Multifocal motor neuropathy
Multifocal motor neuropathy also called MMN or multifocal motor neuropathy with conduction block (MMNCB), is a rare, acquired, immune-mediated demyelinating motor neuropathy characterized by progressive asymmetric slowly progressive muscle weakness, fasciculations, and cramping, without significant sensory involvement 1. Multifocal motor neuropathy typically involves upper limbs more than the lower limbs 2. Electrodiagnostic studies often reveal an asymmetric motor neuropathy with characteristic conduction block. Serum IgM anti-ganglioside antibodies (anti-GM1) are present in the majority of the patients 3.
Clinically, multifocal motor neuropathy with conduction block may resemble amyotrophic lateral sclerosis (ALS) with predominant lower motor neuron involvement, but muscle atrophy and more rapid progression are lacking 1. Duration of disease prior to diagnosis ranges from several months to more than 15 years.
Unlike ALS, MMN usually responds to treatment with intravenous immunoglobulin (IVIg), subcutaneous immunoglobulins or cyclophosphamide, even after many years of duration 4. In some cases, the symptoms are so mild that patients do not require any treatment 3. However, most patients may develop a progressive worsening of strength, especially in the hands and arms, which can induce difficulties to perform even simple daily tasks such as writing, washing, or dressing. However, these patients may benefit from drug treatments as they often have a favorable response to intravenous immunoglobulin (IVIG) 5.
Multifocal motor neuropathy is a rare disorder with an estimated worldwide prevalence of less than 1 per 100,000 people 3. The reported prevalence of MMN is 0.65 cases per 100,000 population in Austria 6 and 0.29 in Japan per 100,000 people 7. It is 2.7 times more common in men as compared to women 2. Furthermore, MMN disease mean age of onset is 40 years and is diagnosed, especially in adults in their third to fifth decades, although it is also reported in children as young as 6 years of age and in the elderly 8. Eighty percent of patients are aged 20-50 years at presentation. Rarely, children as young as 6 years may be affected 9.
Figure 1. Multifocal motor neuropathy (typical weakness of extension of individual fingers in multifocal motor neuropathy)
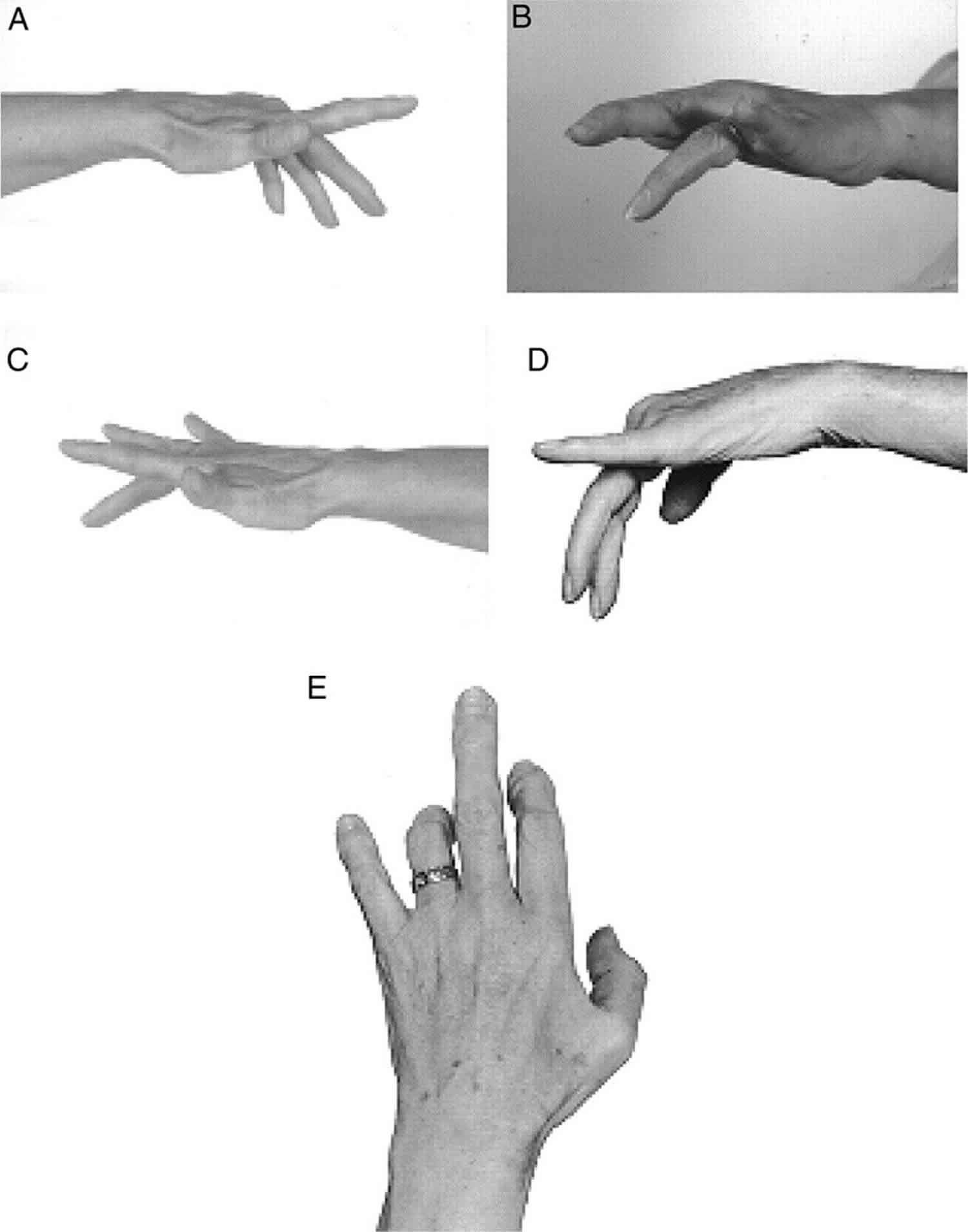
Multifocal motor neuropathy causes
Multifocal motor neuropathy is considered an immune-mediated motor neuropathy without a known cause. Rarely, MMN may develop following treatment with tumor necrosis factor (TNF) – α antagonists 11. A high prevalence of serum IgM anti-GM1 antibodies in these patients and a favorable response to intravenous immunoglobulin (IVIg) support this consideration. The abundance of GM1 in the myelin of motor nerves as compared to sensory nerves can explain the characteristic motor involvement in MMN.
Earlier it was believed that the conduction block seen on electrophysiological studies in MMN is due to severe focal demyelination, but increasing evidence suggests that anti-GM1 antibodies cause sodium and potassium channel dysfunction at or around the node of Ranvier of myelinated motor axons that play an important part in the conduction block. Hence the term ‘nodo-paranodopathy’ is also sometimes used 12. Complement activation probably plays a fundamental role in the pathogenesis of MMN disease 13.
Multifocal motor neuropathy pathophysiology
Anti-GM1 antibodies are proposed to play a major role in the pathophysiology of MMNCB. Titers of anti-GM1 antibodies are frequently elevated (>50%), but their role is still not well understood, even though they remain a useful marker for the diagnosis of multifocal motor neuropathy. GM1 is present in greater concentration in peripheral motor nerves as compared to the sensory nerves. Even in peripheral motor nerves, the greater concentration of GM1 is present around the nodes of Ranvier. Although the exact function of GM1 is unknown, they are considered to stabilize and cluster the ion channels around the nodes of Ranvier that are important for propagation of action potential. The disruption of these ion-channels resulting in decreased action potential propagation manifests as conduction block and decreased conduction velocity on the electrophysiologic studies.
Anti-GM1 antibodies, although common, are not present in all cases of MMNCB. In the absence of anti-GM1 antibodies, the pathophysiology of motor nerve dysfunction is controversial. These patients may either have low undetectable titers of anti-GM1, or probably different antibodies are present that are directed against different antigens. However, the clinical characteristics are the same in MMNCB patients with or without anti-GM1 antibodies 14.
Multifocal motor neuropathy symptoms
Typically, affected persons with multifocal motor neuropathy (MMN) present with a slowly progressive, asymmetric, predominantly distal muscle weakness developing over years. Weakness usually starts in a distribution of a single peripheral nerve with unilateral wrist drop, foot drop, finger weakness or grip weakness, but muscle atrophy is minimal if present at all. Initial involvement of the distal upper limb muscles is most common, leg weakness may also occur. Transient exacerbation may be aggravated by the cold or may occur during pregnancy. Sensory symptoms of pain and tingling are not uncommon but are typically minimal. Some patients may complain of muscle cramps, twitching, fasciculation, or excessive fatigue. The involvement of cranial nerves, bulbar, and respiratory muscles is unusual in these patients 12.
Physical examination
On physical examination, the most remarkable finding is asymmetric muscle weakness in the distribution of individual peripheral nerves that is out of proportion to muscle atrophy. Fasciculations may be present. For instance, with radial nerve involvement, the wrist and finger extensors are more affected than triceps. Muscles of the same myotome may be spared innervated by a different nerve. For example, although both abductor pollicis brevis and abductor digiti minimi are of the same myotome (C8-T1), only abductor pollicis brevis will be affected if the median nerve is involved. Muscle atrophy may be present late in the course of the disease and is often disproportionately mild as compared to the weakness. Deep tendon reflexes may be normal or asymmetrically reduced. Upper motor neuron signs are typically absent, differentiating from amyotrophic lateral sclerosis (ALS). Myokymia may be seen in the affected muscles 12.
Cranial nerves
Cranial nerves are rarely affected, but this may be an uncommon initial manifestation of MMN. Cranial nerve involvement may be limited to the twelfth cranial nerve.
Speech is normal.
Deep tendon reflexes
Deep tendon reflexes are typically absent (particularly in affected limbs) or normal. However, brisk tendon reflexes were found in 9% of patients, even in the weakened limbs.
Motor strength
Asymmetric weakness may occur in a nonmyotomal pattern, usually in the distribution of individual nerves. The upper limbs, particularly the hands, are more commonly involved than the lower limbs. Weakness of respiratory muscles is very rare 15. Weakness frequently worsens with exposure to cold 16.
Muscles innervated by motor nerves with persistent conduction block are usually weak.
Muscle atrophy
Atrophy may be present in weak muscles, but it is usually fairly mild. Late in the disease course atrophy may be more prevalent.
Upper motor neuron signs
These signs are absent.
Muscle tone
Tone is decreased or normal. No clonus, extensor plantar response, or pseudobulbar palsy is present. Pathologic reflexes (eg, Babinski, Hoffman) are not present.
Sensory examination
Sensory examination should be normal, and the sensory loss may be suggestive of Lewis-Sumner syndrome (multifocal acquired demyelinating sensory and motor neuropathy [MADSAM]).
Coordination
Coordination is normal.
Gait
Gait is usually normal, unless more prominent involvement of lower extremity muscles occurs.
Fasciculations and cramping
These are common and may occur outside of the distribution of clinically affected nerves.
Other
No rash or gynecomastia is present.
Multifocal motor neuropathy complications
Multifocal motor neuropathy is rarely fatal due to the sparing of cranial and respiratory muscles. Most complications are treatment-related.
- Intravenous immunoglobulin (IVIG) may cause thromboembolic events (myocardial infarctions, stroke, or deep venous thrombosis), renal failure, anaphylactic reactions, aseptic meningitis, and rarely, transfusion-related acute lung injury.
- Cyclophosphamide may cause bone marrow suppression, hemorrhagic cystitis, and interstitial pneumonitis 5.
Multifocal motor neuropathy diagnosis
Electrodiagnostic evaluation may document the presence of asymptomatic conduction blocks in other clinically unaffected nerves, and it may document more extensive involvement in patients with relatively few symptoms. Positive serology for anti-GM1 antibodies is supportive of the diagnosis of MMN, particularly higher titers.
In 2010, the European Federation of Neurological Societies and Peripheral Nerve Society Task Force revised the following diagnostic criteria to help in the diagnosis of MMN 17.
Core Criteria (both must be present)
- Slowly progressive, focal, asymmetric limb weakness, that is, motor involvement in the motor nerve distribution of at least two nerves, for at least 1 month (usually more than 6 months). If symptoms and signs are present only in the distribution of one nerve, only a possible diagnosis can be made.
- No objective sensory abnormalities except for minor vibration sense abnormalities in the lower limbs.
Supportive Clinical Criteria
- Predominant upper limb involvement
- Decreased or absent tendon reflexes in the affected limb
- Absence of cranial nerve involvement
- Cramps and fasciculations in the affected limb
- Response to immunomodulatory treatment
Exclusion Criteria
- Upper motor neuron signs, such as spasticity, clonus, extensor plantar response
- Marked bulbar involvement
- Sensory impairment more marked than minor vibration loss in the lower limbs
- Diffuse symmetric weakness during the initial weeks
Electrophysiological studies
In multifocal motor neuropathy, electrophysiological studies are consistent with a motor neuropathy with normal sensory nerve conduction studies. There may be evidence of demyelination marked by prolonged motor nerve latencies and slow conduction velocities. F-wave responses may be absent or prolonged. However, the hallmark finding of MMNCB is a motor conduction block and/or temporal dispersion. When the conduction block is absent or cannot be assessed by routine nerve conduction studies due to a more proximal location, the diagnosis is termed as MMN. Needle electromyography findings may reveal reduced recruitment of muscle unit action potentials in weak muscles due to proximal conduction blocks 18.
Conduction block is defined as a more than 50% reduction in compound muscle action potential (CMAP) amplitude or area between proximal and distal nerve stimulation sites and is often indicative of an acquired etiology. In MMNCB, the conduction block characteristically occurs at non-compressible sites. Therefore, the presence of conduction block across the known nerve entrapment sites (e.g., the median nerve at the wrist) cannot be used in the diagnosis of MMNCB. Besides MMNCB, conduction block is also seen in acquired demyelinating conditions, e.g., chronic inflammatory demyelinating polyneuropathy (CIDP) and Guillain-Barre syndrome (GBS) 18.
Clinical and electrodiagnostic criteria
Definite MMN, as follows 19:
- Weakness without objective sensory loss in the distribution of 2 or more nerves is present for more than 1 month
- Definite motor conduction block is present in at least one motor nerve with normal sensory nerve conductions
- Exclusion criteria not present.
Exclusion criteria:
- Upper motor neuron signs, such as spasticity, clonus, extensor plantar response
- Marked bulbar involvement
- Sensory involvement more marked than minor vibration loss in the lower limbs
- Diffuse symmetric weakness during initial weeks
Probable MMN, as follows:
- Weakness without objective sensory loss in the distribution of 2 or more nerves
- The presence of probable motor conduction block in 2 or more motor nerve segments with normal sensory nerve conduction studies
- Exclusion criteria not present
OR:
- Weakness without objective sensory loss in the distribution of one nerve
- The presence of probable motor conduction block in one motor nerve segments with normal sensory nerve conduction studies
- Exclusion criteria are not present.
- At least 2 supportive criteria met
Supportive criteria, as follows:
- Elevated titers of serum IgM anti-GM1 antibodies
- Increased cerebrospinal fluid (CSF) protein (< 1 g/L)
- MRI shows increased signal on T2-weighted imaging of brachial plexus with diffuse nerve swelling
- Objective clinical improvement following intravenous immunoglobulin (IVIG) treatment.
Laboratory investigations
Routine blood and urine laboratory investigations are unremarkable in patients with MMN. The cerebrospinal fluid (CSF) protein level is often normal or mildly increased. It is one of the important features to differentiate MMN from chronic inflammatory demyelinating polyneuropathy (CIDP) 18. Creatine kinase (CK) is frequently elevated (< 3 times the upper limit of reference range).
Anti-GM1 ganglioside antibodies are present in about half of the cases of MMN. However, the anti-GM1 titers do not correlate with the treatment. Anti-GM1 antibodies are not specific for MMN and are also seen in acute motor axonal neuropathy (AMAN) variant of Guillain-Barre syndrome, but their presence supports the diagnosis 18. The sensitivity of antibody detection can be enhanced by testing for GM1/galactocerebroside (GM1/GalC) complexes.
Imaging studies
Nerve ultrasound and magnetic resonance imaging (MRI) can offer important diagnostic data, especially when electrophysiological investigations are not conclusive. The diagnosis of MMN, indeed, can be a challenge when conduction blocks are not detectable, or in the case of advanced axonal loss. Spinal MRI may reveal T2 hyperintense signals in the brachial plexus with or without contrast enhancement in about half of the patients with MMN and CIDP. In MMN, these signals are asymmetric and often unilateral, but these are bilateral and symmetrical in chronic inflammatory demyelinating polyneuropathy (CIDP) 20.
In MMN, an increase in the cross-sectional area of the median and ulnar nerves may be seen with high-resolution ultrasound (HRUS). This finding is helpful when a clinical distinction is difficult with amyotrophic lateral sclerosis (ALS) in which the nerve diameters are typically reduced 18.
Multifocal motor neuropathy treatment
Multifocal motor neuropathy (MMN) is associated with slowly progressive weakness, but most patients are able to remain productive and employed. However, gradual progression may lead to significant disability. Physical and occupational therapy may be helpful in individual cases.
The use of intravenous immunoglobulin (IVIg) represents the main pharmacological treatment option for MMNCB. Of note, indeed, more than 80-90% of the patients respond to IVIG, but frequently long-term maintenance IVIG infusions are required to prevent worsening of symptoms 21. The response in muscle strength improvement is, however, short-term, and only 20% of patients achieve prolonged remission. Most of the patients need periodic IVIg infusions. In spite of regular IVIg infusions, motor deficits may slowly progress due to secondary axonal damage. In a Cochrane review 21, IVIG was considered superior to placebo in the treatment of multifocal motor neuropathy with conduction block.
IVIG is initially administered at a dose of 0.4 g/kg/day for five consecutive days for a total dose of 2 g/kg. Some clinicians administer IVIG in 2 days by administering at 1 g/kg per day. The follow-up maintenance IVIg infusion dose ranges from 0.4 g/kg once weekly to 2 g/kg every 8 weeks depending upon the patient’s condition. Because immunoglobulin is generally administered for long periods, often for years, the subcutaneous route has been investigated. Clinical investigations showed that the subcutaneous immunoglobulin (SCIG) is safe, equally effective, feasible, and the patients can self-administer themselves at home 22. The dose of subcutaneous immunoglobulin (SCIG) is the same as that of IVIG 23
In non-responders, the treatment options are limited. Different immunomodulatory agents such as cyclophosphamide, mycophenolate mofetil, azathioprine, and rituximab, have been reported in the literature with variable results 5. Oral cyclophosphamide has been reported effective in sustaining disease remission and reducing intravenous immunoglobulin (IVIg) frequency but has significant adverse effects 24. In 2007 25, an randomized controlled trial comprising of 28 patients did not reveal a significant difference when mycophenolate mofetil was combined with IVIG as compared to IVIG alone in patients with MMN. Recent reports describe effective treatment with cyclosporine and rituximab in a small number of patients, but additional data are needed before these would be recommended for treatment of MMN. Multiple comparative randomized controlled trials are needed to establish the efficacy of immunomodulatory drugs in MMN.
Cyclophosphamide may be used in combination with plasmapheresis. Lack of benefit was reported for 1 patient who received high-dose cyclophosphamide treatment followed by autologous stem cell transplantation 26.
Other treatments used with variable success include interferon-beta and azathioprine.
Corticosteroids and plasmapheresis are ineffective in patients with MMN 5.
Multifocal motor neuropathy prognosis
The prognosis of multifocal motor neuropathy is usually good. Approximately 80% of patients respond to IVIG treatment. About 20% of the patients achieve long term remission, while the remaining require periodic IVIG or subcutaneous immunoglobulin (SCIG) treatments. Even in non-responders, muscle weakness progresses slowly, and the majority of the patients are able to perform activities of daily living. In one study, more than 94% remained employed 5. In 2015 27, researchers from the PeriNomS Study Group validated the Rasch-built Overall Disability Scale for MMN. It is a 25-item tool developed for monitoring the course of disease and response to treatment 27.
References- Multifocal Motor Neuropathy With Conduction Blocks. https://emedicine.medscape.com/article/1174021-overview
- Lawson VH, Arnold WD. Multifocal motor neuropathy: a review of pathogenesis, diagnosis, and treatment. Neuropsychiatr Dis Treat. 2014;10:567-76.
- Hameed S, Cascella M. Multifocal Motor Neuropathy (MMN) [Updated 2020 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK554524
- European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society–first revision. J Peripher Nerv Syst. 2010 Dec. 15(4):295-301.
- Jinka M, Chaudhry V. Treatment of multifocal motor neuropathy. Curr Treat Options Neurol. 2014 Feb;16(2):269.
- Löscher WN, Oberreiter EM, Erdler M, Quasthoff S, Culea V, Berek K, Embacher N, Grinzinger S, Hess I, Höger FS, Horlings CGC, Huemer M, Jecel J, Kleindienst W, Laich E, Müller P, Oel D, Örtl W, Lenzenweger E, Rath J, Stadler K, Stieglbauer K, Thaler-Wolf C, Wanschitz J, Zimprich F, Cetin H, Topakian R. Multifocal motor neuropathy in Austria: a nationwide survey of clinical features and response to treatment. J. Neurol. 2018 Dec;265(12):2834-2840.
- Miyashiro A, Matsui N, Shimatani Y, Nodera H, Izumi Y, Kuwabara S, Imai T, Baba M, Komori T, Sonoo M, Mezaki T, Kawamata J, Hitomi T, Kawamata J, Hitomi T, Kohara N, Arimura K, Hashimoto S, Arisawa K, Kusunoki S, Kaji R., Japanese Multifocal Motor Neuropathy Study Group. Are multifocal motor neuropathy patients underdiagnosed? An epidemiological survey in Japan. Muscle Nerve. 2014 Mar;49(3):357-61.
- Kamata A, Muramatsu K, Sawaura N, Makioka N, Ogata T, Kuwashima M, Arakawa H. Demyelinating neuropathy in a 6-year-old girl with autism spectrum disorder. Pediatr Int. 2017 Aug;59(8):951-954.
- Ishigaki H, Hiraide T, Miyagi Y, Hayashi T, Matsubayashi T, Shimoda A, et al. Childhood-Onset Multifocal Motor Neuropathy With Immunoglobulin M Antibodies to Gangliosides GM1 and GM2: A Case Report and Review of the Literature. Pediatr Neurol. 2016 Sep. 62:51-7.
- Multifocal motor neuropathy. The diagnostic spectrum and response to treatment. Mark Slee, Arul Selvan, Michael Donaghy. Neurology Oct 2007, 69 (17) 1680-1687; DOI: 10.1212/01.wnl.0000277697.55288.d0
- Cocito D, Bergamasco B, Tavella A, Poglio F, Paolasso I, Costa P, et al. Multifocal motor neuropathy during treatment with infliximab. J Peripher Nerv Syst. 2005 Dec. 10(4):386-7.
- Yeh WZ, Dyck PJ, van den Berg LH, Kiernan MC, Taylor BV. Multifocal motor neuropathy: controversies and priorities. J. Neurol. Neurosurg. Psychiatry. 2020 Feb;91(2):140-148.
- Kieseier BC, Mathey EK, Sommer C, Hartung HP. Immune-mediated neuropathies. Nat Rev Dis Primers. 2018 Oct 11;4(1):31.
- Léger JM, Guimarães-Costa R, Iancu Ferfoglia R. The pathogenesis of multifocal motor neuropathy and an update on current management options. Ther Adv Neurol Disord. 2015 May;8(3):109-22.
- Boonyapisit K, Katirji B. Multifocal motor neuropathy presenting with respiratory failure. Muscle Nerve. 2000 Dec. 23(12):1887-90.
- Straver DC, van Asseldonk JT, Notermans NC, Wokke JH, van den Berg LH, Franssen H. Cold paresis in multifocal motor neuropathy. J Neurol. 2011 Feb. 258(2):212-7.
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society–first revision. J. Peripher. Nerv. Syst. 2010 Dec;15(4):295-301.
- Beadon K, Guimarães-Costa R, Léger JM. Multifocal motor neuropathy. Curr. Opin. Neurol. 2018 Oct;31(5):559-564.
- Bromberg MB, Franssen H. Practical rules for electrodiagnosis in suspected multifocal motor neuropathy. J Clin Neuromuscul Dis. 2015 Mar. 16 (3):141-52.
- Jongbloed BA, Haakma W, Goedee HS, Bos JW, Bos C, Hendrikse J, Van Den Berg LH, Van Der Pol WL. Comparative study of peripheral nerve Mri and ultrasound in multifocal motor neuropathy and amyotrophic lateral sclerosis. Muscle Nerve. 2016 Dec;54(6):1133-1135.
- {Best Evidence} Umapathi T, Hughes RA, Nobile-Orazio E, Léger JM. Immunosuppressant and immunomodulatory treatments for multifocal motor neuropathy. Cochrane Database Syst Rev. 2012 Apr 18. 4:CD003217
- Harbo T, Andersen H, Jakobsen J. Long-term therapy with high doses of subcutaneous immunoglobulin in multifocal motor neuropathy. Neurology. 2010 Oct 12. 75(15):1377-80.
- Eftimov F, Vermeulen M, de Haan RJ, van den Berg LH, van Schaik IN. Subcutaneous immunoglobulin therapy for multifocal motor neuropathy. J. Peripher. Nerv. Syst. 2009 Jun;14(2):93-100.
- Meucci N, Cappellari A, Barbieri S, Scarlato G, Nobile-Orazio E. Long term effect of intravenous immunoglobulins and oral cyclophosphamide in multifocal motor neuropathy. J. Neurol. Neurosurg. Psychiatry. 1997 Dec;63(6):765-9.
- Piepers S, Van den Berg-Vos R, Van der Pol WL, Franssen H, Wokke J, Van den Berg L. Mycophenolate mofetil as adjunctive therapy for MMN patients: a randomized, controlled trial. Brain. 2007 Aug;130(Pt 8):2004-10.
- Axelson HW, Oberg G, Askmark H. No benefit of treatment with cyclophosphamide and autologous blood stem cell transplantation in multifocal motor neuropathy. Acta Neurol Scand. 2008 Jun. 117(6):432-4.
- Vanhoutte EK, Faber CG, van Nes SI, Cats EA, Van der Pol WL, Gorson KC, van Doorn PA, Cornblath DR, van den Berg LH, Merkies IS., PeriNomS Study Group. Rasch-built Overall Disability Scale for Multifocal motor neuropathy (MMN-RODS(©) ). J. Peripher. Nerv. Syst. 2015 Sep;20(3):296-305.





{kind=link}