What is lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a rare progressive multisystem disease that affects the lungs, the kidneys, and the lymph nodes, found almost exclusively in women of childbearing age (the years between puberty and menopause when it is possible to become pregnant), usually in their 30s and 40s 1, 2, 3, 4, 5, 6, 7, 8, 9. Lymphangioleiomyomatosis (LAM) can occur in older women as well, although this is less common. Lymphangioleiomyomatosis (LAM) is characterized by an abnormal growth of spindle-shaped and epithelioid LAM cells, which express alpha-smooth muscle actin (αSMA), especially in the lungs, lymphatic system, and kidneys 1, 2, 10. Abnormal growth of these cells can lead to cystic lung destruction, accumulation of lymph rich fluid in the chest and abdomen, and growth of benign tumors in the kidneys.
Extrapulmonary lymphangioleiomyomatosis (LAM) manifestations frequently include renal angiomyolipomas (AMLs) and lymphatic abnormalities 1, 2. Empirically, lymphangioleiomyomatosis (LAM) is defined as a low-grade metastasizing neoplasm whose cell (or cells) of origin is uncertain 11, 12, 13. LAM has been reported to recur in transplanted lungs, consistent with a metastatic mechanism for the disease 14, 15, but has not been reported to cause graft failure or to jeopardize eligibility for transplant. Recent analyses of single-cell gene expression profiles indicate that LAM cells may originate in the uterus 16 and/or lung mesenchyme 17. However, the similarity of lymphangioleiomyomatosis (LAM) with cancer molecular portraits and the extent of lymphangioleiomyomatosis (LAM) disease heterogeneity are not yet fully understood 18, 19, 20, 21, 22.
There are two types of lymphangioleiomyomatosis (LAM) 23:
- Lymphangioleiomyomatosis (LAM) that occurs alone and sporadically is called isolated or sporadic lymphangioleiomyomatosis (S-LAM).
- When lymphangioleiomyomatosis occurs in association with a rare disease called tuberous sclerosis complex (TSC), it is called tuberous sclerosis complex-lymphangioleiomyomatosis (TSC-LAM). Tuberous sclerosis complex (TSC) is an autosomal-dominant multisystem disorder caused by mutations in the tumor suppressor genes TSC1 on chromosome 9q34 and TSC2 on chromosome 16p13 24, 25, 26, 27, 28, 29, 30, 31. Tuberous sclerosis complex (TSC) is inherited in an autosomal dominant pattern. Tuberous sclerosis complex (TSC) is characterized by the growth of benign tumors throughout the body, including in the heart, brain, and kidneys. Tuberous sclerosis complex (TSC) involves abnormalities of the skin (hypomelanotic macules, confetti skin lesions, facial angiofibromas, shagreen patches, fibrous cephalic plaques, ungual fibromas); brain (subependymal nodules, cortical tubers, and subependymal giant cell astrocytomas [SEGAs], seizures, intellectual disability / developmental delay, psychiatric illness); kidney (angiomyolipomas, cysts, renal cell carcinomas); heart (rhabdomyomas, arrhythmias); and lungs (lymphangioleiomyomatosis [LAM], multifocal micronodular pneumonocyte hyperplasia). Certain symptoms develop before to birth, such as heart tumors (rhabdomyoma). Other symptoms become more obvious in childhood, such as developmental delay and skin changes. Lung and kidney tumors are more likely to develop in adulthood. Central nervous system tumors are the leading cause of morbidity and mortality; renal disease is the second leading cause of early death. Tuberous sclerosis complex (TSC) is diagnosed based on a clinical exam, medical tests such as imaging studies, and genetic testing. Treatment is based on managing the symptoms, and includes medications and surgery. Mutations of TSC1 or TSC2 cause abnormal activation of the mechanistic target of rapamycin (mTOR), which is the basis for the current, U.S. Food and Drug Administration-approved, standard of care use of an mTOR inhibitor for lung and kidney disease 32. Rapamycin has greatly improved outcomes for women with LAM, but some patients continue to lose lung function, albeit at a lower rate, whilst receiving treatment 33, 34. Disease heterogeneity might contribute to the differences in clinical benefit of rapamycin 35. For enlarging subependymal giant cell astrocytomas (SEGAs]): mTOR inhibitors; neurosurgery when size causes life-threatening neurologic symptoms 31. For seizures: vigabatrin and other anti-seizure drugs, and on occasion, epilepsy surgery 31. For renal angiomyolipomas (AMLs) >4 cm, or >3 cm and growing rapidly: mTOR inhibitors are the recommended first line of therapy with secondary therapy options being embolization, renal sparing surgery, or ablative therapy 31. For facial angiofibromas: topical mTOR inhibitors. For symptomatic cardiac rhabdomyomas: surgical intervention or consideration of mTOR inhibitor therapy. For lymphangioleiomyomatosis (LAM): mTOR inhibitors. For TSC-associated neuropsychiatric disorder (TAND): refer to a suitable professional to provide appropriate treatment, which may include Applied Behavior Analysis (ABA) therapy and consideration of medication for those with ADHD.
The cause of lymphangioleiomyomatosis (LAM) is not well understood, however, most sporadic lymphangioleiomyomatosis (S-LAM) is caused by mutations (changes) in the structure of one of two tuberous sclerosis complex (TSC) tumor suppressor genes, called TSC1 and TSC2. Somatic inactivation of tumor suppressor genes TSC2 or much less commonly TSC1 in an unknown cell type(s) appears to be sufficient for sporadic lymphangioleiomyomatosis (S-LAM) development 19. Abnormal muscle-like cells that carry these mutations appear and grow out of control in the lungs, kidneys, or lymph nodes. These LAM cells may initially develop in the uterus or elsewhere, but when they travel to the lungs, they produce the respiratory symptoms of LAM.
Researchers believe female hormone estrogen plays a role, because lymphangioleiomyomatosis (LAM) almost always occurs in females between puberty and menopause and very rarely develops before puberty or after menopause. Lymphangioleiomyomatosis (LAM) also gets worse during pregnancy and after using medicines with estrogen, such as birth control. After menopause, LAM sometimes stops getting worse.
Doctors may diagnose lymphangioleiomyomatosis with imaging tests such as high-resolution computed tomography (HRCT) scans and blood tests for vascular endothelial growth factor D (VEGF-D). Other tests and procedures may be needed to diagnose lymphangioleiomyomatosis.
Lymphangioleiomyomatosis occurs in approximately 30 percent of women with tuberous sclerosis complex (TSC). Sporadic lymphangioleiomyomatosis (S-LAM), which occurs without tuberous sclerosis complex, is estimated to affect 3.3 to 7.4 per million women worldwide. Lymphangioleiomyomatosis may be underdiagnosed because its symptoms are similar to those of other lung disorders such as asthma, bronchitis, and chronic obstructive pulmonary disease.
Signs and symptoms of lymphangioleiomyomatosis most often appear during a woman’s thirties. Affected women have an uncontrolled growth (proliferation) of specialized cells (smooth muscle-like lymphangioleiomyomatosis cells) in certain organs of the body, especially the lungs, kidney and lymphatics, resulting in the formation of lung cysts and the destruction of normal lung tissue. They may also have an accumulation of fluid in the cavity around the lungs (chylothorax). The lung abnormalities resulting from lymphangioleiomyomatosis may cause difficulty breathing (dyspnea), chest pain, and coughing, which may bring up blood (hemoptysis), especially following periods of exercise or exertion. Many women with lymphangioleiomyomatosis have recurrent episodes of collapsed lung (spontaneous pneumothorax) or fluid accumulation around the lungs (pleural effusion). The lung problems may be progressive and in some cases, may result in chronic respiratory failure, without lung transplantation, may eventually lead to limitations in activities of daily living, the need for oxygen therapy, and respiratory failure. Although lymphangioleiomyomatosis cells are not considered cancerous, they may spread between tissues (metastasize). As a result, the condition may recur even after lung transplantation.
The lymphatic system consists of a network of vessels that transport lymph fluid and immune cells throughout the body. Lymph fluid helps exchange immune cells, proteins, and other substances between the blood and tissues.
Women with lymphangioleiomyomatosis may develop cysts in the lymphatic vessels of the chest and abdomen. These cysts are called lymphangioleiomyomas. Affected women may also develop tumors called angiomyolipomas made up of lymphangioleiomyomatosis cells, fat cells, and blood vessels. Angiomyolipomas usually develop in the kidneys. Internal bleeding is a common complication of angiomyolipomas.
Lymphangioleiomyomatosis has no cure. Doctors treat lymphangioleiomyomatosis with sirolimus (rapamycin), a medicine that stabilizes lung function, treats an abnormal fluid buildup in the lung called chylothorax, and improves overall quality of life. They may also prescribe other medicines or therapies to control other symptoms or complications.
It is important to get routine follow-up care because the disease tends to worsen over time. How quickly the disease worsens varies. More than half of women who have lymphangioleiomyomatosis will develop a serious condition called pneumothorax, or collapsed lung, that requires immediate treatment. Over time, lymphangioleiomyomatosis may cause permanent damage to the lungs and cause potentially fatal respiratory failure. Lung transplant is a treatment option for some women whose lungs have been severely damaged by lymphangioleiomyomatosis.
Lymphangioleiomyomatosis key facts
- Lymphangioleiomyomatosis primarily affects your lungs and your breathing. Occasionally, it is associated with tuberous sclerosis complex (TSC). When lymphangioleiomyomatosis is a part of tuberous sclerosis complex, it is more common to have kidney tumors called angiomyolipomas and other benign tumors in the abdomen and, in rare instances, involvement of the brain that may cause seizures and require monitoring.
- Lymphangioleiomyomatosis is a rare disease affecting women of child-bearing age, usually in their 30s and 40s.
- Lymphangioleiomyomatosis can shorten your lifespan, though over the last few decades, some patients are living longer than ever before. It is possible that lymphangioleiomyomatosis can make you lose your lung function to the point of needing a lung transplant.
- There are no behavioral conditions or risk factors that are known to cause lymphangioleiomyomatosis.
- Although female hormones and pregnancy can aggravate lymphangioleiomyomatosis, this is not considered a cause or risk factor for the development of lymphangioleiomyomatosis.
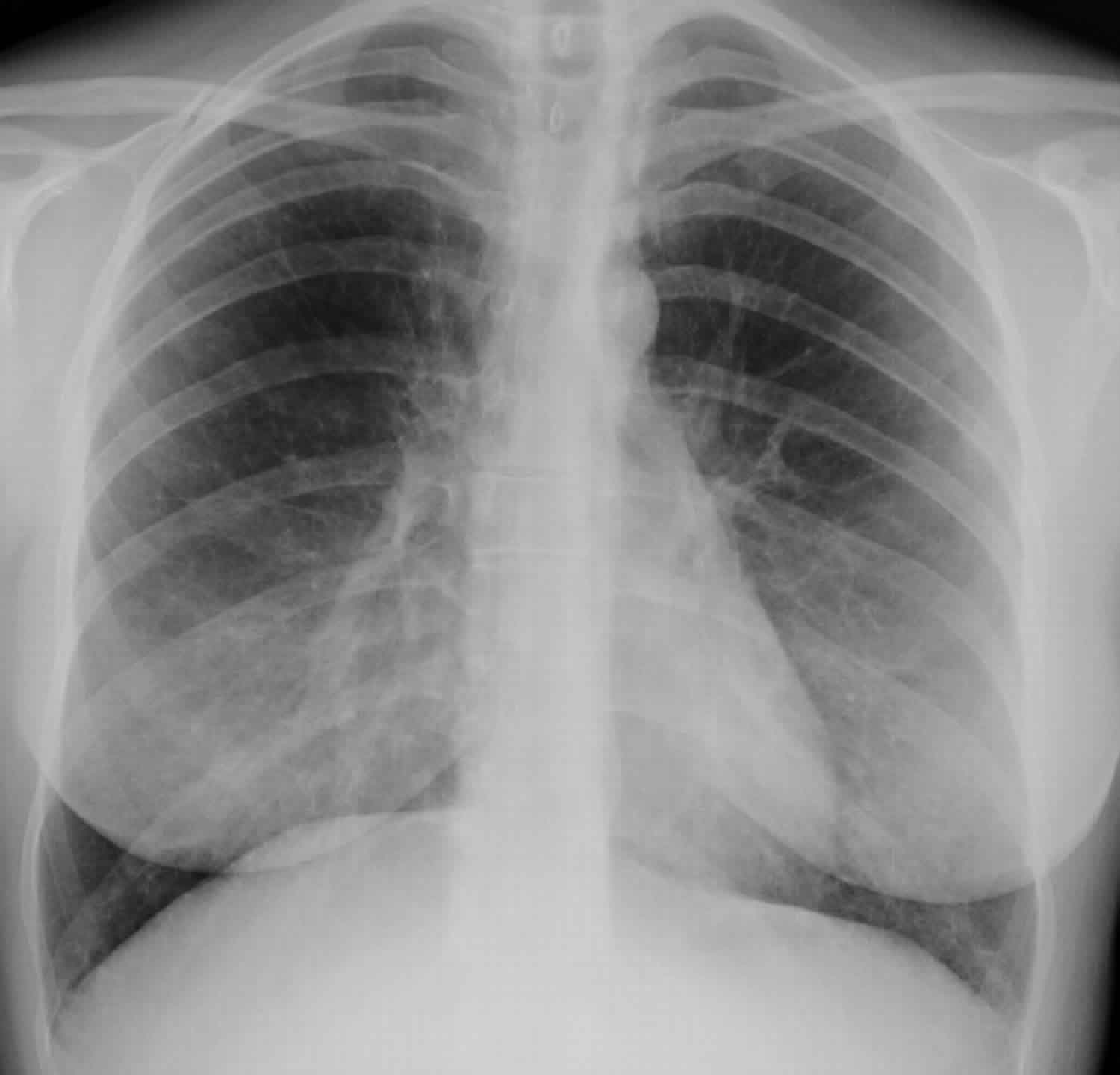
Figure 1. Lymphangioleiomyomatosis

Lymphangioleiomyomatosis causes
The exact cause of lymphangioleiomyomatosis (LAM) is not well understood. Since lymphangioleiomyomatosis occurs almost exclusively in females of reproductive age, it is possible that an association exists between female hormones (i.e., estrogen and progesterone) and lymphangioleiomyomatosis (LAM). Some researchers are studying the connection between a rare genetic disorder known as tuberous sclerosis complex (TSC) and lymphangioleiomyomatosis. Approximately 30 percent of women with tuberous sclerosis complex (TSC) show evidence of pulmonary lymphangioleiomyomatosis, suggesting that these disorders may share a common cause or origin 23. Tuberous sclerosis complex (TSC) is caused by mutations in one of two genes known as the TSC1 gene or TSC2 gene 31. TSC genes function as tumor suppressor genes. Mutations in TSC2 account for the majority (75%) of tuberous sclerosis complex (TSC) cases 36.
Mutations in the TSC1 gene or more commonly, the TSC2 gene, cause lymphangioleiomyomatosis 37. The TSC1 and TSC2 genes provide instructions for making the proteins hamartin and tuberin, respectively. The TSC1 and TSC2 genes are linked to a complex called TBC1D7, which regulates the kinase mechanistic target of the rapamycin called mTOR 38. Within cells, the proteins hamartin and tuberin likely help regulate cell growth and size. The proteins act as tumor suppressors, which normally prevent cells from growing and dividing too fast or in an uncontrolled way.
When both copies of the TSC1 gene are mutated in a particular cell, that cell cannot produce any functional hamartin; cells with two altered copies of the TSC2 gene are unable to produce any functional tuberin. The loss of these proteins allows the cell to grow and divide in an uncontrolled way, resulting in the tumors and cysts associated with lymphangioleiomyomatosis.
Sporadic lymphangioleiomyomatosis (S-LAM) is not inherited. Instead, researchers suggest that it is caused by a random mutation (e.g., mutations that occur in peripheral tissues after conception) in the TSC1 or TSC2 gene that occurs very early in development. As a result, some of the body’s cells have a normal version of the gene, while others have the mutated version. This situation is called mosaicism. When a mutation occurs in the other copy of the TSC1 or TSC2 gene in certain cells during a woman’s lifetime (a somatic mutation), she may develop lymphangioleiomyomatosis. These women typically have no history of this disorder in their family. These mutations were not found in the blood or the normal lung or normal kidney cells of affected individuals. Sporadic lymphangioleiomyomatosis (S-LAM) affects almost only premenopausal women, although some rare cases were reported to occur in men 39. These data suggest that tuberous sclerosis complex (TSC) mutations are a cause of lymphangioleiomyomatosis even in women who do not have the heritable disorder, tuberous sclerosis complex.
Estrogen is thought to play a role in lymphangioleiomyomatosis because the condition 40, 41:
- Primarily affects women.
- Worsens in a pattern that matches up with a women’s menstrual cycle, during pregnancy, and after using medicines such as birth control that contain estrogen.
- Has been known to stop worsening in some women who have entered menopause.
The symptoms associated with lymphangioleiomyomatosis occur due to the spread and accumulation (proliferation) of lymphangioleiomyomatosis cells into various organs of the body, especially the lungs. This abnormal proliferation results in the formation of cysts as well as the obstruction of affected airways (breathing tubes), blood vessels, and lymph vessels. Infiltration of the lung with lymphangioleiomyomatosis cells may result in progressive breathing difficulties, lung collapse, and interference with the lung’s ability to deliver oxygen to the rest of the body.
Obstruction of blood vessels may result in bleeding in the lungs (pulmonary hemorrhage) and coughing up of blood (hemoptysis). Obstruction of lymph vessels may result in chylous effusions and ascites. Lymph vessels are part of the lymphatic system, a circulatory network of vessels, ducts, and nodes that filter and distribute lymph and blood cells throughout the body. Lymph is a fluid that circulates through the body cleaning and strengthening tissue. After performing its functions, lymph is drained away through the lymphatic system.
Risk factors for lymphangioleiomyomatosis
Although female hormones and pregnancy can aggravate lymphangioleiomyomatosis, this is not considered a cause or risk factor for the development of lymphangioleiomyomatosis. Often lymphangioleiomyomatosis is a result of a random genetic mutation or occurs in association with an inherited disorder such as tuberous sclerosis complex (TSC). Women with tuberous sclerosis complex (TSC) have an increased risk of developing tuberous sclerosis complex-lymphangioleiomyomatosis.
In rare cases, lymphangioleiomyomatosis has been reported in men.
Lymphangioleiomyomatosis prevention
There is no way to prevent lymphangioleiomyomatosis (LAM). If you have tuberous sclerosis complex (TSC), your healthcare provider may recommend that you not smoke or take medicine with estrogen. This may help slow down the development of LAM. Medicines may stop abnormal LAM cells from increasing in number and may reduce their size. If you have TSC-LAM, your doctor may recommend genetic counseling before you get pregnant to help you understand the risk of passing tuberous sclerosis complex (TSC) and LAM on to your children.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Lymphangioleiomyomatosis pathology
Lymphangioleiomyomatosis is caused by abnormal growth of the smooth muscle cells in the lungs. The triggers for this process are not well understood, but hormone therapy can aggravate the process. Estrogen is thought to play an important role in the development of the sporadic form of lymphangioleiomyomatosis through stimulation of cell growth that is mediated by estrogen receptors alpha (ERa) 42. Although the mechanism is not entirely understood, researchers hypothesize that estrogen can cause modulation of growth signaling pathways that could lead to uncontrolled cell growth 40. The observation that patient with lymphangioleiomyomatosis have progression of the disease during pregnancy and oral estrogen use supports this. Estrogen use is also known to cause worsening of the disease and in vitro studies have shown that lymphangioleiomyomatosis cells express increased estrogen receptors alpha (ERa) 43. Mutations in tuberous sclerosis complex-2 gene (TSC2) have been proven to have mutations in patients with sporadic lymphangioleiomyomatosis (S-LAM) 44. The TSC2 gene along with TSC1 encodes proteins responsible for inhibition of the mammalian target of rapamycin (mTOR) which is an intracellular signaling pathway the regulated cell proliferation 45. It affects these 2 genes in the hereditary form of lymphangioleiomyomatosis which occurs with tuberous sclerosis.
Mutations in the TSC1 gene or more commonly, the TSC2 gene, cause lymphangioleiomyomatosis 37. The TSC1 and TSC2 genes provide instructions for making the proteins hamartin and tuberin, respectively. The TSC1 and TSC2 genes are linked to a complex called TBC1D7, which regulates the kinase mechanistic target of the rapamycin called mTOR 38. Within cells, the proteins hamartin and tuberin likely help regulate cell growth and size. The proteins act as tumor suppressors, which normally prevent cells from growing and dividing too fast or in an uncontrolled way.
The mutations in the TSC genes, such as TSC2 mutation, have been linked to causing overactivation of the mTOR pathway leading to defects in smooth muscle cell proliferation and the formation of benign tumors (angiomyolipomas) in multiple organs that are commonly seen in LAM 46, 47. The mTOR pathway is important for intracellular phosphorylation and extracellular signal-regulated kinase (ERK) hence serving as a regulator for the growth and proliferation of cells. Thus, the targeted inhibition of the mTOR pathway plays a key role in the development of the new therapy for patients with tuberous sclerosis complex (TSC) 38.
Lymphangioleiomyomatosis symptoms
The uncontrolled growth of lymphangioleiomyomatosis cells and their effect on nearby body tissues causes the signs, symptoms, and complications of lymphangioleiomyomatosis. Symptoms tend to start when women are between the ages of 20 and 40. The symptoms of lymphangioleiomyomatosis vary from case to case. Specific symptoms depend upon which organs are affected by the lymphangioleiomyomatosis cells. In most cases, the initial symptoms involve the lung and include shortness of breath, especially upon exercise or exertion. Affected individuals may also experience episodes of chest pain, coughing, wheezing, and coughing up small amounts of blood (hemoptysis).
Usually, tuberous sclerosis complex-lymphangioleiomyomatosis (TSC-LAM) is milder than sporadic lymphangioleiomyomatosis (S-LAM) and may not cause symptoms affecting the lungs. However, the severity of the disease varies from patient to patient and it is still possible for some women with sporadic lymphangioleiomyomatosis (S-LAM) to also have mild disease without symptoms and some women with tuberous sclerosis complex-lymphangioleiomyomatosis (TSC-LAM) to have more severe disease with worse symptoms and complications.
Lymphangioleiomyomatosis (LAM) may be suspected when a young female patient has any of the below symptoms.
The most common lymphangioleiomyomatosis symptoms are:
- Chest pain or aches that may worsen when you breathe in.
- Fatigue or extreme tiredness that may affect your overall quality of life.
- Frequent cough which can produce bloody phlegm or mucus in your lungs
- Shortness of breath (dyspnea) that at first may occur only during high-energy activities but over time may happen after simple activities such as dressing and showering.
- Severe pain or tightness in the chest and shortness of breath, which can be symptoms of a collapsed lung (pneumothorax).
- Wheezing or a whistling sound when you breathe.
You may feel the following symptoms in other parts of the body that are caused by LAM cells:
- Abdominal pain: Many people who have LAM get benign (noncancerous) tumors in their kidneys. If these tumors grow large enough, they can lead to bleeding in the kidneys or fluid buildup, which can cause pain in the abdomen.
- Lymph nodes that may be larger than normal (lymphadenopathy): Usually, these larger-than-normal lymph nodes develop in body locations where they cannot be felt, such as the chest. Rarely, larger-than-normal lymph nodes may develop in places where they can be felt, such as in the neck or under the arms.
Lymphangioleiomyomatosis is most commonly detected when a younger woman develops a “pneumothorax”, where the lung “pops” and air accumulates around the lung inside the chest wall, causing the lung to collapse. In patients with lymphangioleiomyomatosis, this occurs due to the bursting of the cysts that are formed in the lungs. Pneumothorax causes sharp chest pains and shortness of breath, which may be mild to severe. lymphangioleiomyomatosis can also be associated with accumulation of a milky fluid around the lungs called “chyle”.
Signs of lymphangioleiomyomatosis are:
- Lung cysts detected by chest imaging tests
- Increased VEGF-D levels in the blood. VEGF-D is a vascular growth factor involved in tumor spread.
- Reduced lung function
- Reduced oxygen levels in the blood
Some individuals may experience fluid (e.g. chyle) accumulation in the chest cavity around the lungs (pleural effusion). Chyle is a fat-laden cloudy fluid that is absorbed during digestion by the lymphatic vessels located around the intestine. Chyle normally flows through lymphatic vessels into the upper chest (thoracic duct) and is then deposited into veins, where it mixes with blood. In some people with lymphangioleiomyomatosis, the lymphatic vessels may rupture or become blocked (obstructed), causing chyle to accumulate in the chest cavity (chylothorax). In some cases, chyle may accumulate in the abdomen causing an increase in girth, a condition called chylous ascites. Involvement of the axial lympatics may also lead to formation of lymphangioleiomyomas, chyle-filled lymphatic structures within the chest and abdomen.
Some individuals with lymphangioleiomyomatosis may experience collapse of a lung that is unrelated to trauma (spontaneous pneumothorax). The symptoms of a collapsed lung may include sudden, sharp chest pain; difficult, rapid breathing; rapid heartbeat (tachycardia); low blood pressure (hypotension); profuse sweating (diaphoresis); dizziness; and/or lack of normal chest movement on the affected side of the chest. A collapsed lung may recur in some individuals.
Approximately 50 percent of individuals with lymphangioleiomyomatosis develop angiomyolipomas, which are benign tumors made up of fat, blood vessels and smooth muscle like cells. These tumors affect the kidneys or abdomen and often do not cause symptoms (asymptomatic). In some cases, they may cause flank pain, blood in the urine (hematuria) or bleeding into the abdomen (abdominal hemorrhaging).
Symptoms of lymphangioleiomyomatosis may become progressively worse as lymphangioleiomyomatosis cell proliferation continues, eventually resulting in chronic life-threatening respiratory failure. The symptoms of lymphangioleiomyomatosis often worsen during pregnancy. Many cases of lymphangioleiomyomatosis are associated with osteoporosis, a condition characterized by progressive thinning of the bones.
Tuberous sclerosis complex (TSC)
Tuberous sclerosis complex (TSC) exhibits both inter- and intrafamilial variability in clinical findings. Females tend to have milder disease than males 48, 49. Any organ system can be involved in tuberous sclerosis complex (TSC).
Tuberous sclerosis complex (TSC) should be suspected in individuals with either one major clinical feature or two or more minor features, as listed below 23:
- Major features
- Angiofibromas (≥3) or fibrous cephalic plaque
- Cardiac rhabdomyoma
- Multiple cortical tubers and/or radial migration lines
- Hypomelanotic macules (≥3 macules that are at least 5 mm in diameter)
- Lymphangioleiomyomatosis (LAM)
- Multiple retinal nodular hamartomas
- Renal angiomyolipoma (≥2)
- Shagreen patch
- Subependymal giant cell astrocytoma (SEGA)
- Subependymal nodules (SENs) (≥2)
- Ungual fibromas (≥2)
- Minor features
- Sclerotic bone lesions
- “Confetti” skin lesions (numerous 1- to 3-mm hypopigmented macules scattered over regions of the body such as the arms and legs)
- Dental enamel pits (>3)
- Intraoral fibromas (≥2)
- Multiple renal cysts
- Nonrenal hamartomas
- Retinal achromic patch
- *Note: The combination of LAM and angiomyolipomas without other features does not meet the clinical diagnostic criteria for a definite diagnosis of tuberous sclerosis complex (TSC).
Skin
The skin is affected in virtually 100% of individuals with tuberous sclerosis complex (TSC). Skin lesions include: hypomelanotic macules (~90% of individuals), confetti skin lesions (frequency varies widely from 3% of children to ≤58% overall), facial angiofibromas (~75%), shagreen patches (~50%), fibrous cephalic plaques, and ungual fibromas (20% overall but ≤80% in older affected adults) [Northrup et al 2013]. Among the skin lesions, the facial angiofibromas cause the most disfigurement. None of the skin lesions results in serious medical problems.
Central nervous system (CNS)
Central nervous system (CNS) tumors are the leading cause of morbidity and mortality in tuberous sclerosis complex (TSC). The brain lesions of tuberous sclerosis complex (TSC), including subependymal nodules (SENs), cortical tubers, and subependymal giant cell astrocytomas (SEGAs), can be distinguished with neuroimaging studies. Subependymal nodules (SENs) occur in 80% of individuals and cortical tubers in approximately 90%. Subependymal giant cell astrocytomas (SEGAs) occur in 5%-15% of all individuals with tuberous sclerosis complex (TSC) 50. These giant cell astrocytomas may enlarge, causing pressure and obstruction and resulting in significant morbidity and mortality.
Seizures
More than 80% of individuals with tuberous sclerosis complex (TSC) have been reported to have seizures, although this percentage may reflect ascertainment bias of more severely involved individuals. Tuberous sclerosis complex (TSC) is a known cause of infantile spasms. At least 50% of individuals have developmental delay or intellectual disability. The leading cause of premature death (32.5%) among individuals with tuberous sclerosis complex (TSC) is a complication of severe intellectual disability (e.g., status epilepticus and bronchopneumonia) 51.
Tuberous Sclerosis-Associated Neuropsychiatric Disorder (TAND)
Tuberous Sclerosis-Associated Neuropsychiatric Disorder (TAND) refers to the interrelated functional and clinical manifestations of brain dysfunction common in individuals with tuberous sclerosis complex (TSC), including behavioral, psychiatric, intellectual, academic, neuropsychological, and psychosocial difficulties 52. Although more than 90% of children and adults with tuberous sclerosis complex (TSC) will experience one or more TAND concerns in their lifetime, only 20% ever receive evaluation and intervention for them 53, 54.
- Autism spectrum disorder (ASD). Individuals with tuberous sclerosis complex (TSC) are at high risk for autism spectrum disorder (ASD), with estimates running from 16% to 61% 55, 56, 57, 58, 59, 60, 61, 62, compared to a less than 2% risk in the general population. Signs of autism spectrum disorder (ASD) in individuals with tuberous sclerosis complex (TSC) emerge as early as age nine months 63. Individuals with tuberous sclerosis complex (TSC) who have subependymal giant cell astrocytomas (SEGAs) are nearly twice as likely to have autism spectrum disorder (ASD) 64, and treatment with everolimus has been found to reduce SEGA size, seizures, and features of autism spectrum disorder (ASD) 65, 66. Neurofunctional impairments closely associated with autism spectrum disorder (ASD), including impaired language pathways 67 and atypical face processing 61, have been noted in persons with tuberous sclerosis complex (TSC). Children with tuberous sclerosis complex (TSC) and autism spectrum disorder (ASD) are at higher risk for global cognitive impairment than are children with tuberous sclerosis complex (TSC) who do not have autism spectrum disorder (ASD) 68. The autism spectrum disorder (ASD) profile in toddlers with tuberous sclerosis complex (TSC) has been found to have “complete convergence” with young children with nonsyndromic autism spectrum disorder (ASD) 69.
- Attention deficit hyperactivity disorder (ADHD). Attention deficit hyperactivity disorder (ADHD) is another common and potentially seriously debilitating condition closely associated with tuberous sclerosis complex (TSC). Estimates of ADHD prevalence in individuals with tuberous sclerosis complex (TSC) range from 21% to 50% 55, 70, 71, 72, 59, 62. Deficits in attention (particularly in dual-task performance), cognitive flexibility, and memory have also been noted in neuropsychological studies of children and adults with tuberous sclerosis complex (TSC) 73, 74, 75, 76, 54.
- Learning and cognitive impairment. Individuals with tuberous sclerosis complex (TSC) are at high risk for having intellectual disability, with prevalence rates estimated between 44% and 64% 77, 78, 79.
- Approximately 36%-58% of children with tuberous sclerosis complex (TSC) have serious academic difficulties (e.g., learning disabilities) requiring intervention 80, 76, 62.
- The risk of learning and cognitive impairment increases significantly if seizure activity is not controlled. A number of investigations have demonstrated that a history of infantile spasms and/or poor seizure control in general is associated with lower intellectual ability 77, 78, 81, 82. In a small sample (n=6), Humphrey et al 83 demonstrated a dramatic dose-dependent relationship between seizure activity and intellectual impairment: estimated intelligence quotient (IQ) dropped from 92 (prior to infantile spasms) to 73 (if infantile spasms duration was <1 month) to 62 (if infantile spasms duration was >1 month). These findings underscore the crucial need for adequate seizure control in individuals with tuberous sclerosis complex (TSC).
- Disruptive behaviors and emotional problems. Disruptive behaviors and emotional problems are another cluster of debilitating conditions associated with tuberous sclerosis complex (TSC). Aggression has been noted in many individuals with tuberous sclerosis complex (TSC) (13%-58%) 58, 72, 84, 59, 85, 86 as has self-injurious behavior (27%-41%) 58, 85, 86. Individuals with tuberous sclerosis complex (TSC) are also at high risk for anxiety (9%-48%) and depression (6%-43%) 58, 72, 84, 59, 85, 86, 62.
All individuals with tuberous sclerosis complex (TSC) should be assessed for the presence of TAND, given that it has been closely associated with clinical outcome and quality of life 53. The TAND Checklist 54, a simple paper-and-pencil screening questionnaire available at no cost, is a promising tool to address the significant gap between clinical need associated with TAND and those receiving intervention for these needs 54, 87. Given that unaddressed TAND concerns contribute significantly to poor outcome, and that individuals with tuberous sclerosis complex (TSC) have a very high health care resource utilization 88, 89, the importance of recognizing and addressing TAND concerns cannot be overestimated.
Kidneys
Renal disease is the second leading cause of early death (27.5%) in individuals with tuberous sclerosis complex (TSC) 51. An estimated 80% of children with tuberous sclerosis complex (TSC) have an identifiable renal lesion by a mean age of 10.5 years 90.
Five different renal lesions occur in tuberous sclerosis complex (TSC): benign angiomyolipoma (70% of affected individuals); epithelial cysts (20%-30%) 48, 49; oncocytoma (benign adenomatous hamartoma) (<1%); malignant angiomyolipoma (<1%); and renal cell carcinoma (<3%) 91.
- Benign angiomyolipomas comprise abnormal blood vessels, sheets of smooth muscle, and mature adipose tissue. In children, angiomyolipomas tend to increase in size or number over time. Benign angiomyolipomas can cause life-threatening bleeding and can replace renal parenchyma, leading to end-stage renal disease (ESRD).
- Renal cysts have an epithelial lining of hypertrophic hyperplastic eosinophilic cells. Some affected individuals have features of both TSC caused by deletion of TSC2 and autosomal dominant polycystic kidney disease (ADPKD) caused by deletion of PKD1. In these individuals, progressive enlargement of the cysts may compress functional parenchyma and lead to ESRD 92. Individuals with the TSC2/PKD1 contiguous gene deletion syndrome are also at risk of developing the complications of ADPKD, which include cystic lesions in other organs (e.g., the liver) and Berry aneurysms.
- Malignant angiomyolipoma and renal cell carcinoma (RCC) may result in death. Although rare, these two tumors are much more common in individuals with tuberous sclerosis complex (TSC) than in the general population 93. It is estimated that 2%-5% of persons with tuberous sclerosis complex (TSC) will develop renal cell carcinoma (RCC). The age of diagnosis of renal cell carcinoma (RCC) in those with tuberous sclerosis complex (TSC) is 28-30 years – much earlier than the age of diagnosis for sporadic renal cell carcinoma (RCC) 30, 94. Note: Common imaging techniques may not distinguish fat-poor angiomyolipomas from renal cell carcinoma (RCC). Immunologic staining for HMB-45 for angiomyolipomas and cytokeratin for renal cell carcinoma (RCC) is recommended.
Heart
Cardiac rhabdomyomas are present in 47%-67% of individuals with tuberous sclerosis complex (TSC) 95, 96, 48. Cardiac rhabdomyomas tumors have been documented to regress with time and eventually disappear. The cardiac rhabdomyomas are often largest during the neonatal period. If cardiac outflow obstruction does not occur at birth, the individual is unlikely to have health problems from these tumors later. However, a small number of individuals have arrhythmias postulated to result from rests of persistent cells left after the rhabdomyomas regress.
Lung
Lymphangioleiomyomatosis (LAM) of the lung primarily affects women and has been estimated to occur in approximately 30%-40% of females with tuberous sclerosis complex (TSC); however, a recent study suggests that the diagnosis of LAM is age dependent and occurs in up to 80% of women with tuberous sclerosis complex (TSC) by age 40 years 97. Approximately 5%-10% of women with tuberous sclerosis complex (TSC) present with symptomatic LAM 98. Cystic findings consistent with LAM are observed in 10%-12% of males with tuberous sclerosis complex (TSC) 50.
- The mean age of diagnosis for LAM in those with tuberous sclerosis complex (TSC) is 28 years, compared to 35 years for sporadic LAM.
- Individuals with tuberous sclerosis complex (TSC)-associated LAM as well as sporadic LAM may present with shortness of breath or hemoptysis. Chest radiographs reveal a diffuse reticular pattern and CT examination shows diffuse interstitial changes with infiltrates and cystic changes. Pneumothorax and chylothorax may occur in individuals affected by LAM. Some individuals progress to respiratory failure and death.
- It is suggested that LAM associated with tuberous sclerosis complex (TSC) is milder than sporadic LAM because persons with tuberous sclerosis complex (TSC) and LAM account for only about 15% of registrants in the LAM Foundation 99. Furthermore, persons with tuberous sclerosis complex (TSC) and LAM have less severe lung cysts than persons with sporadic LAM 100.
Multifocal micronodular pneumonocyte hyperplasia (MMPH), characterized by multiple nodular proliferations of type 2 pneumocytes, was first described in association with tuberous sclerosis complex (TSC) in 1991 101. While multifocal micronodular pneumonocyte hyperplasia (MMPH) does not have known prognostic or physiologic consequences, there have been at least two reports of respiratory failure associated with multifocal micronodular pneumonocyte hyperplasia (MMPH) 102, 103. The precise prevalence of multifocal micronodular pneumonocyte hyperplasia (MMPH) in individuals with tuberous sclerosis complex (TSC) is not known but may be as high as 40%-58% 104, 46. Males and females are equally likely to have multifocal micronodular pneumonocyte hyperplasia (MMPH), and it may occur in the presence or absence of LAM in persons with tuberous sclerosis complex (TSC). Multifocal micronodular pneumonocyte hyperplasia (MMPH) can be confused with atypical adenomatous hyperplasia, which is a premalignant lesion that is not clearly associated with tuberous sclerosis complex (TSC).
Eye
The retinal lesions of tuberous sclerosis complex (TSC) include hamartomas (elevated mulberry lesions or plaque-like lesions) observed in 30%-50% of individuals with tuberous sclerosis complex (TSC). These lesions are relatively rare in the general population with a recent case series of 3573 healthy term newborns identifying only two with these lesions 105. Achromic patches (similar to the hypopigmented skin lesions) have been noted to occur in 39% of individuals with tuberous sclerosis complex (TSC), while the general population incidence is 1:20,000 106. Although these lesions are usually asymptomatic, a few persons with tuberous sclerosis complex (TSC) have had progressively enlarging retinal astrocytic hamartomas with total exudative retinal detachment and neovascular glaucoma 107.
Extrarenal Angiomyolipomas (AMLs)
Although rare, extrarenal angiomyolipomas have been reported 108. In a retrospective study of sonographic and CT images, Fricke et al 109 identified eight hepatic angiomyolipomas (AMLs) in 62 individuals with tuberous sclerosis complex (TSC) (13%).
Neuroendocrine Tumors (NETs)
Dworakowska & Grossman 110 summarized case reports of persons with tuberous sclerosis complex (TSC) who had neuroendocrine Tumors (NETs); the majority of tumors were pituitary adenomas (ACTH adenoma and GH adenoma), parathyroid adenomas and hyperplasia, and pancreatic adenomas (insulinoma and islet cell neoplasm). More recently single case reports have included gastrinoma, pheochromocytoma, and carcinoids. Several individuals had a TSC2 pathogenic variant and/or loss of heterozygosity in the islet cell neoplasms.
Lymphangioleiomyomatosis complications
When a person has lymphangioleiomyomatosis, abnormal muscle-like cells begin to grow out of control in certain organs or tissues, especially the lungs, lymph nodes, and kidneys. Over time, these lymphangioleiomyomatosis cells can destroy the normal lung tissue. As a result, air no longer moves freely in and out of the lungs. In some cases, this means the lungs can’t supply enough oxygen to the body’s other organs.
All patients with lymphangioleiomyomatosis should be advised to avoid the use of estrogen-containing oral contraceptive pills given the increased rate of worsening of the symptoms with their use. Progesterone based oral contraceptives appear to be safe and could be an alternative. Pregnancy can also increase the risk of worsening of symptoms and complications, but patients with lymphangioleiomyomatosis reported to have multiple pregnancies without complications. Patients with lymphangioleiomyomatosis should receive influenza and pneumococcal vaccination.
Possible complications of lymphangioleiomyomatosis include any of the following:
- Angiomyolipomas and other tumors. Many women who have lymphangioleiomyomatosis get tumors in their kidneys, called angiomyolipomas. Women who have lymphangioleiomyomatosis also may develop large tumors in the lymph node or growths in other organs such as the liver.
- Blood in the urine. This may occur in women who have kidney tumors.
- Enlarged lymph nodes. These usually occur in the abdomen or the chest. Very rarely, enlarged lymph nodes may occur in locations where they can be felt, such as the neck or under the arms.
- Pleural effusions. This condition can occur if bodily fluids collect in the space between the lung and the chest wall. Often the fluid contains a milky substance called chylothorax. The excess fluid in the chest may cause shortness of breath because the lung has less room to expand.
- Pneumothorax or collapsed lung. This potentially life-threatening condition occurs when air leaks out of the lung and into the space between the lung and chest wall, an area called the pleural space. In lymphangioleiomyomatosis, a pneumothorax can occur if lung cysts rupture through the lining of a lung. Air that collects in the space between the lung and chest wall must be removed to reinflate the lung. A collapsed lung can cause pain and shortness of breath. Sometimes one lung will collapse repeatedly. Pneumothorax usually requires urgent medical care and treatment.
- Swelling or the build-up of fluid. This can happen in the abdomen, pelvic area, legs, ankles or feet. Pain may also occur with the swelling.
Lymphangioleiomyomatosis diagnosis
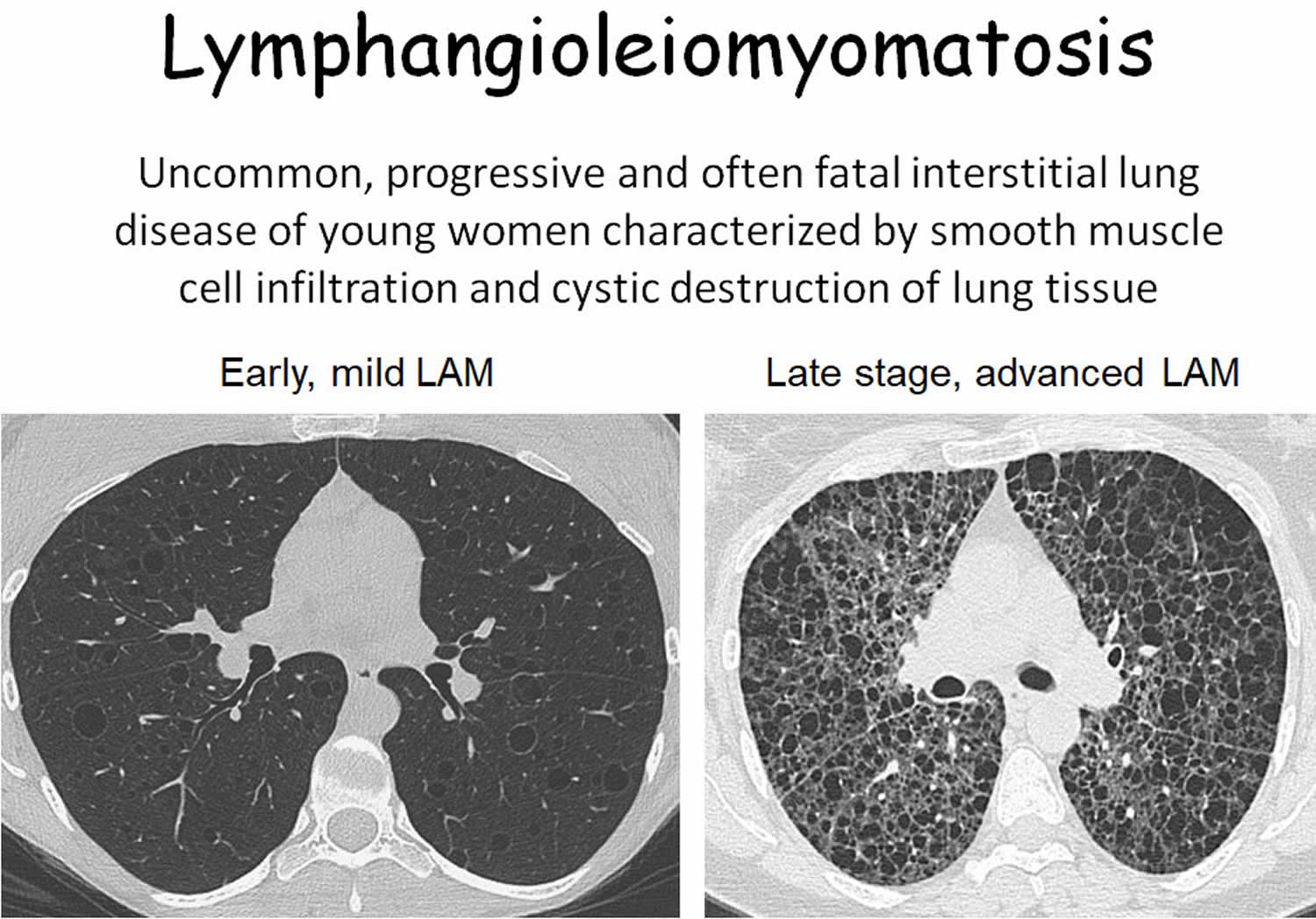
To help diagnose your condition, you may want to see a pulmonologist, a doctor who specializes in lung diseases and conditions, who has experience providing care to patients with lymphangioleiomyomatosis (LAM). The diagnosis of lymphangioleiomyomatosis may be confirmed by a thorough clinical evaluation that includes a detailed patient history and a variety of specialized tests including radiographic (X-ray) studies, lung function tests, VEGF-D blood tests and/or surgical removal and microscopic study of affected tissue (biopsy). In about 80 percent of people with lymphangioleiomyomatosis, CT studies reveal cysts in the lungs in or around one or both lungs (pleural effusion). CT studies of the abdomen may reveal angiomyolipomas or lymphangioleiomyomas that support a diagnosis of lymphangioleiomyomatosis.
Surgical open lung biopsy is one definitive way to confirm the diagnosis of lymphangioleiomyomatosis. These biopsy tissue samples verify the presence of lymphangioleiomyomatosis cells.
Lung function tests
- Lung function tests. For lung function tests, you breathe through a mouthpiece into a machine called a spirometer. The spirometer measures the amount of air you breathe in and out. Other lung function tests can show how much air your lungs can hold and how well your lungs deliver oxygen to your blood.
- Arterial blood gas tests. Your doctor may take a blood sample from an artery in your wrist to measure your blood oxygen levels and to determine if you need oxygen therapy.
- Pulse oximetry. For this test, a small sensor is attached to your finger or ear. The sensor uses light to estimate how much oxygen is in your blood.
- Six-minute walk test. This test measures the distance you can walk in six minutes. It can help determine if you need oxygen therapy while exercising.
Imaging tests
- Chest x ray. A chest x ray creates a picture of the structures in your chest, such as your heart and lungs. The test can show a collapsed lung or fluid in your chest. In the early stages of lymphangioleiomyomatosis, your chest x rays may look normal. As the disease gets worse, the x rays may detect cysts in your lungs and assess how cysts change over time. Your healthcare provider may use a chest X-ray to look for complications of LAM, such as fluid in your lungs or air in the chest cavity that collapses your lung (a pneumothorax).
- High-resolution CT (HRCT) scan. The most useful imaging test for diagnosing lymphangioleiomyomatosis is a high-resolution CT (HRCT) scan of the chest. A high-resolution computed tomography (HRCT) scan creates a computer-generated three-dimensional (3D) picture of your lungs. The three-dimensional (3D) picture shows more detail than the pictures from a chest x ray. A high-resolution CT scan can show cysts, excess fluid, a collapsed lung, and enlarged lymph nodes. The test also can show how much normal lung tissue has been replaced by the lymphangioleiomyomatosis cysts. High-resolution CT scans of your abdomen and pelvis can show whether you have growths in your kidneys, other abdominal organs, or lymph nodes. Your healthcare provider may diagnose you with LAM if the chest HRCT shows thin-walled cysts (fluid-filled pockets) in your lungs.
Blood tests for vascular endothelial growth factor D (VEGF-D)
Your doctor may take a blood sample from a vein in your arm to measure vascular endothelial growth factor D (VEGF-D) levels. Higher levels of VEGF-D can stimulate the growth of new blood vessels, which can enable the cells that cause LAM cells to spread in the body. The VEGF-D blood tests test may help confirm the diagnosis of lymphangioleiomyomatosis in patients whose HRCT scans show lung cysts that suggest a patient has lymphangioleiomyomatosis. About 70% of patients with LAM have raised VEGF-D levels.
VEGF-D levels of 800 pg/mL (picograms per milliliter) or more can help your doctor confirm that you have lymphangioleiomyomatosis. Because you may still have lymphangioleiomyomatosis if your levels are less than 800 pg/mL, your doctor may have you undergo other diagnostic procedures that look for lymphangioleiomyomatosis cells.
Procedures that look for lymphangioleiomyomatosis cells
If lung function, imaging, or blood VEGF-D tests cannot diagnose lymphangioleiomyomatosis, your doctor may recommend one of the following procedures to collect tissue samples that can be used to detect lymphangioleiomyomatosis cells.
- Video-assisted thoracoscopic surgery (VATS). In this procedure, your doctor inserts a small, lighted tube into little cuts made in your chest wall. This lets him or her look inside your chest and snip out a few small pieces of lung tissue. Video-assisted thoracoscopic surgery (VATS) is done in a hospital. The procedure isn’t major surgery, but it does require general anesthesia to make you sleep during the procedure.
- Open lung biopsy. In this procedure, your doctor removes a few small pieces of lung tissue through a cut made in your chest wall between your ribs. An open lung biopsy is done in a hospital. You’ll be given medicine to make you sleep during the procedure. Open lung biopsies are rarely done anymore because the recovery time is much longer than the recovery time from VATS.
- Transbronchial biopsy. In this procedure, your doctor inserts a long, narrow, flexible, lighted tube down your windpipe and into your lungs. He or she then snips out bits of lung tissue using a tiny device. This procedure usually is done in a hospital. Your mouth and throat are numbed to prevent pain. Because only a small amount of tissue is collected, it is possible that this test will not provide enough information.
- Thoracentesis. Thoracentesis uses a needle to collect fluid samples from the lining of your lungs. If your chest imaging tests show that you have fluid building up in the space between the lung and the chest wall called pleural effusions, your doctor may order thoracentesis to collect this pleural fluid for analysis.
- Other biopsies. Your doctor also can diagnose lymphangioleiomyomatosis using the results from other tissue biopsies, such as biopsies of lymph nodes or abdominal or pelvic lesions.
Other tests
If your chest imaging tests show that you have pleural effusions, your doctor may order a pleural fluid analysis. For this test, a fluid sample is taken from the pleural space, which is a thin space between two layers of tissue that line the lungs and chest cavity. Doctors use a procedure called thoracentesis to collect the fluid sample. The fluid is studied for the milky substance called chylothorax.
If you’re diagnosed with sporadic lymphangioleiomyomatosis (S-LAM), your doctor may advise you to have a computed tomography (CT) scan or magnetic resonance imaging (MRI) scan of your head. These tests can help screen for underlying tuberous sclerosis complex (TSC), a condition that can also cause kidney growths and lung cysts. If a woman who has cysts in her lungs is found to have tuberous sclerosis complex (TSC), the doctor will diagnose tuberous sclerosis complex-associated lymphangioleiomyomatosis or TSC–LAM.
Genetic testing identifies mutations (changes) in your TSC1 and TSC2 genes to help your doctor confirm that you have LAM and not another lung disease.
Lymphangioleiomyomatosis treatment
LAM is a progressive disease, meaning it gets worse over time. While there is no cure for lymphangioleiomyomatosis, doctors treat lymphangioleiomyomatosis with sirolimus (rapamycin), an mTOR inhibitor, a medicine that stabilizes lung function, treats an abnormal fluid buildup in the lung called chylothorax, and improves overall quality of life. They may also prescribe other medicines or therapies to control other symptoms or complications. For example, bronchodilators are medicines that can help relax the muscles around the airways if you are wheezing or having trouble breathing.
You may want to see a pulmonologist doctor who specializes in lung diseases and conditions, especially lymphangioleiomyomatosis, to help treat your condition. You may also want to ask your doctor about the latest American Thoracic Society clinical guidelines (http://www.thoracic.org/about/newsroom/press-releases/resources/lam-guidelines.pdf) for diagnosis and management of lymphangioleiomyomatosis, which include new treatment recommendations.
Medicines
Your doctor may prescribe sirolimus (rapamycin) to treat your condition, or bronchodilators or oxygen therapy to help you breathe better. Lung function tests can sometimes show whether these medicines are likely to help you.
- Sirolimus (rapamycin). Sirolimus is the only FDA-approved drug to treat lymphangioleiomyomatosis. Sirolimus is a mTOR inhibitor that controls the abnormal proliferation and growth of smooth muscle cells in the lung tissue and is effective and safe. Studies 111 have shown that sirolimus helps regulate the abnormal growth and movement of lymphangioleiomyomatosis cells. Sirolimus stabilizes lung function and improves quality of life, shrinks abnormal kidney and lymph node growths, and reduces abnormal fluid in the lung called chylothorax. The latest clinical guidelines recommend sirolimus for people with lymphangioleiomyomatosis if they have abnormal or declining lung function or in some patients with lymphangioleiomyomatosis who have pleural effusions containing chylothorax. Sirolimus may also reduce the size of angiomyolipomas and therefore may be a treatment option before procedures that remove or shrink kidney tumors. Sirolimus does have side effects, some of which can be serious. If you have lymphangioleiomyomatosis, talk with your doctor about the benefits and risks of this medicine, and whether it’s an option for you.
- Everolimus a different mTOR inhibitor is an alternative in patients who cannot tolerate sirolimus or have allergies. Everolimus is approved by the FDA for treating benign brain tumors associated with TSC, is also sometimes used to treat LAM.
- Bronchodilators. If you’re having trouble breathing or are wheezing, your doctor may prescribe bronchodilators. These medicines relax the muscles around the airways. This helps the airways open up, making it easier for you to breathe. Bronchodilators are used for symptomatic control and relief of symptoms in patients who show reversibility on lung function tests. Usually, beta-agonists and anticholinergic used in the treatment. Studies have shown benefit and reversibility of airflow obstruction in patients with lymphangioleiomyomatosis treated with albuterol and ipratropium after adjusting for smoking and asthmatic status 112.
- Oxygen Therapy. If the level of oxygen in your blood is low, your doctor may recommend oxygen therapy to increase the amount of oxygen your lungs receive and deliver to your blood. Oxygen usually is given through nasal prongs or a mask. At first, you may need oxygen only while exercising. It also may help to use it while sleeping. Over time, you may need full-time oxygen therapy.
The latest clinical guidelines, which are based on currently available clinical data, do not recommend the following medicines to treat lymphangioleiomyomatosis:
- Doxycycline, an antibiotic that did not show a clinical benefit when used to treat lymphangioleiomyomatosis.
- Hormone therapies including certain medicines—progestins, gonadotrophin-releasing hormone (GnRH) agonists, selective estrogen receptor modulators (SERMs) such as tamoxifen. While these therapies had previously been used to treat lymphangioleiomyomatosis, newer analyses suggest they do not produce clinical benefit in all patients with lymphangioleiomyomatosis. If you are taking any of these hormone medicines, talk to your doctor to see if you need to discontinue their use.
Respiratory rehabilitation
Studies have shown that respiratory rehabilitation in patients with decreased functional capacity and respiratory symptoms benefit from it. It encourages all health care professionals to consider patients with lymphangioleiomyomatosis to have respiratory rehabilitation to increase endurance and improve quality of life 113.
Procedures that remove air or fluid from the chest or abdomen
Several procedures can remove excess air or fluid from your chest or abdomen. Removing fluid from your chest by thoracentesis or from your abdomen by paracentesis may help relieve discomfort and shortness of breath.
Your doctor often can remove the fluid with a needle and syringe. If large amounts of fluid build-up in your chest, your doctor may have to insert a tube into your chest to remove the fluid.
Removing air from your chest may relieve shortness of breath and chest pain caused by a pneumothorax, or collapsed lung. Your doctor usually can remove the air with a tube. The tube is inserted into your chest between your side ribs. Often, the tube is attached to a suction device. If this procedure doesn’t work, or if your lungs repeatedly collapse, you may need surgery.
If fluid or air often leak into your chest, your doctor may recommend a procedure called pleurodesis to prevent repeat episodes. Your doctor may inject a chemical at the site of the leakage (chemical pleurodesis using talc is recommended). The chemical fuses your lung and chest wall together, which removes the space for leakage.
Your doctor may do this procedure at your bedside in the hospital. You will be given medicine to prevent pain. The procedure also can be done in an operating room using video-assisted thoracoscopy. In this case, you will be given medicine to make you sleep during the procedure.
Procedures that remove or shrink kidney tumors
Kidney tumors, or angiomyolipomas, often don’t cause symptoms, but sometimes they can cause ongoing pain or bleeding. If this happens, you may need surgery to remove some of them. If bleeding isn’t too severe, a radiologist often can block the blood vessels feeding the kidney tumors. This may cause them to shrink.
Lung transplant
Some patients who have severe lung damage due to advanced lymphangioleiomyomatosis may be eligible for lung transplants. While lung transplants can improve lung function and quality of life for eligible patients, they have a high risk of complications, including infections and rejection of the transplanted lung by the body.
Studies suggest that more than three-quarters of women with lymphangioleiomyomatosis who receive a lung transplant survive for at least 3 years. In a few cases, doctors have found lymphangioleiomyomatosis cells in the newly transplanted lungs, raising a question to the curative role of lung transplantation. However, the lymphangioleiomyomatosis cells generally don’t stop the transplanted lung from working.
Monitor your condition
If you have lymphangioleiomyomatosis, it is important for you to have routine follow-up care so your doctor can monitor your condition to see if it is stable or worsening. lymphangioleiomyomatosis has no cure, and the disease tends to worsen over time. How quickly the disease worsens varies between patients.
In the early stages of lymphangioleiomyomatosis, you usually can do your normal daily activities. These may include attending school, going to work, and doing common physical activities such as walking up stairs. In the later stages of lymphangioleiomyomatosis, you may find it harder to be active and you may need oxygen therapy.
Serious and possibly life-threatening complications can occur if you have lymphangioleiomyomatosis. More than half of women who have lymphangioleiomyomatosis develop pneumothorax, or collapsed lung. lymphangioleiomyomatosis may cause death from respiratory failure. Read more about possible signs, symptoms and complications of lymphangioleiomyomatosis.
Lung transplant is a treatment option for patients whose lungs have been damaged by lymphangioleiomyomatosis.
Receive other medical care
In addition to monitoring your condition, your doctor may recommend emotional support to improve your quality of life, vaccines to prevent lung infections, lifestyle changes to improve your overall health and avoid some complications, tests or medicines to care for your bones, and pregnancy and birth control options.
Vaccines for lung health
Take steps to care for your lungs. For example, talk with your doctor about getting a pneumococcal pneumonia vaccine and a yearly influenza or flu shot.
Lifestyle changes
If you have lymphangioleiomyomatosis, taking good care of your health is important. Your doctor may recommend you adopt the following healthy lifestyle changes, many of which are also heart-healthy.
- Make healthy eating choices.
- Be physically active.
- Get plenty of rest.
- Quit smoking. Talk to your doctor about programs and products that can help you quit smoking. If you have trouble quitting smoking on your own, consider joining a support group. Many hospitals, workplaces, and community groups offer classes to help people quit smoking. Ask your family members and friends to support you in your efforts to quit.
Also, check with your doctor before traveling by air or traveling to areas where medical attention isn’t readily available. Also, talk to your doctor before traveling to places where the amount of oxygen in the air is low.
Care for your bones
Some women who have lymphangioleiomyomatosis may be at risk for osteoporosis if they have undergone permanent hormonal therapy such as oophorectomy or are receiving certain hormone therapy medicines. This is in part because many hormone therapies affect estrogen, and estrogen is important for keeping bones strong. While newer clinical guidelines do not recommend hormone therapies for the treatment of lymphangioleiomyomatosis, your doctor may order tests to measure your bone density if you have previously had an oophorectomy or are still on hormone therapies for other conditions. If you have lost bone density, your doctor may prescribe medicines or calcium and vitamin D supplements to prevent more bone loss.
Pregnancy and birth control planning
Because hormone changes during pregnancy can worsen lymphangioleiomyomatosis, it is important to talk to your pulmonologist and obstetrician before you get pregnant.
Most doctors don’t recommend birth control pills containing estrogen to women who have lymphangioleiomyomatosis because estrogen is thought to contribute to or worsen lymphangioleiomyomatosis. If you have lymphangioleiomyomatosis, talk to your doctors about birth control options.
Living with lymphangioleiomyomatosis
LAM may lead to serious and life-threatening health problems.
- Collapsed lung: This condition, also known as a pneumothorax, can happen when air leaks into the pleural space in the lungs. Reinflating the lung requires a tube that is inserted into the chest between the ribs. Often, the tube is attached to a suction device. If this procedure does not work or if your lungs repeatedly collapse, you may need surgery to re-expand the lung to normal size. Your doctor may recommend a surgical procedure called pleurodesis to help the lungs and chest cavity stick together and prevent future lung collapses.
- Kidney or other tumors: Most kidney tumors are benign (noncancerous), but sometimes they can cause symptoms such as pain or bleeding. If this happens, you may need surgery to remove them. If the bleeding is not too severe, a doctor called a radiologist may be able to use medical imaging equipment to block the blood vessels feeding the kidney tumors. This may cause them to shrink. Women who have LAM may also develop large tumors in the lymph nodes or in other organs, such as the liver.
- Osteoporosis: This condition causes bones to become weak and break easily. Your doctor may order tests to measure your bone density. If you have lost bone density, your doctor may prescribe medicines or calcium and vitamin D supplements to prevent more bone loss.
- Pleural effusions: These may occur if body fluids collect in the pleural space between the lung and the chest wall. The excess fluid in the chest may cause shortness of breath, because the lung has less room to expand. One form of pleural effusion is known as a chylothorax, which is caused when a certain type of lymphatic fluid (chyle) accumulates in the pleural space.
It is important that you follow your healthcare provider’s instructions for your treatment and see your doctor regularly to monitor your health.
- Follow your treatment plan. It may take several months for your body to respond to treatment. If you stop taking your medicines, your symptoms may return or get worse.
- Get regular vaccinations for lung health. Talk with your doctor about getting a pneumonia vaccine and a yearly flu shot.
- Take care of your mental health. Living with LAM may cause fear, anxiety, depression, and stress. Your healthcare provider can evaluate how your condition is affecting your activity level and your mental health. To help improve your quality of life, your doctor may recommend steps you can take.
- Get counseling, particularly cognitive behavioral therapy.
- Join a patient support group, which may help you adjust to living with LAM. You can see how other patients manage similar symptoms and their condition. Talk with your doctor about local support groups or check with an area medical center.
- Seek support from family and friends, which can help relieve stress and anxiety. Let your loved ones know how you feel and what they can do to help you.
- Take medicines or other treatments. Your doctor may recommend medicines, such as antidepressants, or other treatments that can help improve your quality of life.
- Adopt healthy lifestyle changes. If you have LAM, it is important that you take good care of your health. Your healthcare provider may recommend that you adopt the following healthy lifestyle changes:
- Choose healthy foods. Eating more fruits, vegetables, and whole grains and fewer saturated fats and added sugars can improve your overall health. A healthy diet can promote weight loss or help you maintain a healthy weight.
- Get physical activity. Physical activity improves bone mineral density, muscle strength, flexibility, and posture. You may also benefit from rehabilitation and aerobic exercise, or training to raise the amount of oxygen in your blood. Before starting any exercise program, ask your doctor about what level of physical activity is right for you.
- Quit smoking. If you smoke, stop. Also, try to avoid other lung irritants, such as dust, chemicals, and secondhand smoke.
Your doctor may recommend these lifestyle changes as part of a larger pulmonary rehabilitation program, which they can oversee.
Prevent complications over your lifetime
- Avoid air travel because it increases your risk of developing pneumothorax that collapses the lung. Symptoms include sudden shortness of breath or severe chest pain. Talk to your provider before air travel or before traveling to places at a higher elevation, where there is less oxygen in the air.
- Avoid scuba diving, which may raise your risk of complications.
- See your doctor regularly so they can keep track of any side effects you report and monitor the long-term use of LAM medications.
Pregnancy and birth control planning
Women with LAM can become pregnant, but hormone changes during pregnancy can worsen LAM. Also, women with LAM may be less fertile and have a harder time becoming pregnant. Pregnancy with LAM increases the risk for pneumothorax and chylothorax, may lead to more lung function decline, and raises the chance of preterm delivery. The long-term effects of the medicine sirolimus on mother and child also needs additional study. While many women with LAM have full-term pregnancies, it is important to talk to your healthcare provider if you are thinking of becoming pregnant.
Most doctors do not recommend birth control pills containing estrogen to women who have LAM, because estrogen is thought to contribute to or worsen LAM. If you have LAM and are interested in birth control, talk to your healthcare provider about your options.
LAM lung disease life expectancy
The clinical course of lymphangioleiomyomatosis (LAM) vary from case to case. Not long ago, doctors thought women who had lymphangioleiomyomatosis (LAM) wouldn’t live more than 10 years after diagnosis. With earlier diagnosis and newer treatment, patients with lymphangioleiomyomatosis (LAM) are living longer, with some living more than 20 years after diagnosis, with a reported 10-year survival rate of 80–90% 114, 115, 116, 117, 118, 119.
The median transplant-free survival in patients with lymphangioleiomyomatosis (LAM) estimated to be 29 years from symptom onset and 23 years from diagnosis 120. A study including 401 patients with LAM showed that the 10-year transplant-free survival rate was 86% 114. Another study conducted on 217 patients with LAM, also reported 5-year and 10-year transplant-free survival rates of 94% and 85%, respectively 116. Furthermore, the 5-year and 10-year survival rates of 173 Japanese patients with LAM were 91% and 76%, respectively 117. Patients with LAM experience shortness of breath (dyspnea) and a progressive decline in lung function, both of which eventually result in the need for lung transplantation. Johnson et al. 121 found that the median time from symptom onset to grade 3 shortness of breath (dyspnea) according to the Medical Research Council Dyspnea Scale, was 9.3 years in patients from the United Kingdom (UK) with LAM (n = 72). Given the possibility of lymphangioleiomyomatosis (LAM) recurrence after lung transplantation, many experts now consider lymphangioleiomyomatosis (LAM) to be a low-grade cancer 11, 12, 13.
Several prognostic factors in patients with LAM have been reported in previous studies 114, 116, 117, 122, 123. In a previous study, lower forced expiratory volume in one second (FEV1), 6-minute walk distance (6MWD) and diffusing lung capacity for carbon monoxide (DLCO) were found to be poor prognostic factors for transplantation-free survival in the age-adjusted analysis 116. Of a total of 46 Japanese patients with LAM, the non-survivors were found to have a lower forced expiratory volume in one second/forced vital capacity (FEV1/FVC) ratio and a higher total lung capacity (TLC) than the survivors 122. Furthermore, delayed symptom onset and the presence of angiomyolipoma (AML) were associated with better survival in patients with LAM, whereas home oxygen use was associated with poorer survival 114.
For your follow-up care, you may want to see a pulmonologist, a doctor who specializes in lung diseases and conditions, and who has experience providing care to patients with lymphangioleiomyomatosis. Your doctor will monitor your condition to see if it is stable or getting worse and causing serious complications. Also, your doctor may recommend other medical care, including vaccines, lifestyle changes, and pregnancy and birth control planning.
References- Johnson SR, Cordier JF, Lazor R, Cottin V, Costabel U, Harari S, Reynaud-Gaubert M, Boehler A, Brauner M, Popper H, Bonetti F, Kingswood C; Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010 Jan;35(1):14-26. doi: 10.1183/09031936.00076209
- McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, Steagall WK, Johnson SR, Sahn SA, Ryu JH, Strange C, Seyama K, Sullivan EJ, Kotloff RM, Downey GP, Chapman JT, Han MK, D’Armiento JM, Inoue Y, Henske EP, Bissler JJ, Colby TV, Kinder BW, Wikenheiser-Brokamp KA, Brown KK, Cordier JF, Meyer C, Cottin V, Brozek JL, Smith K, Wilson KC, Moss J; ATS/JRS Committee on Lymphangioleiomyomatosis. Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guidelines: Lymphangioleiomyomatosis Diagnosis and Management. Am J Respir Crit Care Med. 2016 Sep 15;194(6):748-61. doi: 10.1164/rccm.201607-1384ST
- Johnson SR. Lymphangioleiomyomatosis. Eur Respir J. 2006 May;27(5):1056-65. https://erj.ersjournals.com/content/27/5/1056
- Gupta N, Finlay GA, Kotloff RM, Strange C, Wilson KC, Young LR, Taveira-DaSilva AM, Johnson SR, Cottin V, Sahn SA, et al. Lymphangioleiomyomatosis Diagnosis and Management: High-Resolution Chest Computed Tomography, Transbronchial Lung Biopsy, and Pleural Disease Management. An Official American Thoracic Society/Japanese Respiratory Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2017;196:1337–1348. doi: 10.1164/rccm.201709-1965ST
- Harari S, Torre O, Moss J. Lymphangioleiomyomatosis: what do we know and what are we looking for? Eur Respir Rev. 2011;20:34–44. doi: 10.1183/09059180.00011010
- Lymphangioleiomyomatosis. https://rarediseases.org/rare-diseases/lymphangioleiomyomatosis
- What is LAM? https://www.thelamfoundation.org/learn-about-lam/what-is-lam
- Lymphangioleiomyomatosis (LAM). https://www.lung.org/lung-health-diseases/lung-disease-lookup/lymphangioleiomyomatosis-lam/learn-about-lymphangioleiomyomatosis-lam
- What Is LAM? https://www.nhlbi.nih.gov/health/lam
- Espín R, Baiges A, Blommaert E, Herranz C, Roman A, Saez B, Ancochea J, Valenzuela C, Ussetti P, Laporta R, Rodríguez-Portal JA, van Moorsel CHM, van der Vis JJ, Quanjel MJR, Villar-Piqué A, Diaz-Lucena D, Llorens F, Casanova Á, Molina-Molina M, Plass M, Mateo F, Moss J, Pujana MA. Heterogeneity and Cancer-Related Features in Lymphangioleiomyomatosis Cells and Tissue. Mol Cancer Res. 2021 Nov;19(11):1840-1853. doi: 10.1158/1541-7786.MCR-21-0220
- Henske EP, McCormack FX. Lymphangioleiomyomatosis – a wolf in sheep’s clothing. J Clin Invest. 2012 Nov;122(11):3807-16. doi: 10.1172/JCI58709
- Krymskaya VP, McCormack FX. Lymphangioleiomyomatosis: A Monogenic Model of Malignancy. Annu Rev Med. 2017 Jan 14;68:69-83. doi: 10.1146/annurev-med-050715-104245
- McCormack FX, Travis WD, Colby TV, Henske EP, Moss J. Lymphangioleiomyomatosis: calling it what it is: a low-grade, destructive, metastasizing neoplasm. Am J Respir Crit Care Med. 2012 Dec 15;186(12):1210-2. doi: 10.1164/rccm.201205-0848OE
- Karbowniczek M, Astrinidis A, Balsara BR, Testa JR, Lium JH, Colby TV, McCormack FX, Henske EP. Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med. 2003 Apr 1;167(7):976-82. doi: 10.1164/rccm.200208-969OC
- Bittmann I, Rolf B, Amann G, Löhrs U. Recurrence of lymphangioleiomyomatosis after single lung transplantation: new insights into pathogenesis. Hum Pathol. 2003 Jan;34(1):95-8. doi: 10.1053/hupa.2003.50
- Guo M, Yu JJ, Perl AK, Wikenheiser-Brokamp KA, Riccetti M, Zhang EY, Sudha P, Adam M, Potter A, Kopras EJ, Giannikou K, Potter SS, Sherman S, Hammes SR, Kwiatkowski DJ, Whitsett JA, McCormack FX, Xu Y. Single-Cell Transcriptomic Analysis Identifies a Unique Pulmonary Lymphangioleiomyomatosis Cell. Am J Respir Crit Care Med. 2020 Nov 15;202(10):1373-1387. doi: 10.1164/rccm.201912-2445OC
- Obraztsova K, Basil MC, Rue R, Sivakumar A, Lin SM, Mukhitov AR, Gritsiuta AI, Evans JF, Kopp M, Katzen J, Robichaud A, Atochina-Vasserman EN, Li S, Carl J, Babu A, Morley MP, Cantu E, Beers MF, Frank DB, Morrisey EE, Krymskaya VP. mTORC1 activation in lung mesenchyme drives sex- and age-dependent pulmonary structure and function decline. Nat Commun. 2020 Nov 6;11(1):5640. doi: 10.1038/s41467-020-18979-4
- Smolarek TA, Wessner LL, McCormack FX, Mylet JC, Menon AG, Henske EP. Evidence that lymphangiomyomatosis is caused by TSC2 mutations: chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet. 1998 Apr;62(4):810-5. doi: 10.1086/301804
- Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000 May 23;97(11):6085-90. doi: 10.1073/pnas.97.11.6085
- Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003 Dec;38(4):376-81. doi: 10.1002/gcc.10252
- Cai X, Pacheco-Rodriguez G, Fan QY, Haughey M, Samsel L, El-Chemaly S, Wu HP, McCoy JP, Steagall WK, Lin JP, Darling TN, Moss J. Phenotypic characterization of disseminated cells with TSC2 loss of heterozygosity in patients with lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2010 Dec 1;182(11):1410-8. doi: 10.1164/rccm.201003-0489OC
- Crooks DM, Pacheco-Rodriguez G, DeCastro RM, McCoy JP Jr, Wang JA, Kumaki F, Darling T, Moss J. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2004 Dec 14;101(50):17462-7. doi: 10.1073/pnas.0407971101
- Northrup H, Aronow ME, Bebin EM, Bissler J, Darling TN, de Vries PJ, Frost MD, Fuchs Z, Gosnell ES, Gupta N, Jansen AC, Jóźwiak S, Kingswood JC, Knilans TK, McCormack FX, Pounders A, Roberds SL, Rodriguez-Buritica DF, Roth J, Sampson JR, Sparagana S, Thiele EA, Weiner HL, Wheless JW, Towbin AJ, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr Neurol. 2021 Oct;123:50-66. doi: 10.1016/j.pediatrneurol.2021.07.011
- van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, et al. Science. 1997;277:805–808.
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993 Dec 31;75(7):1305-15. doi: 10.1016/0092-8674(93)90618-z
- Carbonara C, Longa L, Grosso E, Borrone C, Garrè MG, Brisigotti M, Migone N. 9q34 loss of heterozygosity in a tuberous sclerosis astrocytoma suggests a growth suppressor-like activity also for the TSC1 gene. Hum Mol Genet. 1994 Oct;3(10):1829-32. doi: 10.1093/hmg/3.10.1829
- Green AJ, Johnson PH, Yates JR. The tuberous sclerosis gene on chromosome 9q34 acts as a growth suppressor. Hum Mol Genet. 1994 Oct;3(10):1833-4. doi: 10.1093/hmg/3.10.1833
- Green AJ, Smith M, Yates JR. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet. 1994 Feb;6(2):193-6. doi: 10.1038/ng0294-193
- Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, Hornigold N, van Slegtenhorst M, Welsh CT, Kwiatkowski DJ. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996 Aug;59(2):400-6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1914733/pdf/ajhg00021-0127.pdf
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006 Sep 28;355(13):1345-56. doi: 10.1056/NEJMra055323
- Northrup H, Koenig MK, Pearson DA, et al. Tuberous Sclerosis Complex. 1999 Jul 13 [Updated 2021 Dec 9]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1220
- McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP 3rd, Goldberg HJ, Young LR, Kinder BW, Downey GP, Sullivan EJ, Colby TV, McKay RT, Cohen MM, Korbee L, Taveira-DaSilva AM, Lee HS, Krischer JP, Trapnell BC; National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011 Apr 28;364(17):1595-606. doi: 10.1056/NEJMoa1100391
- Young L, Lee HS, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP 3rd, Goldberg HJ, Downey GP, Swigris JJ, Taveira-DaSilva AM, Krischer JP, Trapnell BC, McCormack FX; MILES Trial Group. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013 Aug;1(6):445-52. doi: 10.1016/S2213-2600(13)70090-0
- Bee J, Fuller S, Miller S, Johnson SR. Lung function response and side effects to rapamycin for lymphangioleiomyomatosis: a prospective national cohort study. Thorax. 2018 Apr;73(4):369-375. doi: 10.1136/thoraxjnl-2017-210872
- Miller S, Stewart ID, Clements D, Soomro I, Babaei-Jadidi R, Johnson SR. Evolution of lung pathology in lymphangioleiomyomatosis: associations with disease course and treatment response. J Pathol Clin Res. 2020 Jul;6(3):215-226. doi: 10.1002/cjp2.162
- Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, Lin L, Krueger D, Franz DN, Thiele EA, Sahin M, Kwiatkowski DJ. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet. 2015 Nov 5;11(11):e1005637. doi: 10.1371/journal.pgen.1005637
- Huang J, Xu W, Liu P, Liu Y, Shen C, Liu S, Wang Y, Wang J, Zhang T, He Y, Cheng C, Yang L, Zhang W, Tian X, Xu KF. Gene mutations in sporadic lymphangioleiomyomatosis and genotype-phenotype correlation analysis. BMC Pulm Med. 2022 Sep 18;22(1):354. doi: 10.1186/s12890-022-02154-0
- McCarthy C, Gupta N, Johnson SR, Yu JJ, McCormack FX. Lymphangioleiomyomatosis: pathogenesis, clinical features, diagnosis, and management. Lancet Respir Med. 2021 Nov;9(11):1313-1327. doi: 10.1016/S2213-2600(21)00228-9
- Wakida K, Watanabe Y, Kumasaka T, Seyama K, Mitani K, Hiraki T, Kamimura G, Nagata T, Nakamura Y, Sato M. Lymphangioleiomyomatosis in a Male. Ann Thorac Surg. 2015 Sep;100(3):1105-7. doi: 10.1016/j.athoracsur.2014.11.069
- Yu J, Astrinidis A, Howard S, Henske EP. Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am. J. Physiol. Lung Cell Mol. Physiol. 2004 Apr;286(4):L694-700.
- Shaw BM, Kopras E, Gupta N. Menstrual Cycle-related Respiratory Symptom Variability in Patients with Lymphangioleiomyomatosis. Ann Am Thorac Soc. 2022 Sep;19(9):1619-1621. doi: 10.1513/AnnalsATS.202202-144RL
- Khaddour K, Sankari A, Shayuk M. Lymphangioleiomyomatosis. [Updated 2023 Jun 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK534231
- Prizant H, Hammes SR. Minireview: Lymphangioleiomyomatosis (LAM): The “Other” Steroid-Sensitive Cancer. Endocrinology. 2016 Sep;157(9):3374-83.
- Yu J, Astrinidis A, Henske EP. Chromosome 16 loss of heterozygosity in tuberous sclerosis and sporadic lymphangiomyomatosis. Am. J. Respir. Crit. Care Med. 2001 Oct 15;164(8 Pt 1):1537-40.
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011 Jan;12(1):21-35.
- Muzykewicz DA, Sharma A, Muse V, Numis AL, Rajagopal J, Thiele EA. TSC1 and TSC2 mutations in patients with lymphangioleiomyomatosis and tuberous sclerosis complex. J Med Genet. 2009 Jul;46(7):465-8. doi: 10.1136/jmg.2008.065342
- Curatolo P, Specchio N, Aronica E. Advances in the genetics and neuropathology of tuberous sclerosis complex: edging closer to targeted therapy. Lancet Neurol. 2022 Sep;21(9):843-856. doi: 10.1016/S1474-4422(22)00213-7
- Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, Zonnenberg B, Verhoef S, Halley D, van den Ouweland A. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005 Jun;13(6):731-41. doi: 10.1038/sj.ejhg.5201402
- Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, Delgado MR, Wheless JW, Baumgartner JE, Roa BB, Wilson CM, Smith-Knuppel TK, Cheung MY, Whittemore VH, King TM, Northrup H. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007 Feb;9(2):88-100. doi: 10.1097/gim.0b013e31803068c7
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013 Oct;49(4):243-54. doi: 10.1016/j.pediatrneurol.2013.08.001
- Shepherd CW, Gomez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991 Aug;66(8):792-6. doi: 10.1016/s0025-6196(12)61196-3
- de Vries PJ. Neurodevelopmental, psychiatric, and cognitive aspects of tuberous sclerosis complex. In: Kwiatkowski DJ, Whittemore VH, Thiele EA, eds. Tuberous Sclerosis Complex: Genes, Clinical Features, and Therapeutics. Weinheim, Germany: Wiley-Blackwell; 2010a:229267
- Krueger DA. Management of CNS-related Disease Manifestations in Patients With Tuberous Sclerosis Complex. Curr Treat Options Neurol. 2013 Oct;15(5):618-33. doi: 10.1007/s11940-013-0249-2
- de Vries PJ, Whittemore VH, Leclezio L, Byars AW, Dunn D, Ess KC, Hook D, King BH, Sahin M, Jansen A. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol. 2015 Jan;52(1):25-35. doi: 10.1016/j.pediatrneurol.2014.10.004
- Gillberg IC, Gillberg C, Ahlsén G. Autistic behaviour and attention deficits in tuberous sclerosis: a population-based study. Dev Med Child Neurol. 1994 Jan;36(1):50-6. doi: 10.1111/j.1469-8749.1994.tb11765.x
- Bolton PF, Park RJ, Higgins JN, Griffiths PD, Pickles A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. 2002 Jun;125(Pt 6):1247-55. doi: 10.1093/brain/awf124
- Wong V. Study of the relationship between tuberous sclerosis complex and autistic disorder. J Child Neurol. 2006 Mar;21(3):199-204. doi: 10.2310/7010.2006.00046
- de Vries PJ, Hunt A, Bolton PF. The psychopathologies of children and adolescents with tuberous sclerosis complex (TSC): a postal survey of UK families. Eur Child Adolesc Psychiatry. 2007 Feb;16(1):16-24. doi: 10.1007/s00787-006-0570-3
- Chung TK, Lynch ER, Fiser CJ, Nelson DA, Agricola K, Tudor C, Franz DN, Krueger DA. Psychiatric comorbidity and treatment response in patients with tuberous sclerosis complex. Ann Clin Psychiatry. 2011 Nov;23(4):263-9.
- Numis AL, Major P, Montenegro MA, Muzykewicz DA, Pulsifer MB, Thiele EA. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology. 2011 Mar 15;76(11):981-7. doi: 10.1212/WNL.0b013e3182104347
- Spurling Jeste S, Wu JY, Senturk D, Varcin K, Ko J, McCarthy B, Shimizu C, Dies K, Vogel-Farley V, Sahin M, Nelson CA 3rd. Early developmental trajectories associated with ASD in infants with tuberous sclerosis complex. Neurology. 2014 Jul 8;83(2):160-8. doi: 10.1212/WNL.0000000000000568
- Kingswood JC, d’Augères GB, Belousova E, et al.; TOSCA consortium and TOSCA investigators. TuberOus SClerosis registry to increase disease Awareness (TOSCA) – baseline data on 2093 patients. Orphanet J Rare Dis. 2017 Jan 5;12(1):2. doi: 10.1186/s13023-016-0553-5
- McDonald NM, Varcin KJ, Bhatt R, Wu JY, Sahin M, Nelson CA 3rd, Jeste SS. Early autism symptoms in infants with tuberous sclerosis complex. Autism Res. 2017 Dec;10(12):1981-1990. doi: 10.1002/aur.1846
- Kothare SV, Singh K, Chalifoux JR, Staley BA, Weiner HL, Menzer K, Devinsky O. Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia. 2014 Jul;55(7):1025-9. doi: 10.1111/epi.12680
- Hwang SK, Lee JH, Yang JE, Lim CS, Lee JA, Lee YS, Lee K, Kaang BK. Everolimus improves neuropsychiatric symptoms in a patient with tuberous sclerosis carrying a novel TSC2 mutation. Mol Brain. 2016 May 23;9(1):56. doi: 10.1186/s13041-016-0222-6
- Kilincaslan A, Kok BE, Tekturk P, Yalcinkaya C, Ozkara C, Yapici Z. Beneficial Effects of Everolimus on Autism and Attention-Deficit/Hyperactivity Disorder Symptoms in a Group of Patients with Tuberous Sclerosis Complex. J Child Adolesc Psychopharmacol. 2017 May;27(4):383-388. doi: 10.1089/cap.2016.0100
- Lewis WW, Sahin M, Scherrer B, Peters JM, Suarez RO, Vogel-Farley VK, Jeste SS, Gregas MC, Prabhu SP, Nelson CA 3rd, Warfield SK. Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb Cortex. 2013 Jul;23(7):1526-32. doi: 10.1093/cercor/bhs135
- Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol. 2008 May;23(5):520-5. doi: 10.1177/0883073807309788
- Jeste SS, Varcin KJ, Hellemann GS, Gulsrud AC, Bhatt R, Kasari C, Wu JY, Sahin M, Nelson CA 3rd. Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology. 2016 Aug 23;87(8):766-72. doi: 10.1212/WNL.0000000000003002
- Prather P, de Vries PJ. Behavioral and cognitive aspects of tuberous sclerosis complex. J Child Neurol. 2004 Sep;19(9):666-74. doi: 10.1177/08830738040190090601
- Muzykewicz DA, Newberry P, Danforth N, Halpern EF, Thiele EA. Psychiatric comorbid conditions in a clinic population of 241 patients with tuberous sclerosis complex. Epilepsy Behav. 2007 Dec;11(4):506-13. doi: 10.1016/j.yebeh.2007.07.010
- Kopp CM, Muzykewicz DA, Staley BA, Thiele EA, Pulsifer MB. Behavior problems in children with tuberous sclerosis complex and parental stress. Epilepsy Behav. 2008 Oct;13(3):505-10. doi: 10.1016/j.yebeh.2008.05.010
- Ridler K, Suckling J, Higgins NJ, de Vries PJ, Stephenson CM, Bolton PF, Bullmore ET. Neuroanatomical correlates of memory deficits in tuberous sclerosis complex. Cereb Cortex. 2007 Feb;17(2):261-71. doi: 10.1093/cercor/bhj144
- de Vries PJ, Gardiner J, Bolton PF. Neuropsychological attention deficits in tuberous sclerosis complex (TSC). Am J Med Genet A. 2009 Mar;149A(3):387-95. doi: 10.1002/ajmg.a.32690
- Tierney KM, McCartney DL, Serfontein JR, de Vries PJ. Neuropsychological attention skills and related behaviours in adults with tuberous sclerosis complex. Behav Genet. 2011 May;41(3):437-44. doi: 10.1007/s10519-010-9423-4
- Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015 Jul;14(7):733-45. doi: 10.1016/S1474-4422(15)00069-1
- Joinson C, O’Callaghan FJ, Osborne JP, Martyn C, Harris T, Bolton PF. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003 Feb;33(2):335-44. doi: 10.1017/s0033291702007092
- Goh S, Kwiatkowski DJ, Dorer DJ, Thiele EA. Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology. 2005 Jul 26;65(2):235-8. doi: 10.1212/01.wnl.0000168908.78118.99
- van Eeghen AM, Black ME, Pulsifer MB, Kwiatkowski DJ, Thiele EA. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet. 2012 May;20(5):510-5. doi: 10.1038/ejhg.2011.241
- de Vries PJ. Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics. 2010 Jul;7(3):275-82. doi: 10.1016/j.nurt.2010.05.001
- Bolton PF, Clifford M, Tye C, Maclean C, Humphrey A, le Maréchal K, Higgins JN, Neville BG, Rijsdjik F; Tuberous Sclerosis 2000 Study Group; Yates JR. Intellectual abilities in tuberous sclerosis complex: risk factors and correlates from the Tuberous Sclerosis 2000 Study. Psychol Med. 2015 Aug;45(11):2321-31. doi: 10.1017/S0033291715000264
- Capal JK, Bernardino-Cuesta B, Horn PS, Murray D, Byars AW, Bing NM, Kent B, Pearson DA, Sahin M, Krueger DA; TACERN Study Group. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy Behav. 2017 May;70(Pt A):245-252. doi: 10.1016/j.yebeh.2017.02.007
- Humphrey A, MacLean C, Ploubidis GB, Granader Y, Clifford M, Haslop M, Neville BG, Yates JR, Bolton PF; Tuberous Sclerosis 2000 Study Group. Intellectual development before and after the onset of infantile spasms: a controlled prospective longitudinal study in tuberous sclerosis. Epilepsia. 2014 Jan;55(1):108-16. doi: 10.1111/epi.12484
- Staley BA, Montenegro MA, Major P, Muzykewicz DA, Halpern EF, Kopp CM, Newberry P, Thiele EA. Self-injurious behavior and tuberous sclerosis complex: frequency and possible associations in a population of 257 patients. Epilepsy Behav. 2008 Nov;13(4):650-3. doi: 10.1016/j.yebeh.2008.07.010
- Eden KE, de Vries PJ, Moss J, Richards C, Oliver C. Self-injury and aggression in tuberous sclerosis complex: cross syndrome comparison and associated risk markers. J Neurodev Disord. 2014;6(1):10. doi: 10.1186/1866-1955-6-10
- Wilde L, Eden K, de Vries P, Moss J, Welham A, Oliver C. Self-injury and aggression in adults with tuberous sclerosis complex: Frequency, associated person characteristics, and implications for assessment. Res Dev Disabil. 2017 May;64:119-130. doi: 10.1016/j.ridd.2017.03.007
- Leclezio L, de Vries PJ. Advances in the treatment of tuberous sclerosis complex. Curr Opin Psychiatry. 2015 Mar;28(2):113-20. doi: 10.1097/YCO.0000000000000136
- Lennert B, Farrelly E, Sacco P, Pira G, Frost M. Resource utilization in children with tuberous sclerosis complex and associated seizures: a retrospective chart review study. J Child Neurol. 2013 Apr;28(4):461-9. doi: 10.1177/0883073812448437
- Rentz AM, Skalicky AM, Liu Z, Wheless JW, Dunn DW, Frost MD, Nakagawa J, Magestro M, Prestifilippo J. Tuberous sclerosis complex: a survey of health care resource use and health burden. Pediatr Neurol. 2015 Apr;52(4):435-41. doi: 10.1016/j.pediatrneurol.2014.11.013
- Ewalt DH, Sheffield E, Sparagana SP, Delgado MR, Roach ES. Renal lesion growth in children with tuberous sclerosis complex. J Urol. 1998 Jul;160(1):141-5.
- Patel U, Simpson E, Kingswood JC, Saggar-Malik AK. Tuberose sclerosis complex: analysis of growth rates aids differentiation of renal cell carcinoma from atypical or minimal-fat-containing angiomyolipoma. Clin Radiol. 2005 Jun;60(6):665-73; discussion 663-4. doi: 10.1016/j.crad.2005.01.009
- Martignoni G, Bonetti F, Pea M, Tardanico R, Brunelli M, Eble JN. Renal disease in adults with TSC2/PKD1 contiguous gene syndrome. Am J Surg Pathol. 2002 Feb;26(2):198-205. doi: 10.1097/00000478-200202000-00006
- Pea M, Bonetti F, Martignoni G, Henske EP, Manfrin E, Colato C, Bernstein J. Apparent renal cell carcinomas in tuberous sclerosis are heterogeneous: the identification of malignant epithelioid angiomyolipoma. Am J Surg Pathol. 1998 Feb;22(2):180-7. doi: 10.1097/00000478-199802000-00005
- Borkowska J, Schwartz RA, Kotulska K, Jozwiak S. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol. 2011 Jan;50(1):13-20. doi: 10.1111/j.1365-4632.2010.04727.x
- Jones AC, Shyamsundar MM, Thomas MW, Maynard J, Idziaszczyk S, Tomkins S, Sampson JR, Cheadle JP. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999 May;64(5):1305-15. doi: 10.1086/302381
- Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, Reeve MP, Thiele E, Egelhoff JC, Kasprzyk-Obara J, Domanska-Pakiela D, Kwiatkowski DJ. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001 Jan;68(1):64-80. doi: 10.1086/316951
- Adriaensen ME, Schaefer-Prokop CM, Duyndam DA, Zonnenberg BA, Prokop M. Radiological evidence of lymphangioleiomyomatosis in female and male patients with tuberous sclerosis complex. Clin Radiol. 2011 Jul;66(7):625-8. doi: 10.1016/j.crad.2011.02.009
- Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016 May 26;2:16035. doi: 10.1038/nrdp.2016.35
- McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008 Feb;133(2):507-16. doi: 10.1378/chest.07-0898
- Avila NA, Dwyer AJ, Rabel A, Moss J. Sporadic lymphangioleiomyomatosis and tuberous sclerosis complex with lymphangioleiomyomatosis: comparison of CT features. Radiology. 2007 Jan;242(1):277-85. doi: 10.1148/radiol.2421051767
- Popper HH, Juettner-Smolle FM, Pongratz MG. Micronodular hyperplasia of type II pneumocytes–a new lung lesion associated with tuberous sclerosis. Histopathology. 1991 Apr;18(4):347-54. doi: 10.1111/j.1365-2559.1991.tb00856.x
- Cancellieri A, Poletti V, Corrin B. Respiratory failure due to micronodular type II pneumocyte hyperplasia. Histopathology. 2002 Sep;41(3):263-5. doi: 10.1046/j.1365-2559.2002.01433.x
- Kobashi Y, Sugiu T, Mouri K, Irei T, Nakata M, Oka M. Clinicopathological analysis of multifocal micronodular pneumocyte hyperplasia associated with tuberous sclerosis in Japan. Respirology. 2008 Nov;13(7):1076-81. doi: 10.1111/j.1440-1843.2008.01382.x
- Franz DN, Brody A, Meyer C, Leonard J, Chuck G, Dabora S, Sethuraman G, Colby TV, Kwiatkowski DJ, McCormack FX. Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med. 2001 Aug 15;164(4):661-8. doi: 10.1164/ajrccm.164.4.2011025
- Li LH, Li N, Zhao JY, Fei P, Zhang GM, Mao JB, Rychwalski PJ. Findings of perinatal ocular examination performed on 3573, healthy full-term newborns. Br J Ophthalmol. 2013 May;97(5):588-91. doi: 10.1136/bjophthalmol-2012-302539
- Northrup H, Wheless JW, Bertin TK, Lewis RA. Variability of expression in tuberous sclerosis. J Med Genet. 1993 Jan;30(1):41-3. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1016232/pdf/jmedgene00003-0056.pdf
- Shields JA, Eagle RC Jr, Shields CL, Marr BP. Aggressive retinal astrocytomas in four patients with tuberous sclerosis complex. Trans Am Ophthalmol Soc. 2004;102:139-47; discussion 147-8.
- Elsayes KM, Narra VR, Lewis JS Jr, Brown JJ. Magnetic resonance imaging of adrenal angiomyolipoma. J Comput Assist Tomogr. 2005 Jan-Feb;29(1):80-2. doi: 10.1097/01.rct.0000152863.97865.47
- Fricke BL, Donnelly LF, Casper KA, Bissler JJ. Frequency and imaging appearance of hepatic angiomyolipomas in pediatric and adult patients with tuberous sclerosis. AJR Am J Roentgenol. 2004 Apr;182(4):1027-30. doi: 10.2214/ajr.182.4.1821027
- Dworakowska D, Grossman AB. Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr Relat Cancer. 2009 Mar;16(1):45-58. doi: 10.1677/ERC-08-0142
- McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP, Goldberg HJ, Young LR, Kinder BW, Downey GP, Sullivan EJ, Colby TV, McKay RT, Cohen MM, Korbee L, Taveira-DaSilva AM, Lee HS, Krischer JP, Trapnell BC., National Institutes of Health Rare Lung Diseases Consortium. MILES Trial Group. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med. 2011 Apr 28;364(17):1595-606.
- Taveira-DaSilva AM, Steagall WK, Rabel A, Hathaway O, Harari S, Cassandro R, Stylianou M, Moss J. Reversible airflow obstruction in lymphangioleiomyomatosis. Chest. 2009 Dec;136(6):1596-1603.
- Araujo MS, Baldi BG, Freitas CS, Albuquerque AL, Marques da Silva CC, Kairalla RA, Carvalho CR, Carvalho CR. Pulmonary rehabilitation in lymphangioleiomyomatosis: a controlled clinical trial. Eur. Respir. J. 2016 May;47(5):1452-60.
- Oprescu N, McCormack FX, Byrnes S, Kinder BW. Clinical predictors of mortality and cause of death in lymphangioleiomyomatosis: a population-based registry. Lung. 2013;191:35–42. doi: 10.1007/s00408-012-9419-3
- Johnson SR, Whale CI, Hubbard RB, Lewis SA, Tattersfield AE. Survival and disease progression in UK patients with lymphangioleiomyomatosis. Thorax. 2004;59:800–803. doi: 10.1136/thx.2004.023283
- Gupta N, Lee HS, Ryu JH, Taveira-DaSilva AM, Beck GJ, Lee JC, McCarthy K, Finlay GA, Brown KK, Ruoss SJ, et al. The NHLBI LAM registry: prognostic physiologic and radiologic biomarkers emerge from a 15-year prospective longitudinal analysis. Chest. 2019;155:288–296. doi: 10.1016/j.chest.2018.06.016
- Hayashida M, Seyama K, Inoue Y, Fujimoto K, Kubo K. The epidemiology of lymphangioleiomyomatosis in Japan: a nationwide cross-sectional study of presenting features and prognostic factors. Respirology. 2007;12:523–530. doi: 10.1111/j.1440-1843.2007.01101.x
- Matsui K, Beasley MB, Nelson WK, Barnes PM, Bechtle J, Falk R, Ferrans VJ, Moss J, Travis WD. Prognostic significance of pulmonary lymphangioleiomyomatosis histologic score. Am J Surg Pathol. 2001;25:479–484. doi: 10.1097/00000478-200104000-00007
- Carrington CB, Cugell DW, Gaensler EA, Marks A, Redding RA, Schaaf JT, Tomasian A. Lymphangioleiomyomatosis. Physiologic-pathologic-radiologic correlations. Am Rev Respir Dis. 1977 Dec;116(6):977-95. doi: 10.1164/arrd.1977.116.6.977
- Oprescu N, McCormack FX, Byrnes S, Kinder BW. Clinical predictors of mortality and cause of death in lymphangioleiomyomatosis: a population-based registry. Lung. 2013 Feb;191(1):35-42. doi: 10.1007/s00408-012-9419-3
- Johnson SR, Whale CI, Hubbard RB, Lewis SA, Tattersfield AE. Survival and disease progression in UK patients with lymphangioleiomyomatosis. Thorax. 2004;59:800–803. doi: 10.1136/thx.2004.023283
- Kitaichi M, Nishimura K, Itoh H, Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995;151:527–533. doi: 10.1164/ajrccm.151.2.7842216
- Taveira-DaSilva AM, Stylianou MP, Hedin CJ, Hathaway O, Moss J. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest. 2004;126:1867–1874. doi: 10.1378/chest.126.6.1867





{kind=link}