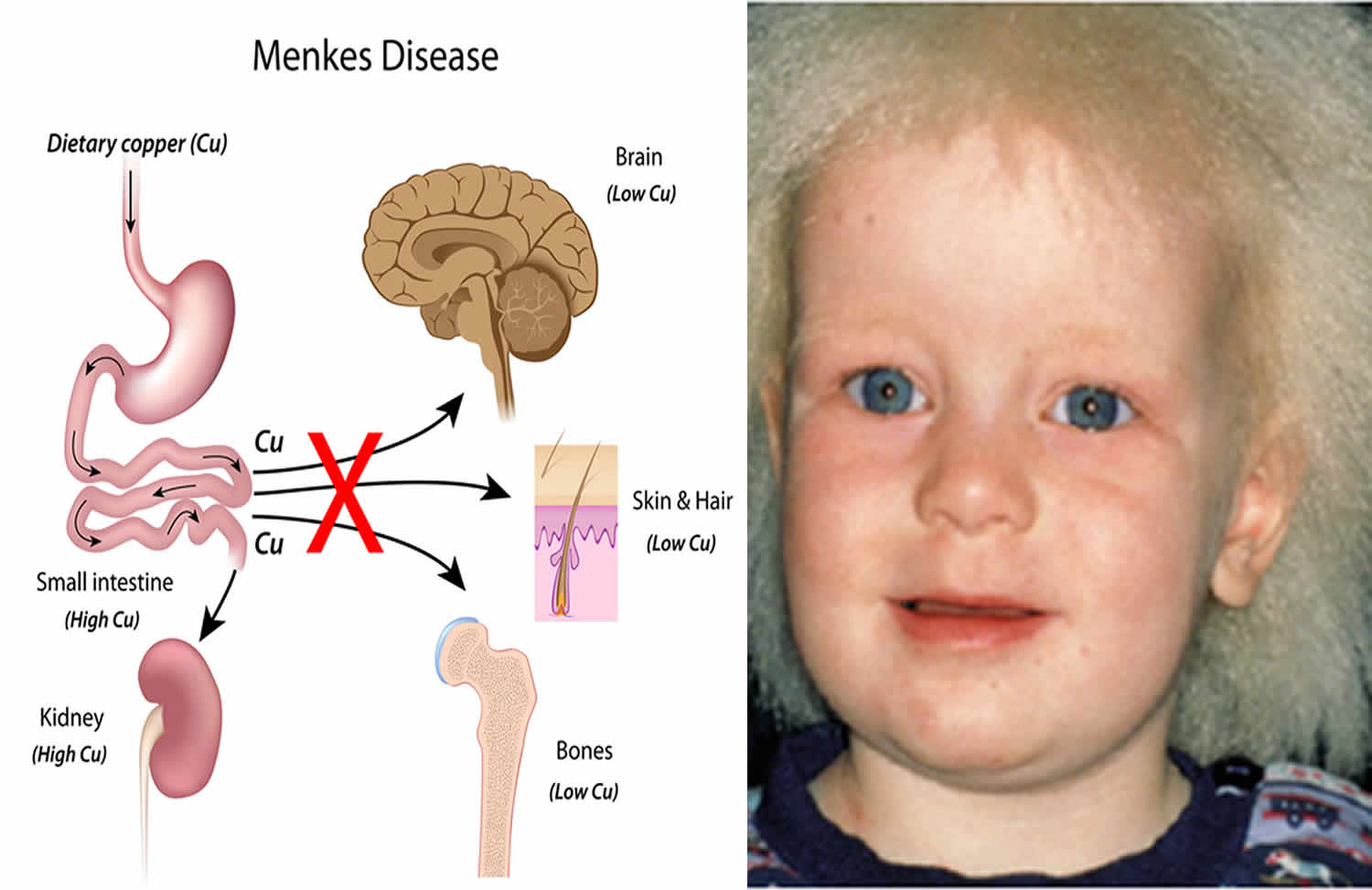
What is Menkes disease
Menkes disease also called copper transport disease, kinky hair disease, steely hair disease or Menkes syndrome, is a rare inherited genetic disorder that affects of copper metabolism in the body. Menkes disease is caused by the failure of the copper transport systems within the cell and then across the cell membrane that is responsible for the symptoms of the disorder. Because of the failure of this transport system, copper is unavailable to various cells where it is essential for the structure and function of various enzymes that control the development of hair, brain, bones, liver and arteries. Menkes disease primarily affects male infants. Copper accumulates at abnormally low levels in the liver and brain, but at higher than normal levels in the kidney and intestinal lining. Affected infants may be born prematurely, but appear healthy at birth and develop normally for 6 to 8 weeks. Then symptoms begin, including floppy muscle tone, seizures, and failure to thrive. Menkes disease is also characterized by subnormal body temperature and strikingly peculiar hair, which is kinky, colorless or steel-colored, and breaks easily. There is often extensive neurodegeneration in the gray matter of the brain. Arteries in the brain may be twisted with frayed and split inner walls. This can lead to rupture or blockage of the arteries. Weakened bones (osteoporosis) may result in fractures. Menkes disease is suspected in males who develop hypotonia, failure to thrive, and seizures between age six and ten weeks. Shortly thereafter, hair changes become manifest: the scalp and (usually) eyebrow hair is short, sparse, coarse, twisted, and often lightly pigmented (white, silver, or gray). The hair is shorter and thinner on the sides and back of the head. The hair can be reminiscent of steel wool cleaning pads. Light microscopic hair analysis reveals pili torti (hair shafts twisting 180°), trichoclasis (transverse fracture of the hair shaft), and trichoptilosis (longitudinal splitting of the hair shaft). Because of the flattening of the normal cylindric structure, the periodicity of the twisting in pili torti is different from that found in naturally curly hair.
Specific clinical features of Menkes syndrome include:
- Distinctive facial features: jowly appearance with sagging cheeks
- Pectus excavatum (midline depression in the bony thorax)
- Skin laxity, particularly on the nape of the neck and trunk
- Umbilical or inguinal hernias
- Hypotonia, neurodevelopmental delays, and failure to thrive, typically manifest by age three to six months
Menkes disease is characterized by sparse, kinky hair; failure to gain weight and grow at the expected rate (failure to thrive); and deterioration of the nervous system. Additional signs and symptoms include weak muscle tone (hypotonia), sagging facial features, seizures, developmental delay, and intellectual disability. Children with Menkes disease typically begin to develop symptoms during infancy and often do not live past age 3. Early treatment with copper may improve the prognosis in some affected individuals. In rare cases, symptoms begin later in childhood.
Menkes disease is an X-linked genetic disorder caused by mutations in the ATP7A gene that is responsible for production of the ATPase enzyme that regulates copper levels in the body. Variants of Menkes that are caused by mutations in the ATP7A gene but result in less severe symptoms include mild Menkes disease and occipital horn syndrome. X-linked genetic disorders are conditions caused by an abnormal gene on the X chromosome and occur mostly in males. Females that have mutations in the ATP7A gene present on one of their X chromosomes are carriers for Menkes disease.
Occipital horn syndrome sometimes called X-linked cutis laxa, is a less severe form of Menkes disease that begins in early to middle childhood. It is characterized by wedge-shaped calcium deposits in a bone at the base of the skull (the occipital bone), coarse hair, and loose skin and joints.
Occipital horn syndrome is suspected in males with:
- Occipital horns (distinctive wedge-shaped calcifications at the site of attachment of the trapezius muscle and the sternocleidomastoid muscle to the occipital bone). These calcifications may be clinically palpable or observed on skull radiographs.
- Lax skin and joints
- Bladder diverticula
- Inguinal hernias
- Vascular tortuosity
- Dysautonomia (chronic diarrhea, orthostatic hypotension)
- Mild cognitive deficits
The incidence of Menkes syndrome and occipital horn syndrome is estimated to be 1 in 100,000 to 1 in 250,000 newborns 1.
Treatment with daily copper injections may improve the outcome in Menkes disease if it begins within days after birth. Other treatment is symptomatic and supportive.
Figure 1. Menkes disease
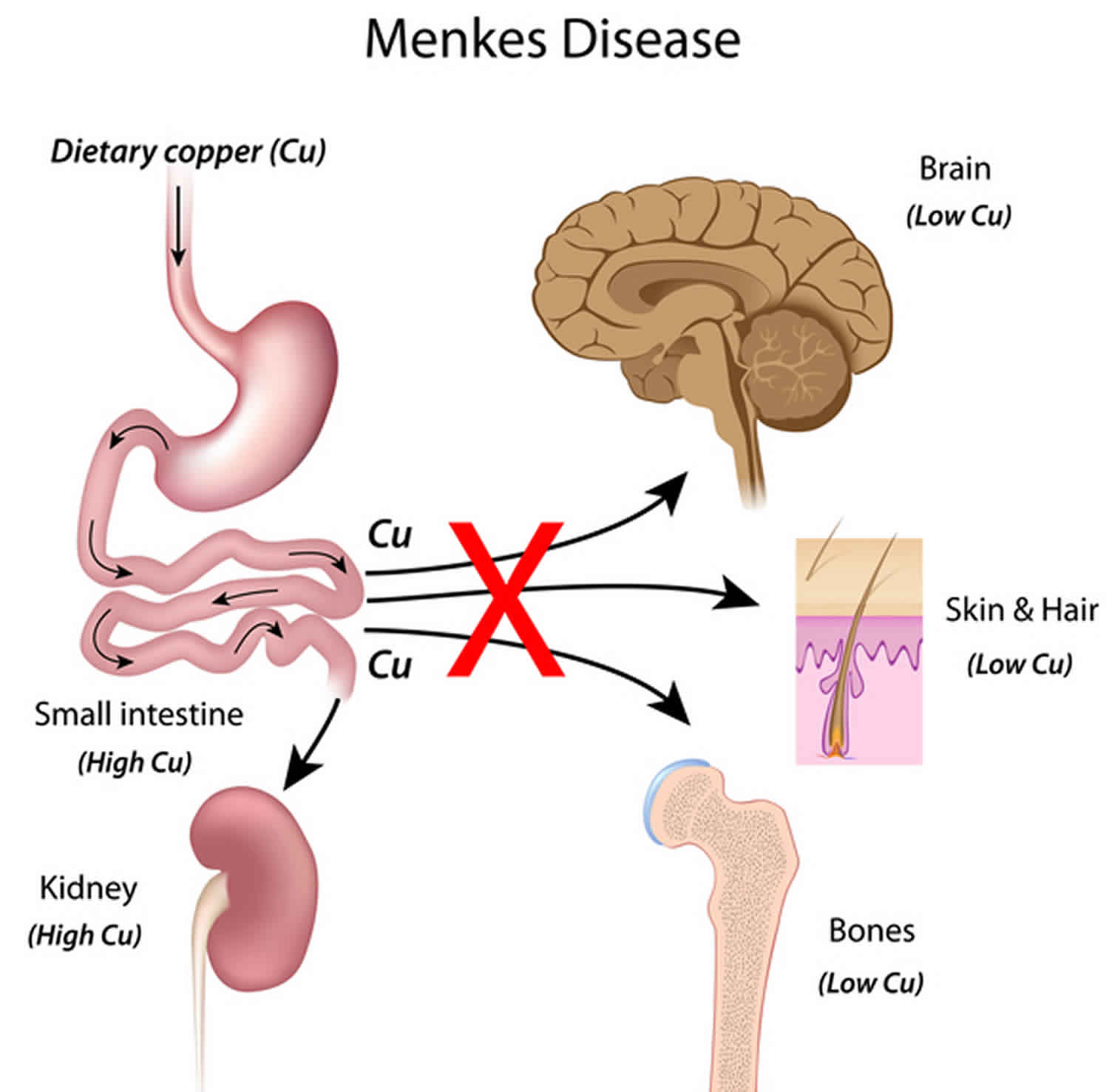
What causes Menkes disease?
Mutations in the ATP7A gene cause Menkes syndrome. The ATP7A gene provides instructions for making a protein that is important for regulating copper levels in the body. Copper is necessary for many cellular functions, but it is toxic when present in excessive amounts. Mutations in the ATP7A gene result in poor distribution of copper to the body’s cells. Copper accumulates in some tissues, such as the small intestine and kidneys, while the brain and other tissues have unusually low levels of copper. The decreased supply of copper can reduce the activity of numerous copper-containing enzymes that are necessary for the structure and function of bone, skin, hair, blood vessels, and the nervous system. The signs and symptoms of Menkes syndrome and occipital horn syndrome are caused by the reduced activity of these copper-containing enzymes.
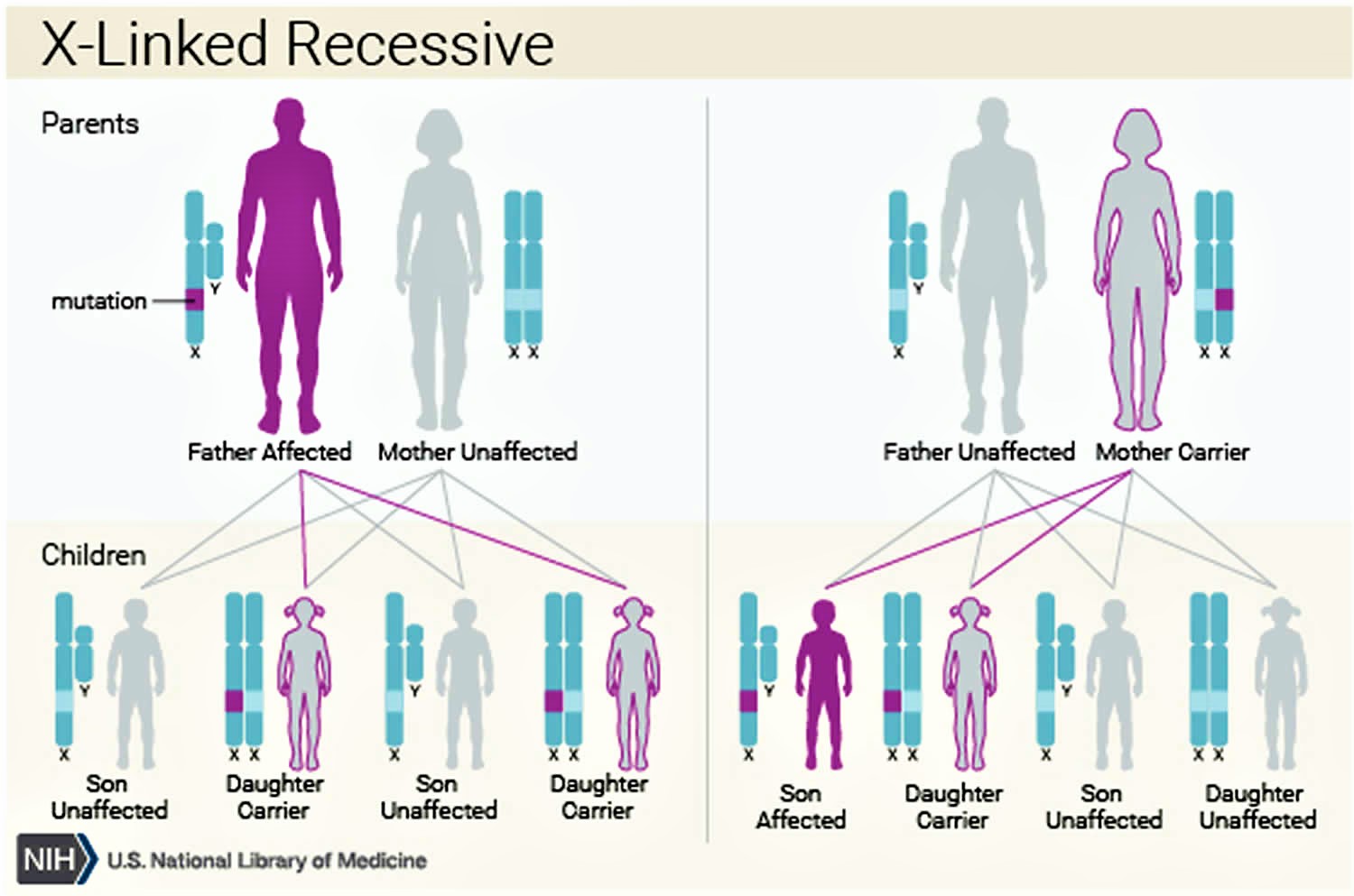
Menkes disease inheritance pattern
Menkes syndrome is inherited in an X-linked recessive pattern. The gene associated with Menkes syndrome is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause Menkes disease. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause Menkes disease. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Carrier females usually do not display symptoms because females have two X chromosomes and one is inactivated so that the genes on that chromosome are nonfunctioning. It is usually the X chromosome with the abnormal gene that is inactivated. Males have one X chromosome that is inherited from their mother and if a male inherits an X chromosome that contains a disease gene he will develop the disease.
Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son.
Males with X-linked disorders pass the disease gene to all of their daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring. Males with Menkes disease have not been reported to live to reproductive age. Fertility of individuals with mild Menkes disease or occipital horn syndrome is not known.
In about one-third of cases, Menkes syndrome is caused by new mutations in the ATP7A gene. People with a new mutation do not have a history of the disorder in their family.
Figure 2. Menkes disease X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Menkes disease symptoms
Menkes disease is characterized by brittle, tangled, sparse, steely or kinky hair that is often white, ivory, or gray in color. Unusual facial features include pudgy cheeks and abnormal eyebrows. Affected infants are often born prematurely. Lower than normal body temperature (hypothermia) and excess bilirubin in the blood (hyperbilirubinemia) may occur causing a yellow appearance (jaundice). Hypothermia may also occur in older infants. In some cases, early symptoms may resolve, and normal or slightly slowed development may proceed for two to three months. Severe developmental delay, loss of early development skills, and convulsions may occur. Brain abnormalities such as a blood clot at the base of the brain (subdural hematoma) and/or rupture or thrombosis of arteries in the brain not infrequently occur. Spastic dementia and seizures may eventually arise. Weakened bones (osteoporosis) are common and can result in fractures. The combination of subdural hematoma and bone fractures may lead to an incorrect diagnosis of child abuse. Emphysema, bladder abnormalities, degeneration of the retina and cysts of the iris have also been described. In rare cases, symptoms are very mild and only a few typical symptoms may appear.
Menkes disease signs and symptoms may include 1:
- Curly, thin hair, dull and discolored hair, rough to the touch, especially in areas subject to friction, and may be noticed as early as 2 months of age
- Trouble gaining weight
- Growth retardation
- Progressive nervous system deterioration
- Weak muscle tone (hypotonia)
- Facial droop
- Seizures
- Intellectual disability
The following may also be observed 2:
- Preterm babies
- Lump in the head when children are born by c-section surgery
- Low temperature (hypothermia)
- Low blood sugar (hypoglycemia)
- Yellow skin and eyes (jaundice) that requires several days of treatment with incubator and light
- Hollowed chest (pectus excavatum)
- Inguinal or umbilical hernia
There is a milder form of the disease called the occipital horn syndrome or X-linked cutis laxa, which begins in children and which also has loose skin and joints.
Menkes disease diagnosis
Menkes disease is diagnosed by measurement of a decreased amount of copper and ceruloplasmin in blood plasma but these tests are not always reliable in the newborn period. A new method of diagnosis that can potentially identify affected infants before copper deficiency affects the brain involves measurement of catecholamine levels in blood plasma. Molecular genetic testing for mutations in the APT7A gene is available to confirm the diagnosis. Carrier testing and prenatal diagnosis are available if a specific ATP7A mutation in an affected family member.
Menkes disease treatment
Treatment is based on the symptoms. It may include inserting a tube through a small opening in the abdomen that delivers nutrition directly to the stomach (gastrostomy tube or G-tube) to ensure an adequate nutrition. Some people who have pouches in the bladder (bladder diverticulum) may need surgery 3.
Early administration of copper may extend the patient’s life and can prevent neurologic damage in some patients (about 30% of cases) or improve the symptoms when it is started very early. The copper is administrated (as copper histidine or copper chloride) into a vein (intravenous (IV)), or by injecting it under the skin (subcutaneous) because when it is given by mouth it is not absorbed by the intestines. Some studies have shown that if copper administration starts before 10 days of life in certain cases there are no neurological problems. Also, the treatment may also result in fewer seizures and less irritability. Other treatment may include 2:
- Penicillamine: To avoid excess of copper during the treatment
- Vitamin C: It may increase the effectiveness of the copper treatment but there is no real evidence of being effective
- Vitamin E: It has been suggested as a treatment perhaps by its antioxidant property
- Insertion of genetic material (DNA) of the normal gene directly into the brain, using virus carrying the DNA (viral gene therapy). This type of treatment is still in research
- L-threo-dihydroxyphenylserine (L-DOPS): Results in the improvement dopamine deficiency beta-hydroxylase (DBH) that is one of the enzymes impaired in the disease.
- Developmental intervention
Treatment with copper chloride and/or L-histidine should be provided by a clinician familiar with their use. Copper chloride and L-histidine solutions of 350-500 µg/day or every other day injected intravenously or subcutaneously increase the serum and cerebrospinal fluid copper levels to the normal range after 6 weeks. This treatment seems to improve symptoms related to copper efflux from the cell. Metaphyseal widening and spurring and periosteal thickening regress. Bone maturation progresses, although mineralization is still defective. The ratio of DOPA/DHPG normalizes. However, the connective-tissue defects do not respond to parenteral copper histidine treatment.
Unfortunately, the neurologic symptoms, once present, are less amenable to treatment. Sheela et al reported that a 15-month-old who was treated with subcutaneous copper supplementation for 30 months became seizure free and the skin and hair darkened, but the child continued to have severe developmental delay 4.
Several investigations are ongoing to find more effective treatments.
Menkes disease prognosis
Since newborn screening for Menkes disease is not available, and early detection is infrequent because the clinical signs of Menkes disease are subtle in the beginning, Menkes disease is rarely treated early enough to make a significant difference. In general, the prognosis for babies with Menkes disease is poor 5. Most untreated patients with the classic form of Menkes disease die by age 3 years.
The major problems in Menkes disease involve the neurologic, gastrointestinal, and connective tissue (including blood vessels) systems. Pneumonia, leading to respiratory failure, is a common cause of death, although some patients with Menkes disease die suddenly in the absence of any apparent acute medical problem 2.
The majority of children with Menkes disease die within the first decade of life (many by age 3 years), but early treatment with copper within the first few months of life appears to be effective in increasing the life expectancy of some patients (from 3 to 13 years of age or more) 6. Also, when treatment is started very early, especially during the first 10 days of life, it may result in improved neurological outcomes 2. Several investigations are ongoing to find more effective treatments.
Kaler et al 7 noted that infants who received treatment early (within 10 +/- 4 days) had a 92% survival after a median follow-up of 4.6 years. Historical controls that were diagnosed at 163 +/- 113 days and treated only had a 13% survival after a median follow-up of 1.8 years. In fact, 2 of their patients (one with a missense mutation and one with a splice junction alteration) who were treated within 10 days of life had normal neurodevelopmental outcomes at age 5 and 7. A child with the same mutation as the 5-year-old but treated later at 22 days of age did have neurologic sequelae but could walk with support and ride a tricycle. Patients with mutations that disrupt the translational reading frame or have a premature termination codon did not do as well. Their treatment regimen consisted of subcutaneous copper histidine injections of 250 micrograms twice a day to 1 year of age and then daily injections to 3 years of age.
Tang et al 8 also reported that 2 infants treated within 25 days of age had near normal neurodevelopment at 7 months and 3 years of age. However, another infant with an identical missense mutation who began treatment at 228 days of age remained at a 2- to 4-month neurodevelopmental level at a chronological age of 2.5 years. This mutation resulted in normal levels of ATP7A transcript, but the mutated protein had abnormally high posttranslational degradation.
Paulsen et al 9 caution that the biochemical profile needs to be examined as reinitiation of protein translation may ameliorate the effects of a large frameshift deletion with a premature termination codon.
Donsante et al 10 also reported that 3 untreated brothers had differing courses of their disease. The 2 older brothers had mild disease and were able to ambulate independently and did not have seizures. The youngest brother was more delayed and had epilepsy, but he also had a cardiac arrest as a neonate. The cardiac arrest may be the cause of the more severe problems in the youngest brother, but he also had lower serum copper levels and the highest DOPA:DHPG ratio. Furthermore, Donsante et al also reported 2 untreated brothers who had different courses. The older brother also had a milder course with the occipital horn phenotype but the younger brother had profound delay and epilepsy. The less affected sibling had higher ATP7A levels but it was not clear why. There was no somatic mosaicism detected in either family.
Menkes disease life expectancy
The majority of children with Menkes disease die within the first decade of life (many by age 3 years), but early treatment with copper within the first few months of life appears to be effective in increasing the life expectancy of some patients (from 3 to 13 years of age or more) 6. Also, when treatment is started very early, especially during the first 10 days of life, it may result in improved neurological outcomes 2. Several investigations are ongoing to find more effective treatments.
References- Menkes syndrome. https://ghr.nlm.nih.gov/condition/menkes-syndrome
- Menkes Disease. https://emedicine.medscape.com/article/1180460-overview
- Kaler SG. ATP7A-Related Copper Transport Disorders. 2003 May 9 [Updated 2016 Aug 18]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1413
- Sheela SR, Latha M, Liu P, et al. Copper-replacement treatment for symptomatic Menkes disease: ethical considerations. Clin Genet. 2005 Sep. 68(3):278-83.
- Menkes Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Menkes-Disease-Information-Page
- National Center for Biotechnology Information (US). Genes and Disease [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1998-. Menkes syndrome. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22216
- Kaler SG, Holmes CS, Goldstein DS, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008 Feb 7. 358(6):605-14.
- Tang J, Donsante A, Desai V, Patronas N, Kaler SG. Clinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727R. Mol Genet Metab. 2008 Nov. 95(3):174-81.
- Paulsen M, Lund C, Akram Z, Winther JR, Horn N, Moller LB. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am J Hum Genet. 2006 Aug. 79(2):214-29.
- Donsante A, Tang J, Godwin SC, et al. Differences in ATP7A gene expression underlie intrafamilial variability in Menkes disease/occipital horn syndrome. J Med Genet. 2007 Aug. 44(8):492-7.





{kind=link}