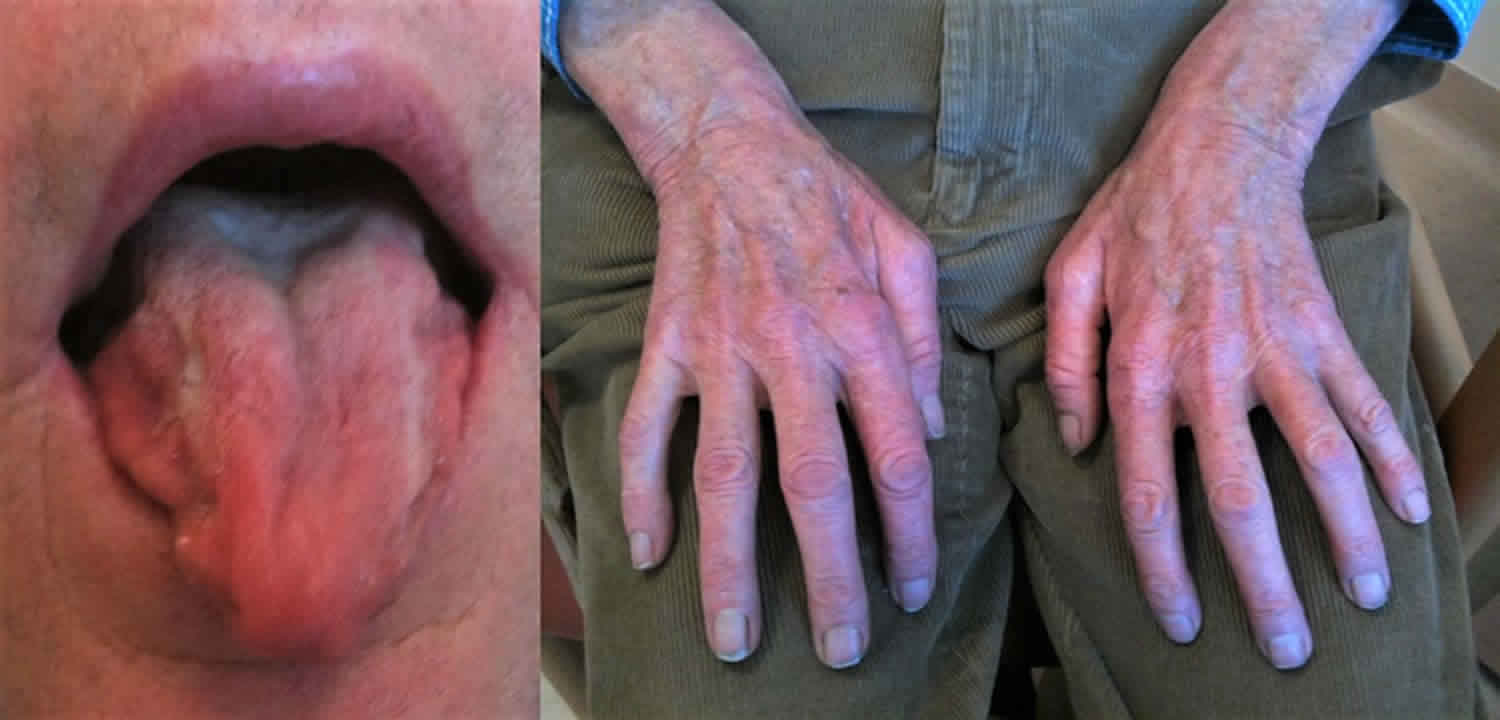
Kennedy’s disease occurs only in males. Kennedy disease is typically an adult-onset disease, where symptoms occur mainly between the ages of 20 and 50, although it has been diagnosed in men from their teens to their 70s 2. Early symptoms include tremor of the outstretched hands, muscle cramps with exertion, and fasciculations (fleeting muscle twitches visible under the skin). Eventually, individuals develop limb weakness which usually begins in the pelvic or shoulder regions. Weakness of the facial and tongue muscles may occur later in the course of the disease and often leads to dysphagia (difficulty in swallowing), dysarthria (slurring of speech), and recurrent aspiration pneumonia. Some individuals develop gynecomastia (excessive enlargement of male breasts) and low sperm count or infertility. Still others develop non-insulin-dependent diabetes mellitus.
Kennedy disease affects fewer than 1 in 350,000 males and is very rare in females, who are protected by their low levels of circulating testosterone, accounting for the sex-limited inheritance pattern in this disorder 3. Kennedy disease has been diagnosed in the USA, Europe, Asia, South America, and Australia. The Japanese population appears to have a very high prevalence of Kennedy Disease because of a founder effect.
Currently there is no known cure for Kennedy’s disease. Treatment is symptomatic and supportive. Physical therapy and rehabilitation to slow muscle weakness and atrophy may prove helpful.
Kennedy’s disease life expectancy is normal, though a small percentage of patients (about 10%) succumb to the disease in their 60’s or 70’s due to swallowing complications (aspiration pneumonia, asphyxiation) resulting from the bulbar weakness.
Kennedy’s disease cause
Kennedy disease or spinal and bulbar muscular atrophy is caused by a change (mutation) in the AR gene that encodes for a protein known as the androgen receptor on the X chromosome. The instructions within every gene consist of different arrangements of four basic chemicals (nucleotide bases) called adenine (A), cytosine (C), guanine (G), and thymine (T). Individuals with the disease have an abnormal section in the AR gene, which is due to an excessive number of CAG trinucleotide repetitions in the DNA sequence. An unaffected individual has 10-35 CAG repeats in the AR gene while a person with Kennedy disease has more than 36 CAG repeats in the gene.
The androgen receptor is in the cytoplasm of a cell where it responds to signals from male sex hormones (androgens). These receptors are abundant in many body tissues such as the skin, kidney, prostate, skeletal muscle, and the central nervous system motor neurons in the spinal cord and brainstem. In an unaffected person, the androgen hormone will bind to the receptor, and then the hormone-receptor complex will translocate into the nucleus, where it will signal genes to increase protein production for various functions.
In Kennedy disease, the exact mechanism for neuronal impairment is unknown, but it has to do with an altered functioning of the mutant androgen receptor.
Kennedy disease is an X-linked genetic disorder that occurs primarily in males. Very rarely, female carriers of the abnormal gene may show symptoms.
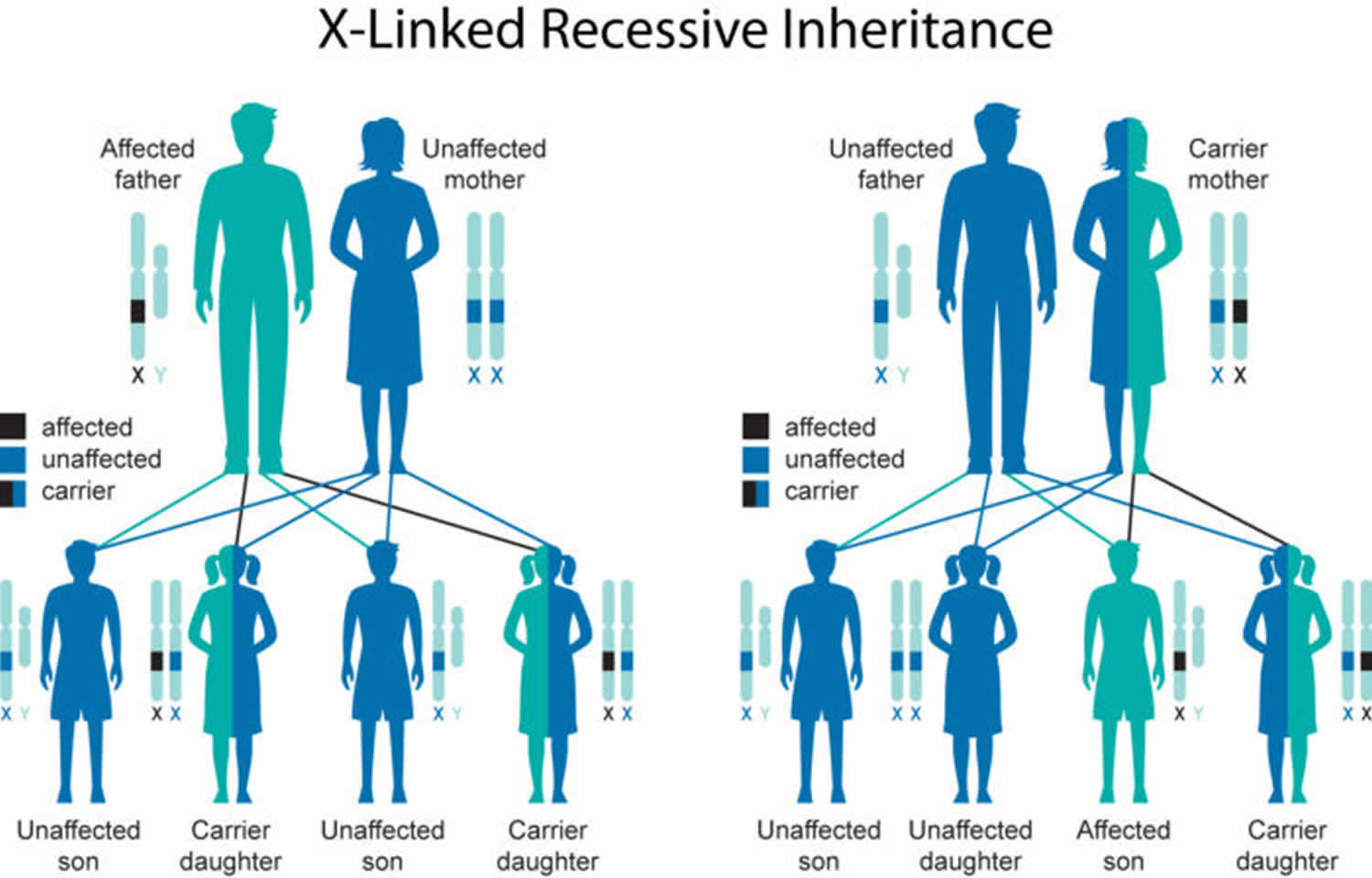
Normal females have two X chromosomes, in which one is an activated chromosome and the other is inactivated. Female carriers for Kennedy disease typically do not show symptoms because the androgen receptor must bind to its ligand, testosterone, to translocate to the nucleus and perform its functions. As females have low circulating levels of testosterone, Kennedy disease female carriers do not activate their mutant androgen receptors, thus rendering the mutant state of the androgen receptor protein innocuous. Males have only one X chromosome and will develop Kennedy disease if they inherit the X chromosome containing the disease gene. Affected males with X-linked disorders will always pass the gene to their daughters, but will only pass their normal Y chromosome to their sons. Therefore, all of the daughters of an affected male will be carriers for the disease, while sons of an affected male will not have the disease. Sons of female carriers have a 50 percent chance of inheriting the disease, while daughters have a 50 percent chance of becoming carriers.
Kennedy’s disease inheritance pattern
Kennedy’s disease is inherited in an X-linked pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome), a mutation in the only copy of the gene in each cell causes the disorder. In most cases, males experience more severe symptoms of the disorder than females (who have two X chromosomes). Females with a mutation in one copy of the AR gene in each cell are typically unaffected. A few females with mutations in both copies of the gene have had mild features related to the condition, including muscle cramps and occasional tremors. Researchers believe that the milder signs and symptoms in females may be related to lower androgen levels.
A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Figure 1. Kennedy’s disease X-linked inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Kennedy’s disease symptoms
Kennedy’s disease affected individuals begin to develop neurological symptoms between 20 to 50 years of age. These early symptoms include:
- Weakness/cramps in arm and leg muscles (proximal > distal)
- Face, mouth, and tongue muscle weakness
- Difficulty with speaking and swallowing (dysphagia)
- Twitching (Fasciculations)
- Tremors and trembling in certain positions
- Enlarged breasts (gynecomastia)
- Numbness
- Infertility
- Testicular atrophy
Kennedy’s disease affects the lower motor neurons that are responsible for the movement of many muscles in the legs, arms, mouth, and throat. Affected individuals will show signs of twitching, often in the tongue and/or hand, followed by muscle weakness and problems with facial muscles. These neurons, which connect the spinal cord to the muscles, become defective and die, so the muscles cannot contract. The destruction of these nerves is the main reason for the numbness, muscle weakness, and inability to control muscle contraction. With lack of normal neuromuscular function, a patient may experience hypertrophied calves in which the calf muscles thicken due to muscle cramps. In some cases, patients may also have one side of the body more affected than the other side.
Kennedy’s disease also affects nerves that control the bulbar muscles, which are important for breathing, speaking, and swallowing. Androgen insensitivity can also occur, sometimes beginning in adolescence and continuing through adulthood, characterized by enlarged breasts, decreased masculine appearance, and infertility. Patients may experience problems such as low sperm count and erectile dysfunction.
Neurologic findings
Neurologic symptoms typically begin between age 30 and 50 years 4. Onset of neurologic symptoms does not usually occur in childhood or adolescence.
Early signs are difficulty with walking and a tendency to fall. Many individuals have muscle cramps, while others complain of an action tremor 5. Deep tendon reflexes are decreased.
After one to two decades of symptoms, most affected individuals have difficulty climbing stairs. With time, atrophy of the proximal and distal musculature becomes evident. About one third of affected individuals require a wheelchair 20 years after the onset of symptoms.
Most individuals eventually show involvement of the bulbar muscles and have difficulty with speech articulation and swallowing. Severely affected individuals (many of whom are nonambulatory) are at risk for aspiration pneumonia and ventilatory failure because of weakness of the bulbar and respiratory musculature 6. This complication is the main life-threatening problem in Kennedy’s disease, and likely becomes an issue for only a minority of individuals. Therefore, the majority of individuals with Kennedy’s disease have a normal life expectancy and do not die from direct complications of their motor neuron disease. Fifteen of 223 persons in the Atsuta et al 6 study died at a mean age of 65 years.
Affected males may also have degeneration of the dorsal root ganglia, leading to mild (usually subclinical) abnormalities in sensory function in the distal extremities 5.
Electrodiagnostic studies are consistent with diffuse denervation atrophy, anterior horn cell loss, and sensory neuronopathy 7.
Histopathology
Degeneration of anterior horn cells in the spinal cord of affected individuals is observed 8. Changes in muscle include evidence of myopathy 9, in addition to neurogenic muscle atrophy. Immunohistochemistry shows inclusions of mutated androgen receptor (AR) protein 10.
Androgen insensitivity
Symptoms of androgen insensitivity typically begin in adolescence with gynecomastia, which is observed frequently in affected males 11. Variability in disease severity and progression occurs both within and between families 12. This is especially true of the androgen insensitivity signs of testicular atrophy and oligospermia/azoospermia with reduced fertility (see Androgen Insensitivity Syndrome). Males with Kennedy’s disease may not be able to grow a thick beard and may have difficulty conceiving.
The androgen insensitivity can be of greater concern to affected individuals than the motor neuron disease, especially early in the course of the disorder 13.
Heterozygotes
Neurologic findings
Females who are carriers of full-penetrance alleles of greater than 38 CAG repeats in AR are usually asymptomatic. While number of carriers have experienced muscle cramps or occasional tremors, female carriers usually do not have significant motor neuron disease 14.
Females who are symptomatic may have an abnormal electromyography 15.
Androgen insensitivity
Kennedy’s disease is a sex-limited disorder, with females protected by having low levels of circulating androgens leading to lower levels of androgen receptor stimulation. In addition, due to X-chromosome inactivation, females have only a portion of actively transcribed full-penetrance alleles (CAG>37), but it is the low level of circulating androgen that likely accounts for limited to absent symptoms in heterozygous female carriers or in females with biallelic full-penetrance AR alleles.
Genotype-phenotype correlations
Studies of the number of CAG repeats in AR alleles in males with Kennedy’s disease have established a correlation between number of CAG repeats and disease severity. In general, CAG repeat number inversely correlates with the age of onset of muscle weakness, difficulty climbing stairs, and wheelchair dependence 16. Thus, males with Kennedy’s disease whose alleles have a larger number of CAG repeats tend to have earlier disease onset and more rapid progression 17. For example, early onset (age 8-15 years) and rapid progression have been described in a family in which affected individuals have alleles of 50-54 CAG repeats 18.
However, these correlations are only generalizations and exceptions have been reported. For example, a male in a family with Kennedy’s disease with AR alleles of 37 CAG repeats (the average number of repeats in affected males) has been reported to be asymptomatic at age 46 years 19. The largest AR repeat expansion reported in a person with Kennedy’s disease is 68 20.
The genotype-phenotype correlation between allelic CAG repeat number and disease severity can only account for about 60% of the variability observed in clinical findings, indicating that other factors in addition to CAG repeat number determine age of disease onset and rate of disease progression. Indeed, relatives with Kennedy’s disease with an identical CAG repeat number may have considerably different disease courses.
Kennedy’s disease diagnosis
A diagnosis of Kennedy disease or spinal and bulbar muscular atrophy is suspected based on physical signs and symptoms, and sometimes family history.
Kennedy’s disease should be suspected in males with the following clinical features and family history.
Clinical features:
- Adolescent-onset signs of androgen insensitivity (e.g., gynecomastia)
- Post-adolescent onset of:
- Spinal lower motor neuron disease with muscle weakness of the limbs or muscle cramps
- Bulbar lower motor neuron disease with fasciculations of the tongue, lips, or perioral region; dysarthria and difficulty swallowing
- No signs of upper motor neuron disease (e.g., hyperreflexia, spasticity)
Family history consistent with X-linked inheritance
Note: Lack of a family history consistent with X-linked inheritance does not preclude the diagnosis.
Diagnosis can be confirmed by molecular genetic testing on a blood sample for CAG trinucleotide repeat expansion in the AR gene. Individuals with greater than 36 CAG trinucleotide repeats in the AR gene are diagnosed with the condition.
Clinical testing and work-up
Annual examinations to assess muscle strength may be appropriate.
Kennedy disease treatment
Currently, there is no known treatment or cure for Kennedy disease. Physical therapy, occupational therapy, and speech therapy are commonly used to adapt to the progressing disease and maintain an individual’s skills. Braces, walkers, and wheel chairs are used for ambulation. Breast reduction surgery is sometimes used as needed in patients with gynecomastia 21. Testosterone is not an appropriate treatment, as it can make the disease worse 22.
Surveillance
Appropriate measures include:
- Strength testing (annually)
- Pulmonary function tests (annually in advanced cases)
Agents and circumstances to avoid
Individuals with a tendency to fall should avoid slippery or rough walking surfaces.
Therapies under investigation
Anti-androgen therapy. There is no consensus or clear evidence as to whether anti-androgen therapy is an effective form of treatment for the neurologic complications.
- Anti-androgen therapy shows promise based on studies in Drosophila and mouse models as well as knowledge of the molecular basis of Kennedy’s disease. For these reasons, a Japanese group 23 performed a clinical trial of leuprorelin in individuals with Kennedy’s disease who were followed over 48 weeks: significant improvement was observed in cricopharyngeal opening duration, but in no other outcome measures. In particular, there was no effect on the primary outcome measure (the ALS Functional Rating Scale or ALSFRS) in the period of randomization. Although the trial was continued as an open label extension, and encouraging results were reported, the conclusion was that this clinical trial did not establish efficacy for anti-androgen therapy in Kennedy’s disease 24.
- A larger subsequent study with swallow function as the primary outcome measure also did not show an overall benefit, except in post hoc analysis of subjects in whom disease duration was less than ten years 25.
- Another anti-androgen therapy approach was attempted 26: individuals with Kennedy’s disease were randomized to placebo or dutasteride, a drug that blocks the conversion of testosterone to dihydrotestosterone (DHT). The rationale was that DHT may mediate many of the toxic effects, and this drug would permit affected individuals to retain the anabolic effects of testosterone, thereby diminishing the side effects of anti-androgen therapy. However, this study did not show a significant effect of dutasteride on the progression of muscle weakness in Kennedy’s disease.
Hence, the utility of anti-androgen therapy as a treatment for Kennedy’s disease remains unclear. Furthermore, it is possible that anti-androgen therapies, even if effective, would need to be administered prior to disease onset or early on in the neurodegenerative process. More importantly, the side effects of anti-androgen therapies would probably far outweigh the therapeutic benefit for most individuals, and likely should be reserved for people with Kennedy’s disease who are wheelchair bound or exhibit pronounced bulbar weakness.
Creatine supplementation. Studies of amyotrophic lateral sclerosis (ALS) suggest that creatine supplementation may temporarily enhance muscle strength and exercise performance in this motor neuron disease 27, prompting speculation that it may offer a similar benefit to individuals with Kennedy’s disease; this remains to be tested.
Experimental therapies in animal models
- Other interventions that have been shown to have benefit in mouse models of Kennedy’s disease include the HSP-90 inhibitors 17-AAG and 17-DMAG, the synthetic curcumin derivative ASC-J9, and insulin-like growth factor 1 28.
- More recently, one group directly examined the role of muscle expression of mutated AR in Kennedy’s disease disease pathogenesis by developing a BAC transgenic mouse model featuring a floxed first exon to permit cell-type specific excision of a human AR transgene 29. They engineered the human AR transgene to carry 121 CAG repeats (BAC fxAR121), and found that BAC fxAR121 mice develop a gender-restricted, progressive neuromuscular phenotype, characterized by weight loss, motor deficits, muscle atrophy, myopathy, and shortened life span. By terminating expression of mutated AR in the skeletal muscles of BAC fxAR121 male mice, this study revealed a crucial role for muscle expression of mutated AR in Kennedy’s disease disease pathogenesis. Hence, this work predicts that muscle-directed therapies hold great promise as definitive treatments for Kennedy’s disease motor neuron degeneration.
- Another study sought to ameliorate toxicity in mouse models of Kennedy’s disease by suppressing polyQ-AR expression using antisense oligonucleotides (ASOs) 30. This investigation developed compounds to specifically target AR expression in the periphery, and using two mouse models, found that peripheral gene suppression of mutated AR rescues deficits in muscle weight, fiber size, and grip strength; reverses changes in muscle gene expression; and extends the life span of mutated males. Interestingly, delivery of an anti-AR ASO to the CNS also elicited a modest improvement in these disease read-outs in a Kennedy’s disease mouse model, but was much less effective than peripheral delivery. Hence, this report, together with the genetic rescue study of Kennedy’s disease 29, strongly suggests that peripheral administration of therapies directed to muscle should be explored in humans with Kennedy’s disease. Preparations are underway to initiate a clinical trial of anti-AR ASO therapy via peripheral delivery in males with Kennedy’s disease.
Kennedy’s disease prognosis
Kennedy’s disease is slowly progressive. Individuals tend to remain ambulatory until late in the disease, although some may be wheelchair-bound during later stages. The life span of individuals with Kennedy’s disease is usually normal.
Kennedy’s disease life expectancy
Kennedy’s disease life expectancy is normal, though a small percentage of patients (about 10%) succumb to the disease in their 60’s or 70’s due to swallowing complications (aspiration pneumonia, asphyxiation) resulting from the bulbar weakness.
References- Alves CN, Braga TKK, Somensi DN, Nascimento BSVD, Lima JAS, Fujihara S. X-linked spinal and bulbar muscular atrophy (Kennedy’s disease): the first case described in the Brazilian Amazon. Einstein (Sao Paulo). 2018;16(2):eRC4011. Published 2018 Jun 7. doi:10.1590/S1679-45082018RC4011 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5995545
- Kennedy’s Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Kennedys-Disease-Information-Page
- Kennedy disease. https://rarediseases.org/rare-diseases/kennedy-disease/
- Rhodes LE, Freeman BK, Auh S, Kokkinis AD, La Pean A, Chen C, Lehky TJ, Shrader JA, Levy EW, Harris-Love M, Di Prospero NA, Fischbeck KH. Clinical features of spinal and bulbar muscular atrophy. Brain. 2009;132:3242–51.
- Grunseich C, Rinaldi C, Fischbeck KH. Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis. 2014b;20:6–9.
- Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M, Tanaka F, Tamakoshi A, Sobue G. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006;129:1446–55.
- Ferrante MA, Wilbourn AJ. The characteristic electrodiagnostic features of Kennedy’s disease. Muscle Nerve. 1997;20:323–9.
- Ogata A, Matsuura T, Tashiro K, Moriwaka F, Demura T, Koyanagi T, Nagashima K. Expression of androgen receptor in X-linked spinal and bulbar muscular atrophy and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1994;57:1274–5.
- Sorarù G, D’Ascenzo C, Polo A, Palmieri A, Baggio L, Vergani L, Gellera C, Moretto G, Pegoraro E, Angelini C. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci. 2008;264:100–5.
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–70.
- Sinnreich M, Sorenson EJ, Klein CJ. Neurologic course, endocrine dysfunction and triplet repeat size in spinal bulbar muscular atrophy. Can J Neurol Sci. 2004;31:378–82.
- Lee JH, Shin JH, Park KP, Kim IJ, Kim CM, Lim JG, Choi YC, Kim DS. Phenotypic variability in Kennedy’s disease: implication of the early diagnostic features. Acta Neurol Scand. 2005;112:57–63.
- Warner CL, Griffin JE, Wilson JD, Jacobs LD, Murray KR, Fischbeck KH, Dickoff D, Griggs RC. X-linked spinomuscular atrophy: a kindred with associated abnormal androgen receptor binding. Neurology. 1992;42:2181–4.
- Mariotti C, Castellotti B, Pareyson D, Testa D, Eoli M, Antozzi C, Silani V, Marconi R, Tezzon F, Siciliano G, Marchini C, Gellera C, Donato SD. Phenotypic manifestations associated with CAG-repeat expansion in the androgen receptor gene in male patients and heterozygous females: a clinical and molecular study of 30 families. Neuromuscul Disord. 2000;10:391–7.
- Sobue G, Doyu M, Kachi T, Yasuda T, Mukai E, Kumagai T, Mitsuma T. Subclinical phenotypic expressions in heterozygous females of X-linked recessive bulbospinal neuronopathy. J Neurol Sci. 1993;117:74–8.
- La Spada AR, Roling DB, Harding AE, Warner CL, Spiegel R, Hausmanowa-Petrusewicz I, Yee WC, Fischbeck KH. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat Genet. 1992;2:301–4.
- Igarashi S, Tanno Y, Onodera O, Yamazaki M, Sato S, Ishikawa A, Miyatani N, Nagashima M, Ishikawa Y, Sahashi K, et al. Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology. 1992;42:2300–2.
- Echaniz-Laguna A, Rousso E, Anheim M, Cossée M, Tranchant C. A family with early-onset and rapidly progressive X-linked spinal and bulbar muscular atrophy. Neurology. 2005;64:1458–60.
- Kuhlenbäumer G, Kress W, Ringelstein EB, Stögbauer F. Thirty-seven CAG repeats in the androgen receptor gene in two healthy individuals. J Neurol. 2001;248:23–6.
- Grunseich C, Kats IR, Bott LC, Rinaldi C, Kokkinis A, Fox D, Chen KL, Schindler AB, Mankodi AK, Shrader JA, Schwartz DP, Lehky TJ, Liu CY, Fischbeck KH. Early onset and novel features in a spinal and bulbar muscular atrophy patient with a 68 CAG repeat. Neuromuscul Disord. 2014a;24:978–81.
- Sperfeld AD, Karitzky J, Brummer D, Schreiber H, Häussler J, Ludolph AC, Hanemann CO. X-linked bulbospinal neuronopathy: Kennedy disease. Arch Neurol. 2002;59:1921–6.
- Goldenberg JN, Bradley WG. Testosterone therapy and the pathogenesis of Kennedy’s disease (X-linked bulbospinal muscular atrophy). J Neurol Sci. 1996;135:158–61.
- Banno H, Katsuno M, Suzuki K, Takeuchi Y, Kawashima M, Suga N, Takamori M, Ito M, Nakamura T, Matsuo K, Yamada S, Oki Y, Adachi H, Minamiyama M, Waza M, Atsuta N, Watanabe H, Fujimoto Y, Nakashima T, Tanaka F, Doyu M, Sobue G. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:140–50.
- Fischbeck KH, Bryan WW. Anti-androgen treatment for spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:119–20.
- Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Yabe I, Sasaki H, Aoki M, Morita M, Nakano I, Kanai K, Ito S, Ishikawa K, Mizusawa H, Yamamoto T, Tsuji S, Hasegawa K, Shimohata T, Nishizawa M, Miyajima H, Kanda F, Watanabe Y, Nakashima K, Tsujino A, Yamashita T, Uchino M, Fujimoto Y, Tanaka F, Sobue G., Japan SBMA Interventional Trial for TAP-144-SR (JASMITT) study group. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:875–84.
- Fernández-Rhodes LE, Kokkinis AD, White MJ, Watts CA, Auh S, Jeffries NO, Shrader JA, Lehky TJ, Li L, Ryder JE, Levy EW, Solomon BI, Harris-Love MO, La Pean A, Schindler AB, Chen C, Di Prospero NA, Fischbeck KH. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. 2011;10:140–7.
- Mazzini L, Balzarini C, Colombo R, Mora G, Pastore I, De Ambrogio R, Caligari M. Effects of creatine supplementation on exercise performance and muscular strength in amyotrophic lateral sclerosis: preliminary results. J Neurol Sci. 2001;191:139–44.
- Fischbeck KH. Developing treatment for spinal and bulbar muscular atrophy. Prog Neurobiol. 2012;99:257–61.
- Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, Tokunaga S, Lopez E, Sopher BL, Bennett CF, Shelton GD, Cleveland DW, La Spada AR. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron. 2014;82:295–307.
- Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S, Yu XX, Cortes CJ, Bennett CF, Monia BP, La Spada AR, Hung G. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014;7:774–84.





{kind=link}