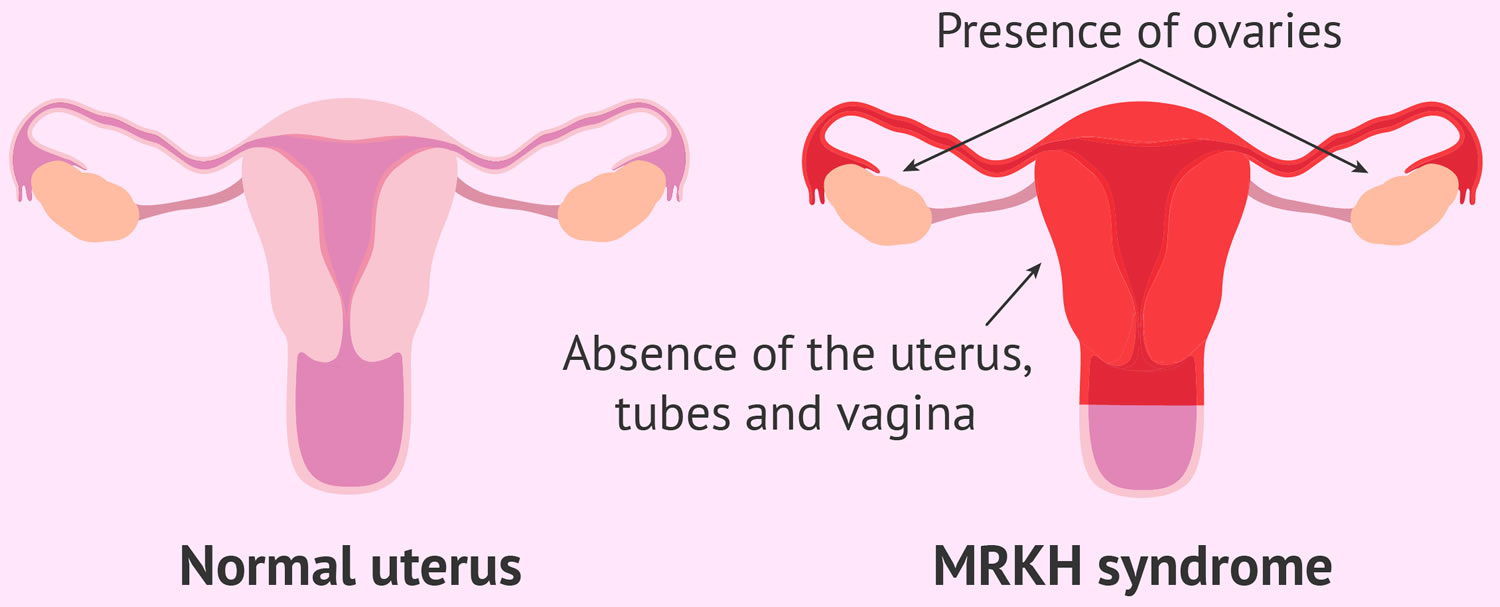
MRKH syndrome
MRKH syndrome is short for Mayer-Rokitansky-Küster-Hauser syndrome (named after the four doctors who first diagnosed the condition), which is also known as Mullerian aplasia, congenital (present at birth) absence of the uterus and vagina, genital renal ear syndrome, or Rokitansky syndrome, is a rare disorder that affects women that causes an incomplete development of the female reproductive tract consisting of congenital absence of the uterus and vagina 1, 2, 3, 4. MRKH syndrome is characterized by the failure of the uterus and the vagina to develop properly in women who have normal fully functional ovaries that can be located in unusual places in the body cavity and normal external genitalia 5. Typically, women with MRKH syndrome lack a fully functional uterus, cervix and upper vaginal canal. Women with MRKH syndrome have normal external genitalia and breast development, and often have a small external vaginal opening, called a ‘dimple’ that looks like a hymen 6. Furthermore, women with MRKH syndrome develop normal secondary sexual characteristics during puberty (e.g., breast development and pubic hair), but do not have a menstrual cycle by age 16 (primary amenorrhea) due to the absent uterus. Often, the failure to begin the menstrual cycle is the initial clinical sign of MRKH syndrome. Women with MRKH syndrome have a female chromosome pattern (46,XX) and normally functioning ovaries that can be located in unusual places in the body cavity. Women with MRKH syndrome also have normal breast and pubic hair development. Although women with MRKH syndrome are usually unable to carry a pregnancy, they may be able to have children through assisted reproductive therapies and gestational surrogacy. Women with MRKH also can have kidney misplacement, hearing loss, and even heart defects 6. Because of the nature of the disorder, MRKH syndrome can cause significant psychological challenges and counseling is recommended 7, 2, 5.
Gestational surrogacy is a process where one person who is often referred to as a gestational carrier and who did not provide the egg used in conception, carries a fetus through pregnancy and gives birth to a baby for another person or couple 8, 9. The person who carries the fetus is called a “surrogate” or “gestational carrier.” The person or couple who are seeking to parent the baby or babies are called the “intended parent(s)” 8, 9. In most cases, at least one intended parent is genetically related to the child, and the surrogate (gestational carrier) is not. This makes gestational surrogacy less legally complicated than other forms of surrogacy because stepparent or second-parent adoption is not required 9.
MRKH syndrome affects approximately 1 in 4,000-5,000 newborn girls 5, 10, 11, 12.
The range and severity of MRKH syndrome can vary greatly and the disorder is generally broken down into MRKH syndrome type 1 also known as isolated uterovaginal aplasia, which occurs as an isolated finding, and MRKH syndrome type 2 associated with extragenital abnormalities, which occurs with abnormalities of additional organ systems including mainly the kidneys (30–40%), the skeleton, hearing loss and/or ringing in the ears (tinnitus) and heart malformations 5, 11, 13. When only reproductive organs are affected, the condition is classified as MRKH syndrome type 1. Some women with MRKH syndrome also have abnormalities in other parts of the body; in these cases, the condition is classified as MRKH syndrome type 2. In MRKH syndrome type 2, the kidneys may be abnormally formed or positioned in different places throughout the abdominal cavity, or one kidney may fail to develop (unilateral renal agenesis). Affected individuals commonly develop skeletal abnormalities, particularly of the spinal bones (vertebrae). Females with MRKH syndrome type 2 may also have hearing loss and/or ringing in the ears (tinnitus) and heart defects. The frequency of MRKH syndrome type 1 and MRKH syndrome type 2 is 56–72% and 28–44%, respectively 14, 15, 16, 17.
By definition, MRKH syndrome only affects females. However, some researchers have noted similar symptoms in males 5. Affected males have exhibited absence or underdevelopment of the Wolffian duct, an organ that is present in a developing embryo that eventually evolves into certain structures such as the tube connecting the testes to the urethra (vas deferens) 5. Affected males may also have low levels of live sperm in their semen (azoospermia), kidney abnormalities, spinal malformations, hearing impairment and additional physical findings 5. This condition is sometimes referred to as ARCS which is short for Azoospermia, Renal anomalies and Cervicothoracic Spine dysplasia 5. The relationship, if any, between ARCS and MRKH syndrome remains unsolved 5. However, rare cases of ARCS and MRKH in the same family have been reported, making both syndromes likely to be of identical genetic origin 5.
The exact cause of MRKH syndrome remains largely unknown and the use of various genomic techniques has allowed the identification of promising recurrent genetic abnormalities in some patients 18, 1, 2, 19, 20. To date, seven deletions and one duplication of chromosomal segments have been identified in several persons affected by MRKH syndrome. These anomalies have been found independently in different persons (i.e., one and only one of these chromosomal anomalies per person). These anomalies are of varying length and can contain one gene or many different genes. This has allowed researchers to hypothesize the involvement of certain genes, which are called candidate genes. The possible genes implicated and the most reported chromosomal regions are: RBM8A gene (1q21.1 or band 21.1 on the long arm of chromosome 1), WNT4 gene (1p31-1p35 or band 31 to 35 on the short arm of chromosome 1), HOXA gene (7p15.3 or band 15.3 on the short arm of chromosome 7), TBX6 gene (16p11 or band 11 short arm of chromosome 16), LHX1 and HNF1B genes (17q12 or band 12 long arm of chromosome 17), 22q11.21, and Xp22 19. Table 1 below presents the chromosomal regions most commonly implicated in MRKH syndrome and congenital uterine anomalies and the suspicious genes involved, as indicated by animal and human studies 19. Table 1 also presents the clinical features associated with defects in the respective genetic locations, the main results of non-human studies regarding these chromosomal regions, and whether they are linked with Type 1 or Type 2 MRKH. These new data demonstrate the genetic origin of the MRKH syndrome. They also show that several different genes defects can cause MRKH syndrome. In this case, MRKH syndrome can be considered as of multigenic origin, meaning that different genes can independently be responsible for the syndrome.
The diagnosis of MRKH syndrome is often made during adolescence when teens and women with MRKH syndrome seek a physician’s care is because they fail to menstruate by age 16 (primary amenorrhea) 6. Median age at referral has been reported to be 17.5 years 14. The first physician most teens see is a local family physician or gynecologist. Most medical doctors have never heard of MRKH, and this lack of knowledge can prolong the time to diagnosis 6. In the USA, women with MRKH syndrome will most likely be referred to several specialists at the nearest teaching hospital. Most patients see at least a geneticist and a gynecologist; some will also see an endocrinologist, a surgeon and possibly a urologist. A few clinics also offer the patient the option to see a clinical psychologist who specializes in reproductive issues. The patient will be examined by these specialists, and an ultrasound, MRI or laproscopic surgery is usually done during this time. This process can take weeks or months, and can be very stressful, traumatic and invasive to the patient 6.
The treatment of MRKH syndrome includes non-invasive vaginal dilations recommended as first-line therapy or, in case of failure or non-compliance with treatment, surgery to create a neovagina (a vagina that is created through surgical procedures) 21, 19. Concerning the fertility of women with MRKH syndrome, gestational surrogacy is an option (but prohibited in most countries); however, the successful clinical trial of uterus transplantation by a Swedish team followed by the first live-birth in September, 2014 in Gothenburg, proofed the first available fertility treatment in MRKH syndrome and uterus transplantation is now being performed in other countries around the world allowing women with MRKH syndrome to carry their own child and achieve biological motherhood 2.
MRKH syndrome cause
The cause of MRKH syndrome is unknown 18, 1, 2. Changes in several genes that are involved in development before birth have been identified in females with MRKH syndrome 2, 19, 20. The possible genes implicated and the most reported chromosomal regions are: RBM8A gene (1q21.1 or band 21.1 on the long arm of chromosome 1), WNT4 gene (1p31-1p35 or band 31 to 35 on the short arm of chromosome 1), HOXA gene (7p15.3 or band 15.3 on the short arm of chromosome 7), TBX6 gene (16p11 or band 11 short arm of chromosome 16), LHX1 and HNF1B genes (17q12 or band 12 long arm of chromosome 17), 22q11.21, and Xp22 19. Table 1 presents the chromosomal regions most commonly implicated in MRKH syndrome and congenital uterine anomalies and the suspicious genes involved, as indicated by animal and human studies 19. Table 1 also presents the clinical features associated with defects in the respective genetic locations, the main results of non-human studies regarding these chromosomal regions, and whether they are linked with Type 1 or Type 2 MRKH. These new data demonstrate the genetic origin of the MRKH syndrome. They also show that several different genes defects can cause MRKH syndrome. In this case, MRKH syndrome can be considered as of multigenic origin, meaning that different genes can independently be responsible for the syndrome.
The reproductive abnormalities of MRKH syndrome are due to incomplete development of the Müllerian duct. This structure in the embryo develops into the uterus, fallopian tubes, cervix, and the upper part of the vagina. The cause of the abnormal development of the Müllerian duct in affected individuals is unknown. Originally, researchers suspected that MRKH syndrome was caused by environmental factors during pregnancy, such as medication or maternal illness. However, subsequent studies have not identified an association with any specific maternal drug use, illness, or other factor. Researchers now suggest that in combination, genetic and environmental factors contribute to the development of MRKH syndrome, although the specific factors are often unknown.
It is also unclear why some affected individuals have abnormalities in parts of the body other than the reproductive system. Certain tissues and organs, such as the kidneys, develop from the same embryonic tissue as the Müllerian duct, and researchers suspect that problems during development could affect these organs as well.
Table 1. The most common genes and chromosomal regions associated with MRKH syndrome
| Chromosome Location | Suspected Genes Involved | Associated Syndromes | Non-Humans Study | MRKH syndrome Phenotype |
|---|---|---|---|---|
| 1q21 | RBM8A | TAR syndrome (thrombocytopenia, absence of the radius) 22, 23, 24, 25 | Drosophila melanogaster: RBM8A encodes Y14 protein, which affects oocyte differentiation and determination of primordial germ cells 26 | Type 1 + 2 |
| 16p11.2 | TBX6 | Autism spectrum disorders, neurological disorders, unaffected persons 25 | Mouse models: Deletion of TBX6 presents skeletal (mainly vertebral) and urinary tract malformations 27, 28 | Type 1 + 2 |
| 17q12 | LHX1 | Anomalies in the embryogenesis, in body axis formation 25, 29 | Mouse model:
Mouse model: | Type 1 + 2 |
| HNF1B | Renal cysts and diabetes 25 | Mouse models:
| ||
| 22q11 | Uncertain (TBX1) | DiGeorge or Velocardiofacial syndrome (heart defects, hypocalcemia, immunodeficiency, typical facial malformations, cognitive and behavioral disorders) | Type 1 + 2 |
Footnote: The most common genes and chromosomal regions associated with MRKH syndrome, their associated clinical presentation, animal studies of these genes, and phenotype of MRKH related to defects in these genes.
[Source 19 ]Inheritance pattern
Evidence of the inheritance pattern of MRKH syndrome remains scarce, due to the fertility restrictions of MRKH syndrome patients in the past; hence, no family trees were available to study 34, 35. The majority of MRKH syndrome occur in females with no history of the disorder in their family.
Less often, MRKH syndrome is passed through generations in families 35, 36. First-degree relatives of MRKH patients seem to have a 1 to 5% risk of congenital uterine anomalies as in most multifactorial disorders 37. The majority of the studies of MRKH syndrome familial cases suggest an autosomal dominant inheritance pattern limited to the female sex, implying that the genetic defect is typically inherited by the father 38. Autosomal dominant inheritance means that one copy of the altered gene in each cell is typically sufficient to cause the disorder, although the gene involved is usually unknown. Wolffian duct hypoplasia, or agenesis, and other defects such as kidney abnormalities, hearing impairment, and skeletal deformities have been reported in the males of these families, similar to MRKH patients 35, 36, 39. Another finding of note is that some medical literatures refer to the presence of discordant monozygotic twins with MRKH syndrome, implying that environmental factors, i.e., epigenetic changes, may play a role in gene expression affecting Müllerian duct development 40, 41, 42, 43, 44, 45, 46, 47, 48.
MRKH syndrome symptoms
The symptoms of MRKH syndrome vary greatly from one woman to another. It is important to note that affected individuals may not have all of the symptoms discussed below. Affected individuals should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
Mayer-Rokitansky-Küster-Hauser syndrome signs and symptoms of vaginal and uterine agenesis often go unnoticed until girls reach their teens, but don’t menstruate (amenorrhea). Some women have monthly cramping or abdominal pain.
Mayer-Rokitansky-Küster-Hauser Type 1
This form of MRKH syndrome is also known as isolated Mullerian aplasia, or Rokitansky sequence 5. The disorder is characterized by the failure of the uterus and the vagina to develop properly. The severity of MRKH syndrome type 1 may vary greatly from one person to another. In most cases, the uterus and/or the vagina have not developed (aplasia); in other rare cases, there may be narrowing (atresia) of the upper portion of the vagina and an underdeveloped or rudimentary uterus. In some cases, the Fallopian tubes may be affected as well. The ovaries of females with MRKH syndrome are unaffected and function normally.
In most cases, the initial symptom of MRKH syndrome type 1 is the failure to begin menstrual cycles (primary amenorrhea). Despite amenorrhea, affected females do experience normal secondary sexual development including breast development, the growth of hair under the arms and in the pubic area, and an increase in body fat around the hips and other areas. Sex steroid levels, female sexual identification, and level of sexual desire (libido) are all also normal. However, because of the absence of the uterus and properly developed fallopian tubes, all affected women are unable to bear children (infertile). Many affected females also experience difficulty while attempting sexual intercourse due to the shortness of the vagina. Some women may also experience pain during intercourse.
MRKH syndrome type 1 is sometimes referred to as Mullerian aplasia because the Mullerian ducts are a dual structure within a growing embryo that ultimately develops into the uterus, Fallopian tubes, cervix and the upper portion of the vagina. It is believed that improper development of tissues derived from the Mullerian ducts occurring during embryogenesis, ultimately causes the symptoms of MRKH syndrome.
Mayer-Rokitansky-Küster-Hauser Type 2
When the abnormalities that characterize MRKH syndrome type 1 occur in association with additional physical findings, the disorder is classified as MRKH syndrome type 2 or Mullerian duct aplasia, Renal dysplasia and Cervical Somite anomalies or short for MURCS association 5. The most common abnormalities associated with MRKH syndrome type 2 are failure of the kidneys to development properly (renal adysplasia) and various skeletal malformations, mainly vertebral. Much less frequent defects include heart malformations and hearing impairment.
Women with MRKH syndrome type 2 may exhibit absence of a kidney (unilateral renal agenesis), malformation of one or the two kidneys (renal dysplasia), underdeveloped (hypoplastic) kidneys and/or improper positioning within the body of one or both kidneys (renal ectopia). Other kidney malformations include pelvic kidney, duplex kidney, and horseshoe kidney 2. Kidney abnormalities can cause growth deficiency, kidney stones, an increased susceptibility to urinary tract infections and abnormal accumulation of urine in the kidney due to obstruction (hydronephrosis).
Kidney malformations are the most frequent extragenital abnormalities in MRKH syndrome occurring in about 30 to 40% in European cohorts 2. Interestingly, Deng et al. 17 reported a lower prevalence of kidney malformations of only 13% in their Chinese cohort suggesting the possibility of inter-ethnic phenotypical variations in MRKH syndrome from European patient cohorts. Pan et al. 49 reported an even lower prevalence of 5% in their Chinese cohort but that study did not include information on the extent of renal examinations performed, which could imply an underestimation of the prevalence. Absence of a kidney (unilateral renal agenesis) is the most frequent anomaly accounting for around half of all kidney malformations associated with MRKH syndrome 2. Notably, absence of a kidney (unilateral renal agenesis) is often associated with complete absence of the ipsilateral Müllerian duct which suggests a close relationship between early kidney and Müllerian duct development 50.
Skeletal malformations are the second most frequent extragenital manifestations in MRKH syndrome type 2 affecting around approximately 10 to 40% depending on the extent of examinations performed and anomalies included 2. Skeletal malformations typically involve the bones (vertebrae) in the spinal column within the neck (cervical vertebrae) and the upper back (thoracic vertebrae) may develop improperly (dysplasia). As a result, some of the vertebrae within the neck may be missing and/or fused, causing shortness of the neck, limited neck motion, and an abnormally low hairline (Klippel-Feil syndrome). In addition, affected females may exhibit asymmetric, fused or wedge vertebrae; malformed or missing ribs; abnormal sideways curvature of the spine (scoliosis); elevation of the shoulder blade (scapula), due to the scapula’s failure to move into the appropriate position during fetal development (Sprengel deformity). Abnormalities of the head and face may also occur including an abnormally small jaw (micrognathia), cleft lip, cleft palate and underdevelopment of one side of the face causing facial asymmetry.
Some affected women develop hearing loss due to the failure of sound waves to be conducted through the middle ear (conductive hearing loss), usually due to structural abnormalities of the middle ear (e.g. external meatus atresia, stapedial ankylosis). Hearing loss may also be due to impaired ability of the auditory nerve to transmit sensory input to the brain (sensorineural hearing loss). The degree of hearing impairment may vary. The ears may be malformed (dysplastic) in some cases. When the ears are involved the disorder may be referred to as genital renal ear syndrome (GRES). Strübbe et al. 51 performed a systematic evaluation of associated anomalies with ear, nose, and throat assessment and reported ear abnormalities in 11% of patients, which could indicate an underestimation.
In rare cases, some females with MRKH syndrome type 2 have had additional physical abnormalities including abnormalities of the hands and/or arms and heart malformations. Abnormalities of the extremities may include absence of a portion of one or more fingers or toes (ectrodactyly), webbing of the fingers or toes (syndactyly), duplicated thumb and absence of the long, thin bone of the forearm (absent radius). Heart abnormalities are reported in < 5% of patients (e.g. pulmonary valve stenosis, atrial septal defect). Heart malformations may include “a hole in the heart” between the two upper chambers of the heart (atrial septal defects), narrowing of the pulmonary valve (pulmonary valvular stenosis) or tetralogy of Fallot, a rare grouping of four different heart defects.
In a small number of patients, MRKH syndrome has been reported with a very severe phenotype: vertebral defect, anal atresia, cardiac malformations, tracheoesophageal fistula or esophageal atresia, renal defect, and limb defect (VACTERL association), while even fewer have been reported with isolated anorectal malformations 52, 53, 14.
Psychological and psychosexual issues in MRKH syndrome
The diagnosis of MRKH syndrome may have profound psychological and/or psychosexual impact and it is a hallmark in the management to counsel the patient and support mental health upon the diagnosis and onwards in life 54, 55, 56. Receiving the diagnosis of MRKH syndrome, many patients experience facing overwhelming issues regarding identity, sexuality and infertility, and the importance of good caring and counselling should not be underestimated 2. The diagnosis of MRKH syndrome is often made during adolescence; a period of sensitive physical and emotional development and vulnerability, which further imposes the physician’s caring and awareness towards the patients’ emotions, reactions and coping strategies. Furthermore, it is important to be aware of potential cultural aspects and their influence on reactions to the diagnosis in patients and their families and peers.
Knowledge of the psychological aspects in MRKH syndrome remains limited but studies have agreed on presence of higher levels of psychological distress in patients compared with women without MRKH syndrome 57, 58. Bargiel-Matusiewicz et al. 59 reported increasingly higher neuroticism following diagnosis as well as low level of problem-focused coping style. Ernst et al. 60 reported on motivators and barriers to disclosure, emotions receiving the diagnosis and its impact onwards in life. Patterson et al. 61 identified four major themes having MRKH syndrome: hindered independence in relation to the mother’s involvement in the care, feelings of being different, difficulties managing intimacy, and the threat to female identity.
Many patients experience difficulties talking about feelings and emotions related to MRKH syndrome. Therefore, patients should be encouraged to gain trust with someone close (parents, partner, friend) with whom thoughts can be shared 60. Sharing thoughts and emotions with other patients can be valuable and group programs for women with MRKH syndrome have been reported to decrease psychological distress 62, 63. Patients should also be encouraged to connect to a peer support group allowing them to learn from other patients’ experiences. There are several support groups established worldwide 64, 65.
Sexuality and sexual activity are complex concepts which should be viewed in the broadest senses and not merely limited to the mechanistic ability to engage penetrative intercourse 66. The World Health Organization (WHO) suggests a holistic approach describing sexuality in biopsychosocial terms. In this view, it is important to recognize the patient’s possibilities and abilities to reach good self-esteem and well-being, establish sexual relations, and empower the patient with coping capabilities to navigate through the psychosexual challenges which may follow having MRKH syndrome 58. While recognizing the challenge for any health-care professional to provide sufficient counselling and support, referral to experts in genital malformations and/or disorders of sexual development, sexologists or psychologists is often relevant and should always be considered.
MRKH syndrome diagnosis
In most cases, females with MRKH syndrome come to the attention of physicians due to the failure of menstrual cycles to begin during puberty (primary amenorrhea). Some may seek medical attention due to fertility problems. In rare cases, multiple congenital malformations and/or symptoms caused by renal abnormalities may lead to a possible diagnosis of MRKH syndrome type 2.
A diagnosis is made based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests such as specialized imaging techniques. Transabdominal ultrasonography must be the first investigation. It may be complemented by magnetic resonance imaging (MRI). An ultrasound records echoes of high-frequency sound waves to produce a detailed image of deep structures within the body. An ultrasound can depict the uterus and vagina. It can also be used to evaluate the kidneys. An ultrasound is a simple, noninvasive procedure that lacks radiation. An MRI uses a magnetic field and radio waves to produce cross-sectional images of particular organs and bodily tissues. It is also noninvasive and is generally more sensitive than an ultrasound. In addition to evaluating the uterus and vagina, an MRI can simultaneously be used to evaluate the kidney and skeleton.
Karyotyping may be performed to rule out other conditions. Karyotyping is used to examine the chromosomes in a sample of cells. Females with MRKH syndrome have a normal 46, XX karyotype.
Establishing an accurate diagnosis of MRKH syndrome also requires the search for other eventually associated malformations, and will also include some biological tests necessary for the differential diagnosis. Once MRKH syndrome is diagnosed, a full check-up must be undertaken to search for associated malformations. Since renal and skeletal abnormalities may not be symptomatic, it is necessary to perform at least a transabdominal ultrasonography and spinal radiography. In case of suspicion of hearing impairment and/or a cardiac anomaly, complementary audiogram and/or heart echography must also be carried out.
In women diagnosed for MRKH syndrome, levels of FSH (plasmatic follicle stimulating hormone), LH (luteinizing hormone) and 17ß-oestradiol are normal, proving the integrity of ovarian function. There is no biological hyperandrogenism, as shown by a normal plasmatic level of testosterone. Hyperandrogenism is the term for the excessive secretion of male sex hormones (androgens) and is caused by a variety of ovarian and adrenal diseases.
Table 2 summarizes the routine diagnostic work-up and typical findings in MRKH syndrome 2. Patients with failure to menstruate by age 16 (primary amenorrhea) should be referred to a gynecology department or a gynecologist with expertise in pediatric/adolescent gynecology or disorders in sex development (DSD) for examination. Physical examination is carried out which may include examination of external genitalia and examination of the introitus/vagina with respect to the patient’s age and motivation (should be avoided in prepubertal adolescents). Transperineal or transabdominal ultrasonography (US) is performed revealing the absence of the uterus and presence of ovaries. Magnetic resonance imaging (MRI) of the internal genitalia is considered the golden standard method for the diagnosis of uterovaginal agenesis in MRKH syndrome and should always be performed when available. MRI is non-invasive and superior to computed tomography (CT) in showing the Müllerian structures in detail (uterine remnants or complete agenesis) including the presence of endometrium in uterine remnants. MRI also shows the ovaries and extragenital malformations, and has high interrater agreement with laparoscopy 67, 50. Therefore, laparoscopy is rarely indicated for diagnostic purposes only, but may be relevant in patients with pain-causing uterine remnants where surgical removal of the tissue is needed 54. Examination of the kidneys by ultrasonography (US) or MRI should be performed to screen for kidney malformations (prevalence of ~ 30%) 50, 14. Detailed screening of other typical extragenital anomalies (skeletal, ear, cardiac etc.), by imaging and otorhinopharyngeal assessment, is not done routinely but should be considered in case of relevant complaints or findings at the physical examination.
More studies are still needed to conclude on the relevance of a full screening of extragenital abnormalities for all patients and its recommendation in clinical practice. Chromosomal analysis by G and Q-banding is often performed to confirm normal female karyotype (46,XX). Chromosomal microarray analysis can be considered for the detection of copy number variations but is not obligate for the diagnosis. Overall test positive rate for imbalances has been reported at about 16 to 20% but the interpretation of the pathogenicity of these findings remains a challenge 3, 68, 69. Other relevant laboratory tests include follicle stimulating hormone (FSH), luteinizing hormone (LH), androgens and estradiol, which are generally considered to be normal in MRKH syndrome 14, 70. However, biochemical (non-clinical) hyperandrogenemia was recently reported in about 50% of patients 71, but this finding requires further validation.
Table 2. Routine diagnostic work-up in MRKH syndrome
| Examination | Typical findings |
|---|---|
| Physical examination including a precautious pelvic exam by an experienced pediatric/adolescent gynecologist. | Normal height, secondary sex characteristics, and hair growth. Normal external genitalia. Short blind-ending vagina (0–3 cm) with no cervix at the apex. No uterus detected by manual palpation. |
| Radiologic examination | |
| US of internal genitalia (transvaginal/−perineal) a | No uterus or vaginal canal. Two functional ovaries. |
| Pelvic MRI scan | Confirms the diagnosis. Determines the presence of rudimentary uterine buds or complete uterovaginal agenesis |
| Renal scan (by ultrasonography or MRI) | Renal abnormalities are found in approximately 30% of patients |
| Consider examinations for other associated malformations (e.g. EOS scan, otorhinopharyngeal assessment and echocardiography | Various skeletal malformations (axis and limbs), hearing impairment and congenital heart defects (rare). |
| Biochemical analysis | |
| Gonadotropins (FSH, LH) | Normal levels following menstrual cycle |
| Estradiol | Normal levels |
| Androgen status | Normal female levels |
| Chromosomal analysis (can be used to differentiate from 46,XY DSDs) | 46,XX |
Footnote: aTransabdominal ultrasonography should be considered in younger patients
Abbreviations: FSH = follicle stimulating hormone; LH = luteinizing hormone; MRI = magnetic resonance imaging; US ultrasonography; DSD = disorders in sex development
[Source 2 ]MRKH syndrome treatment
The treatment of MRKH syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Depending upon the affected individual’s age at diagnosis, pediatricians or internists, gynecologists, kidney specialists (nephrologists), endocrinologist, orthopedic surgeons, plastic surgeons, physical therapists, psychiatrists and other health care professionals may need to work together to ensure a comprehensive approach to treatment.
Women with MRKH syndrome are encouraged to seek counseling after a diagnosis and before treatment because the diagnosis can cause anxiety and extreme psychological distress. Psychological support and counseling both professionally and through support groups is recommended for affected females and their families.
Treatment will usually include appropriate management of the physical findings associated with MRKH syndrome and psychological support for the emotional issues that often accompany the diagnosis.
The treatment of vaginal aplasia consists of creating a neovagina for sexual intercourse. This should be proposed to the women when they are emotionally mature and ready to start sexual activity. Treatment may be either nonsurgical or surgical. Nonsurgical techniques are considered the first-line approach. Vaginal dilators are specially designed plastic tubes that are used to help enlarge or create a vagina. The most common method is known as Franck’s dilator method. With this method, a physician (and then woman herself) applies a vaginal dilator, which progressively stretches and widens the vagina. This daily procedure may be continued for up to six weeks to several months.
Plastic surgery may be necessary to create an artificial vagina (vaginoplasty). There are a variety of different surgical techniques that may be used and there is no consensus as to which technique is best. Females who undergo surgery to create an artificial vagina will most likely need to use vaginal dilators after the surgery to enhance the chance of success.
Because females with MRKH syndrome do not have a functional uterus, they cannot bear children (infertile). However, some affected women have been able to have a child by using in vitro fertilization of their own eggs and surrogate pregnancy. However, because MRKH syndrome appears to be of genetic origin, the risk of passing on the disease to children exists and any decision to conceive should therefore be undertaken after careful consultation with their physicians and appropriate medical personnel.
Females with MRKH syndrome who exhibit absence of one kidney (unilateral renal agenesis) may have an increased susceptibility to urinary tract infections and/or kidney stones (renal calculi). Physicians should carefully monitor affected females for infection and prescribe antibiotics as necessary. Skeletal abnormalities may also require reconstructive surgery, physical therapy, and/or other medical management depending upon the specifics and severity of the bone deformities.
Vaginal dilation
The most commonly used non-invasive method is vaginal self-dilation also known as Frank’s method 72. In Frank’s method, progressive dilators are manually placed on the vaginal apex for 10 to 30 minutes one to three times a day. Thorough counseling to ensure patient readiness and firm instructions in the technique should always precede start of self-dilation. Therapeutical education throughout the treatment is pivotal to achieve success and the patient should be followed closely in order to monitor progress and provide good support and guidance. An alternative non-invasive option is vaginal dilation by intercourse also known as d’Alberton’s method 73 which has been reported with good anatomical and functional outcome compared with self-dilation and surgery 74, 75. d’Alberton’s method (vaginal dilation by intercourse), however, requires regular sexual intercourse with a partner and therefore, this approach is not an option for all patients. Importantly, the three latter methods, which are based on dilation of the vaginal dimple, will provide the vagina with a normal mucosal lining. This may be advantageous in a uterus transplantation situation since this will provide the vagina with a normal vaginal microbiota, which may be of importance for success at embryo transfer as well as for correctly grade rejection by cervical biopsy.
Disadvantages in vaginal self-dilation therapy include the risk of low compliance (especially in younger patients), time consume needed for a satisfactory result, the discomfort that some patients experience, and a low risk of urethral dilation 74, 76.
Since 2002, the American College of Obstetricians and Gynecologists (ACOG) has recommended vaginal self-dilation therapy as first line treatment based on the high overall success rate (90–96%), being non-invasive with a low complication rate, and low costs 54, 77, 78, 79. Due to the risk of low compliance using dilators, the treatment should be supervised and followed by a health professional experienced in this therapy 2. The American College of Obstetricians and Gynecologists recommend that surgery should be reserved for patients experiencing failure with vaginal self-dilation therapy and emphasizes that surgery still requires post-surgical vaginal self-dilation to avoid strictures. Dilation therapy as first choice is also supported by Callens et al. 76, which further suggest laparoscopic Vecchietti vaginoplasty as preferred second-line therapy. Most importantly, thorough counseling regarding expected outcome and possible complications should always precede any attempt for vaginal construction, and it is fundamental to ensure the full maturity and motivation of the patient undergoing such treatment. Moreover, some women choose no treatment, which for some patients might be the right choice 80.
Despite of the accumulated literature and the American College of Obstetricians and Gynecologists (ACOG) recommendation, vaginal self-dilation as first-line therapy for vaginal agenesis is still not widely accepted by surgical experts in the field 2. The rarity of the MRKH syndrome implies that most treating centers only acquire expertise and thus preference for a single procedure. This may result in reporting and publication biases in the available literature concerning outcome and complications and it complicates the initiation of comparative studies including both surgical and non-surgical approaches. Furthermore, most studies differ in terms of follow-up and measuring outcome 76. Only few comparative studies have been reported to date and those studies comparing vaginal self-dilation therapy with surgical procedures generally conclude vaginal self-dilation therapy to be non-inferior to surgery 81, 82, 83, 74, 84, 85, 53. Another common limitation in most previous studies is the focus only on anatomical outcome (neovaginal depth and width) instead of functional outcome and patient satisfaction 86. In conclusion, prospective studies based on therapeutical education within multidisciplinary expert teams using standardized outcome measurements of both anatomical and functional results and complications with long-term follow-up are needed in order to ascertain these preliminary reports for future clinical recommendations.
Infertility treatment
Women with MRKH syndrome belong to the group of females with absolute uterine factor infertility, which comprise those with anatomical absence of a uterus or presence of a non-functional uterus, in terms of implanting an embryo and carrying a pregnancy 2. Other groups of absolute uterine factor infertility that lack a uterus, apart from MRKH syndrome, are women hysterectomized during fertile life because of malignancy (mainly cervical cancer), benign disease (leiomyoma) or as a life-saving procedure because of massive obstetrical bleeding 2. Women with intrauterine adhesions, large inoperable leiomyoma, or some specific uterine malformations also belong to the group of women with absolute uterine factor infertility 2.
The motherhood options for women with MRKH syndrome, and other causes of absolute uterine factor infertility, have traditionally been legal adoption, as international or national adoption. Since the mid-1990s gestational surrogacy (gestational surrogacy), has been an alternative to gain genetic motherhood, and after adoption from the birth-giving mother, also legal motherhood. In this procedure, in vitro fertilization (IVF) is first performed with oocytes from the MRKH woman and her partner’s sperm. An embryo is then placed inside the womb of another women, the gestational surrogate carrier. The gestational surrogacy arrangement may be commercial or altruistic (typically with a close relative (mother, sister) as the carrier) depending on jurisdiction in the specific nation/state, as exemplified by the difference in arrangements in California, with mainly commercial gestational surrogacy, and Canada with exclusively altruistic gestational surrogacy 87. Gestational surrogacy is not allowed in the Nordic countries and in most other parts of the world, the reasons being ethical, religious or legal or a combination of these 88. However, it is well-known that gestational surrogacy is used by many women with MRKH syndrome, residing in countries with non-approval of gestational surrogacy, and concerning Northern Europe the main countries for reproductive tourism concerning gestational surrogacy seem to be USA, Ukraine and Georgia. Until recently India was the main gestational surrogacy country for Nordic couples with female absolute uterine factor infertility, but India has now banned commercial gestational surrogacy 89.
MRKH surgery
MRKH surgical procedures include vaginoplasties using various autografts such as McIndoe vaginoplasty (split-skin graft covering a mold placed in the dissected pouch between the rectum and bladder), Baldwin vaginoplasty (bowel graft), Davydov vaginoplasty (peritoneal graft), and Williams vulvavaginoplasty (labia majora flaps) 90, 91, 92, 93. More recently, vaginoplasties using cultured autologous vulvar tissue 94, 95 and tissue-engineered biomaterial 96 have been suggested. An alternative surgical procedure is the laparoscopic Vecchietti vaginoplasty in which a surgical traction device is placed on the anterior abdominal wall with subperitoneal threads attached to a mold in the vagina 97, 98. It is important to note that there is no quick solution to obtain a functional vagina and surgical options still require continued postoperative vaginal dilation by regular sexual intercourse or vaginal dilators, to ensure satisfactory long-term outcome 2.
In general, disadvantages of the surgical methods are the invasiveness and need of anesthesia, the risk of neovaginal strictures requiring onwards dilation following surgery as well as specific complications related to different graft tissues.
Uterus transplantation
Uterus transplantation has now emerged as the first true infertility treatment for women with MRKH syndrome and giving them full (gestational, genetic, legal) motherhood from start 2. The group in Gothenburg, Sweden, initiated basic uterus transplantation research in animal models already in 1999. The idea to uterus transplantation was given to a young woman with cervical cancer who would undergo a radical hysterectomy 99. Through structured animal-based research 100 and continuous ethics discussions 101 within the research group in Sweden the team optimized the surgical technique, immunosuppression for a uterine graft, rejection diagnosis as well as assuring normal development of the offspring from allogeneic uterus transplantation 102. In 2012, the Swedish team received permission from the ethics board as well as permission from the hospital board to perform a prospective observational study of live donor uterus transplantation, including up to ten procedures 2. The surgeries, performed in 2013, included eight MRKH syndrome patients and one cervical cancer patient as recipients. The donors were five mothers, one sister, one maternal aunt, one mother-in-law and one family friend 103. The surgical outcome of the nine uterus transplantation procedures was that donor surgery was extremely difficult, with surgical durations of 10 to 13 hours, but that the transplantation procedures in the recipients were simpler, with surgical durations of 4 to 5 hours 103. Seven out of nine procedures were surgically successful, with viable grafts showing regular menstruations during the first post-transplantation year. Two out of the seven grafts had to be surgically removed during the initial months, because of vascular thrombosis in one case and intrauterine infection in the other case 103.
The proof-of-concept of uterus transplantation as an infertility treatment to women with MRKH came with the world’s first livebirth after uterus transplantation which took place in September, 2014 in Gothenburg, Sweden 104. Patient number 5, in the cohort of nine women in the original Swedish uterus transplantation study 103 had MRKH syndrome with also unilateral kidney agenesis. She had acquired a neovagina through self-dilation and underwent uterus transplantation at age 35 years, receiving a uterine graft from a 61 year-old, 2-parous, family friend that had been postmenopausal for 7 years before uterus donation. One year after uterus transplantation she underwent transfer with a single embryo and achieved pregnancy, which continued essentially uneventful until the recipient acquired preeclampsia in gestational week 31 + 5, with cesarean section performed the following day. A healthy boy was delivered and his development since then has been normal.
This birth was followed by seven more live births in Sweden 105, 106, until the ninth uterus transplantation baby in the world was born in USA in December 2017, after a live donor uterus transplantation procedure 107. There also exist several births after deceased donor uterus transplantation 108. Today, approximately 75 uterus transplantation procedures have been performed and all but two of these have been performed in MRKH patients 2. Around 25 babies have been born worldwide, with some of the MRKH patients having delivered healthy babies twice 2.
The uterus transplantation surgery of the MRKH women is generally preformed through a sub-umbilical midline incision 2. The vaginal vault is dissected free from the bladder and the rectum and this is possible through cleavage of the uterine remnant in the midline. The external iliac arteries and veins are then dissected free and cleaned from surrounding tissue, to allow for anastomosis 2. Brännström and his team usually also attach fixation sutures to the sacrouterine ligaments, round ligaments and to the cardinal ligaments. The chilled and flushed uterus is then coming from the back-table into the pelvis. End-to-side anastomoses are then performed with the internal iliac segments on the uterine vessels of the graft to the external iliac vessels of the recipient. After reperfusion, the vagina is opened to perform end-to-end vaginal anastomosis, which then is followed by fixation of the uterus to the ligaments.
It is important to point out that already at this early stage of human uterus transplantation, which is still at an experimental phase, the procedure has proved to be an effective fertility treatment with a take-home-baby rate above 80%, in patients with graft survival of more than six months after uterus transplantation 2. It is likely that the efficiency of uterus transplantation will become even greater in the future, in line with the advancement of other medical procedures. Another essential aspect of uterus transplantation, in comparison to other types of solid organ transplants, is that uterus transplantation is the first ephemeral transplantation, where immunosuppression is only for a restricted time, and the medication can be withdrawn when the graft is removed 2. This is typically accomplished within 5 years, when the transplanted woman has given birth to the desired number of children 2. Therefore, the negative, long-term side effects of immunosuppression using calcineurin inhibitors, such as nephrotoxicity, can be avoided. This is important in the context of MRKH syndrome, where several patients have single kidneys.
The immunosuppression used for the transplanted patients is standard induction therapy, similar to what is used for kidney transplantation. The mainstay of maintenance immunosuppression has been the calcineurin inhibitor, tacrolimus 106.
In the future it is likely that minimal invasive surgery, by robotic-assisted laparoscopy, will be the main surgical approach for live donor surgery and some years later also for recipient surgery in uterus transplantation 109. Advantages for the patients will be less tissue trauma, with possibility to decrease the length of hospital stay to 1–2 days and sick-leave to 1–2 weeks.
References- Kyei-Barffour I, Margetts M, Vash-Margita A, Pelosi E. The Embryological Landscape of Mayer-Rokitansky-Kuster-Hauser Syndrome: Genetics and Environmental Factors. Yale J Biol Med. 2021 Dec 29;94(4):657-672. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8686787
- Herlin MK, Petersen MB, Brännström M. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis. 2020 Aug 20;15(1):214. doi: 10.1186/s13023-020-01491-9
- Williams LS, Demir Eksi D, Shen Y, Lossie AC, Chorich LP, Sullivan ME, Phillips JA 3rd, Erman M, Kim HG, Alper OM, Layman LC. Genetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families. Fertil Steril. 2017 Jul;108(1):145-151.e2. doi: 10.1016/j.fertnstert.2017.05.017
- Reindollar RH, Byrd JR, McDonough PG. Delayed sexual development: a study of 252 patients. Am J Obstet Gynecol. 1981 Jun 15;140(4):371-80. doi: 10.1016/0002-9378(81)90029-6
- Mayer-Rokitansky-Küster-Hauser Syndrome. https://rarediseases.org/rare-diseases/mayer-rokitansky-kuster-hauser-syndrome
- Medical Information about MRKH. https://www.beautifulyoumrkh.org/medical-information.html
- Tsitoura A., Michala L. The Sexuality of Adolescents and Young Women with MRKH Syndrome: A Qualitative Study. J. Sex. Med. 2021;18:2012–2019. doi: 10.1016/j.jsxm.2021.09.006
- Gestational Surrogacy Fact Sheet. https://health.ny.gov/community/pregnancy/surrogacy/gestational_surrogacy_fact_sheet.htm
- About Surrogacy. https://surrogate.com/about-surrogacy/types-of-surrogacy/what-is-gestational-surrogacy
- Oppelt PG, Lermann J, Strick R, Dittrich R, Strissel P, Rettig I, Schulze C, Renner SP, Beckmann MW, Brucker S, Rall K, Mueller A. Malformations in a cohort of 284 women with Mayer-Rokitansky-Küster-Hauser syndrome (MRKH). Reprod Biol Endocrinol. 2012 Aug 20;10:57. doi: 10.1186/1477-7827-10-57
- Herlin M., Bjørn A.-M.B., Rasmussen M., Trolle B., Petersen M.B. Prevalence and patient characteristics of Mayer–Rokitansky–Küster–Hauser syndrome: A nationwide registry-based study. Hum. Reprod. 2016;31:2384–2390. doi: 10.1093/humrep/dew220
- Aittomäki K., Eroila H., Kajanoja P. A population-based study of the incidence of müllerian aplasia in Finland. Fertil. Steril. 2001;76:624–625. doi: 10.1016/S0015-0282(01)01963-X
- Blontzos N., Iavazzo C., Vorgias G., Kalinoglou N. Leiomyoma development in Mayer-Rokitansky-Küster-Hauser syndrome: A case report and a narrative review of the literature. Obstet. Gynecol. Sci. 2019;62:294–297. doi: 10.5468/ogs.2019.62.4.294
- Herlin M, Bjørn AM, Rasmussen M, Trolle B, Petersen MB. Prevalence and patient characteristics of Mayer-Rokitansky-Küster-Hauser syndrome: a nationwide registry-based study. Hum Reprod. 2016 Oct;31(10):2384-90. doi: 10.1093/humrep/dew220
- Oppelt P, Renner SP, Kellermann A, Brucker S, Hauser GA, Ludwig KS, Strissel PL, Strick R, Wallwiener D, Beckmann MW. Clinical aspects of Mayer-Rokitansky-Kuester-Hauser syndrome: recommendations for clinical diagnosis and staging. Hum Reprod. 2006 Mar;21(3):792-7. doi: 10.1093/humrep/dei381
- Lalatta F, Motta F, Restelli E, Bellini M, Miozzo M, Gervasini C, Dallapiccola B, Gentilin B, Fedele L. Dysmorphologic assessment in 115 Mayer-Rokitansky-Küster-Hauser patients. Clin Dysmorphol. 2015 Jul;24(3):95-101. doi: 10.1097/MCD.0000000000000087
- Deng S, He Y, Chen N, Zhu L. Spectrum of Type I and Type II Syndromes and Associated Malformations in Chinese Patients with Mayer-Rokitansky-Küster-Hauser Syndrome: A Retrospective Analysis of 274 Cases. J Pediatr Adolesc Gynecol. 2019 Jun;32(3):284-287. doi: 10.1016/j.jpag.2018.07.007
- Chu C, Li L, Li S, Zhou Q, Zheng P, Zhang YD, Duan AH, Lu D, Wu YM. Variants in genes related to development of the urinary system are associated with Mayer-Rokitansky-Küster-Hauser syndrome. Hum Genomics. 2022 Mar 31;16(1):10. doi: 10.1186/s40246-022-00385-0
- Triantafyllidi VE, Mavrogianni D, Kalampalikis A, Litos M, Roidi S, Michala L. Identification of Genetic Causes in Mayer-Rokitansky-Küster-Hauser (MRKH) Syndrome: A Systematic Review of the Literature. Children (Basel). 2022 Jun 27;9(7):961. doi: 10.3390/children9070961
- Miao Y, Wen J, Huang L, Wu J, Zhao Z. Diagnosis and Management of Ovarian Tumor in Mayer-Rokitansky-Küster-Hauser (MRKH) Syndrome. Biomed Res Int. 2018 Mar 12;2018:2369430. doi: 10.1155/2018/2369430
- Creighton S., Crouch N., Deans R., Cutner A., Michala L., Barnett M., Williams C., Liao L.-M. Nonsurgical dilation for vaginal agenesis is promising, but better research is needed. Fertil. Steril. 2012;97:e32. doi: 10.1016/j.fertnstert.2012.03.040
- Ledig S., Schippert C., Strick R., Beckmann M.W., Oppelt P.G., Wieacker P. Recurrent aberrations identified by array-CGH in patients with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil. Steril. 2011;95:1589–1594. doi: 10.1016/j.fertnstert.2010.07.1062
- Cheroki C., Krepischi A., Szuhai K., Brenner V., Kim C., Otto P.A., Rosenberg C. Genomic imbalances associated with mullerian aplasia. J. Med. Genet. 2007;45:228–232. doi: 10.1136/jmg.2007.051839
- McGowan R., Tydeman G., Shapiro D., Craig T., Morrison N., Logan S., Balen A.H., Ahmed S.F., Deeny M., Tolmie J., et al. DNA copy number variations are important in the complex genetic architecture of müllerian disorders. Fertil. Steril. 2015;103:1021–1030.e1. doi: 10.1016/j.fertnstert.2015.01.008
- Ledig S., Wieacker P. Clinical and genetic aspects of Mayer–Rokitansky–Küster–Hauser syndrome. Med. Genet. 2018;30:3–11. doi: 10.1007/s11825-018-0173-7
- Parma D.H., Bennett P.E., Boswell R.E. Mago Nashi and Tsunagi/Y14, respectively, regulate Drosophila germline stem cell differentiation and oocyte specification. Dev. Biol. 2007;308:507–519. doi: 10.1016/j.ydbio.2007.06.007
- Nacke S., Schäfer R., Habré de Angelis M., Mundlos S. Mouse mutant “rib-vertebrae” (rv): A defect in somite polarity. Dev. Dyn. 2000;219:192–200. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1046>3.0.CO;2-9
- White P.H., Farkas D.R., McFadden E.E., Chapman D.L. Defective somite patterning in mouse embryos with reduced levels of Tbx6. Development. 2003;130:1681–1690. doi: 10.1242/dev.00367
- Bozzi F., Bertuzzi S., Strina D., Giannetto C., Vezzoni P., Villa A. The Exon–Intron Structure of HumanLHX1 Gene. Biochem. Biophys. Res. Commun. 1996;229:494–497. doi: 10.1006/bbrc.1996.1832
- Kobayashi A., Shawlot W., Kania A., Behringer R.R. Requirement of Lim1 for female reproductive tract development. Development. 2004;131:539–549. doi: 10.1242/dev.00951
- Shawlot W., Behringer R.R. Requirement for LIml in head-organizer function. Nature. 1995;374:425–430. doi: 10.1038/374425a0
- Tsang T. E., Khoo P. L., Jamieson R. V., Zhou S. X., Ang S. L., Behringer R., Tam P. P. (2001). The allocation and differentiation of mouse primordial germ cells.. Int. J. Dev. Biol. 45: 549-555. https://ijdb.ehu.eus/article/11417898
- Coffinier C., Barra J., Babinet C., Yaniv M. Expression of the vHNF1/HNF1β homeoprotein gene during mouse organogenesis. Mech. Dev. 1999;89:211–213. doi: 10.1016/S0925-4773(99)00221-X
- Wottgen M., Brucker S., Renner S., Strissel P., Strick R., Kellermann A., Wallwiener D., Beckmann M., Oppelt P. Higher incidence of linked malformations in siblings of Mayer-Rokitansky-Kuster-Hauser-syndrome patients. Hum. Reprod. 2008;23:1226–1231. doi: 10.1093/humrep/den059
- Fontana L., Gentilin B., Fedele L., Gervasini C., Miozzo M. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin. Genet. 2017;91:233–246. doi: 10.1111/cge.12883
- Herlin M., Højland A.T., Petersen M.B. Familial occurrence of Mayer-Rokitansky-Küster-Hauser syndrome: A case report and review of the literature. Am. J. Med. Genet. Part A. 2014;164:2276–2286. doi: 10.1002/ajmg.a.36652
- Jacquinet A., Millar D., Lehman A. Etiologies of uterine malformations. Am. J. Med. Genet. Part A. 2016;170:2141–2172. doi: 10.1002/ajmg.a.37775
- Shokeir MH. Aplasia of the Müllerian system: evidence for probable sex-limited autosomal dominant inheritance. Birth Defects Orig Artic Ser. 1978;14(6C):147-65.
- De Cássia M., Pavanello R., Eigier A., Otto P.A., Optiz J.M., Reynolds J.F. Relationship between Mayer-Rokitansky-Küster (MRK) anomaly and hereditary renal adysplasia (HRA) Am. J. Med. Genet. 1988;29:845–849. doi: 10.1002/ajmg.1320290414
- Rall K., Eisenbeis S., Barresi G., Rückner D., Walter M., Poths S., Wallwiener D., Riess O., Bonin M., Brucker S. Mayer-Rokitansky-Küster-Hauser syndrome discordance in monozygotic twins: Matrix metalloproteinase 14, low-density lipoprotein receptor–related protein 10, extracellular matrix, and neoangiogenesis genes identified as candidate genes in a tissue-specific mosaicism. Fertil. Steril. 2015;103:494–502.e3. doi: 10.1016/j.fertnstert.2014.10.053
- Lischke JH, Curtis CH, Lamb EJ. Discordance of vaginal agenesis in monozygotic twins. Obstet Gynecol. 1973 Jun;41(6):920-4.
- Steinkampf M.P., Dharia S.P., Dickerson R.D. Monozygotic twins discordant for vaginal agenesis and bilateral tibial longitudinal deficiency. Fertil. Steril. 2003;80:643–645. doi: 10.1016/S0015-0282(03)00758-1
- Heidenreich W, Pfeiffer A, Kumbnani HK, Scholz W, Zeuner W. Diskordante eineiige Zwillinge mit Mayer-Rokitansky-Küster-Syndrom [Disordant monozygotic twins with Mayer Rokitansky Kütser syndrome (author’s transl)]. Geburtshilfe Frauenheilkd. 1977 Mar;37(3):221-3. German.
- Duru U.A., Laufer M.R. Discordance in Mayer-von Rokitansky-Küster-Hauser Syndrome Noted in Monozygotic Twins. J. Pediatr. Adolesc. Gynecol. 2009;22:e73–e75. doi: 10.1016/j.jpag.2008.07.012
- Regenstein AC, Berkeley AS. Discordance of müllerian agenesis in monozygotic twins. A case report. J Reprod Med. 1991 May;36(5):396-7.
- Milsom S.R., Ogilvie C.M., Jefferies C., Cree L. Discordant Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome in identical twins—A case report and implications for reproduction in MRKH women. Gynecol. Endocrinol. 2015;31:684–687. doi: 10.3109/09513590.2015.1032928
- Maniglio P., Ricciardi E., Laganà A.S., Triolo O., Caserta D. Epigenetic modifications of primordial reproductive tract: A common etiologic pathway for Mayer-Rokitansky-Kuster-Hauser Syndrome and endometriosis? Med. Hypotheses. 2016;90:4–5. doi: 10.1016/j.mehy.2016.02.015
- Rall K., Barresi G., Walter M., Poths S., Haebig K., Schaeferhoff K., Schoenfisch B., Riess O., Wallwiener D., Bonin M., et al. A combination of transcriptome and methylation analyses reveals embryologically-relevant candidate genes in MRKH patients. Orphanet J. Rare Dis. 2011;6:32. doi: 10.1186/1750-1172-6-32
- Pan HX, Luo GN. Phenotypic and clinical aspects of Mayer-Rokitansky-Küster-Hauser syndrome in a Chinese population: an analysis of 594 patients. Fertil Steril. 2016 Oct;106(5):1190-1194. doi: 10.1016/j.fertnstert.2016.06.007
- Preibsch H, Rall K, Wietek BM, Brucker SY, Staebler A, Claussen CD, Siegmann-Luz KC. Clinical value of magnetic resonance imaging in patients with Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: diagnosis of associated malformations, uterine rudiments and intrauterine endometrium. Eur Radiol. 2014 Jul;24(7):1621-7. doi: 10.1007/s00330-014-3156-3
- Strübbe EH, Cremers CW, Willemsen WN, Rolland R, Thijn CJ. The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome without and with associated features: two separate entities? Clin Dysmorphol. 1994 Jul;3(3):192-9.
- Rall K, Eisenbeis S, Henninger V, Henes M, Wallwiener D, Bonin M, Brucker S. Typical and Atypical Associated Findings in a Group of 346 Patients with Mayer-Rokitansky-Kuester-Hauser Syndrome. J Pediatr Adolesc Gynecol. 2015 Oct;28(5):362-8. doi: 10.1016/j.jpag.2014.07.019
- Bjørsum-Meyer T, Herlin M, Qvist N, Petersen MB. Vertebral defect, anal atresia, cardiac defect, tracheoesophageal fistula/esophageal atresia, renal defect, and limb defect association with Mayer-Rokitansky-Küster-Hauser syndrome in co-occurrence: two case reports and a review of the literature. J Med Case Rep. 2016 Dec 21;10(1):374. doi: 10.1186/s13256-016-1127-9
- Committee on Adolescent Health Care. ACOG Committee Opinion No. 728: Müllerian Agenesis: Diagnosis, Management, And Treatment. Obstet Gynecol. 2018 Jan;131(1):e35-e42. doi: 10.1097/AOG.0000000000002458
- Bean EJ, Mazur T, Robinson AD. Mayer-Rokitansky-Küster-Hauser syndrome: sexuality, psychological effects, and quality of life. J Pediatr Adolesc Gynecol. 2009 Dec;22(6):339-46. doi: 10.1016/j.jpag.2008.11.006
- Facchin F, Francini F, Ravani S, Restelli E, Gramegna MG, Vercellini P, Aimi G. Psychological impact and health-related quality-of-life outcomes of Mayer-Rokitansky-Küster-Hauser syndrome: A systematic review and narrative synthesis. J Health Psychol. 2021 Jan;26(1):26-39. doi: 10.1177/1359105319901308
- Heller-Boersma JG, Schmidt UH, Edmonds DK. Psychological distress in women with uterovaginal agenesis (Mayer-Rokitansky-Kuster-Hauser Syndrome, MRKH). Psychosomatics. 2009 May-Jun;50(3):277-81. doi: 10.1176/appi.psy.50.3.277
- Weijenborg PTM, Kluivers KB, Dessens AB, Kate-Booij MJ, Both S. Sexual functioning, sexual esteem, genital self-image and psychological and relational functioning in women with Mayer-Rokitansky-Küster-Hauser syndrome: a case-control study. Hum Reprod. 2019 Sep 29;34(9):1661-1673. doi: 10.1093/humrep/dez130
- Bargiel-Matusiewicz K, Kroemeke A. Personality traits and coping styles in women with Mayer-Rokitansky-Küster-Hauser syndrome. Arch Med Sci. 2015 Dec 10;11(6):1244-9. doi: 10.5114/aoms.2015.56350
- Ernst ME, Sandberg DE, Keegan C, Quint EH, Lossie AC, Yashar BM. The Lived Experience of MRKH: Sharing Health Information with Peers. J Pediatr Adolesc Gynecol. 2016 Apr;29(2):154-8. doi: 10.1016/j.jpag.2015.09.009
- Patterson CJ, Crawford R, Jahoda A. Exploring the psychological impact of Mayer-Rokitansky-Küster-Hauser syndrome on young women: An interpretative phenomenological analysis. J Health Psychol. 2016 Jul;21(7):1228-40. doi: 10.1177/1359105314551077
- Weijenborg PT, ter Kuile MM. The effect of a group programme on women with the Mayer-Rokitansky-Küster-Hauser syndrome. BJOG. 2000 Mar;107(3):365-8. doi: 10.1111/j.1471-0528.2000.tb13232.x
- Heller-Boersma JG, Schmidt UH, Edmonds DK. A randomized controlled trial of a cognitive-behavioural group intervention versus waiting-list control for women with uterovaginal agenesis (Mayer-Rokitansky-Küster-Hauser syndrome: MRKH). Hum Reprod. 2007 Aug;22(8):2296-301. doi: 10.1093/humrep/dem167
- Beautiful You MRKH Foundation. https://www.beautifulyoumrkh.org/home.html
- Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH 3rd, Mazur T, Meyer-Bahlburg HF, Mouriquand P, Quigley CA, Sandberg DE, Vilain E, Witchel S; Global DSD Update Consortium. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm Res Paediatr. 2016;85(3):158-80. doi: 10.1159/000442975. Epub 2016 Jan 28. Erratum in: Horm Res Paediatr. 2016;85(3):180. Koopman, Peter [added]. Erratum in: Horm Res Paediatr. 2016;86(1):70.
- Graugaard C. Sexuality as a health-promoting factor – theoretical and clinical considerations. Nat Rev Urol. 2017 Oct;14(10):577-578. doi: 10.1038/nrurol.2017.117
- Lermann J, Mueller A, Wiesinger E, Häberle L, Brucker S, Wallwiener D, Dittrich R, Renner SP, Beckmann MW, Oppelt PG. Comparison of different diagnostic procedures for the staging of malformations associated with Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril. 2011 Jul;96(1):156-9. doi: 10.1016/j.fertnstert.2011.04.051
- Demir Eksi D, Shen Y, Erman M, Chorich LP, Sullivan ME, Bilekdemir M, Yılmaz E, Luleci G, Kim HG, Alper OM, Layman LC. Copy number variation and regions of homozygosity analysis in patients with MÜLLERIAN aplasia. Mol Cytogenet. 2018 Feb 3;11:13. doi: 10.1186/s13039-018-0359-3
- Sandbacka M, Laivuori H, Freitas É, Halttunen M, Jokimaa V, Morin-Papunen L, Rosenberg C, Aittomäki K. TBX6, LHX1 and copy number variations in the complex genetics of Müllerian aplasia. Orphanet J Rare Dis. 2013 Aug 16;8:125. doi: 10.1186/1750-1172-8-125
- Morcel K, Camborieux L; Programme de Recherches sur les Aplasies Müllériennes; Guerrier D. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007 Mar 14;2:13. doi: 10.1186/1750-1172-2-13
- Henes M, Jurow L, Peter A, Schoenfisch B, Taran FA, Huebner M, Seeger H, Brucker SY, Rall KK. Hyperandrogenemia and ovarian reserve in patients with Mayer-Rokitansky-Küster-Hauser syndrome type 1 and 2: potential influences on ovarian stimulation. Arch Gynecol Obstet. 2018 Feb;297(2):513-520. doi: 10.1007/s00404-017-4596-1
- Frank R. The formation of an artificial vagina without operation. Am J Obs Gynecol. 1938;35:1053–1055.
- D’Alberton A, Santi F. Formation of a neovagina by coitus. Obstet Gynecol. 1972 Nov;40(5):763-4.
- Herlin M, Bay Bjørn AM, Jørgensen LK, Trolle B, Petersen MB. Treatment of vaginal agenesis in Mayer-Rokitansky-Küster-Hauser syndrome in Denmark: a nationwide comparative study of anatomical outcome and complications. Fertil Steril. 2018 Sep;110(4):746-753. doi: 10.1016/j.fertnstert.2018.05.015
- Cheikhelard A, Bidet M, Baptiste A, Viaud M, Fagot C, Khen-Dunlop N, Louis-Sylvestre C, Sarnacki S, Touraine P, Elie C, Aigrain Y, Polak M; French MRKH Study Group. Surgery is not superior to dilation for the management of vaginal agenesis in Mayer-Rokitansky-Küster-Hauser syndrome: a multicenter comparative observational study in 131 patients. Am J Obstet Gynecol. 2018 Sep;219(3):281.e1-281.e9. doi: 10.1016/j.ajog.2018.07.015
- Callens N, De Cuypere G, De Sutter P, Monstrey S, Weyers S, Hoebeke P, Cools M. An update on surgical and non-surgical treatments for vaginal hypoplasia. Hum Reprod Update. 2014 Sep-Oct;20(5):775-801. doi: 10.1093/humupd/dmu024
- ACOG Committee on Adolescent Health Care. ACOG Committee Opinion. Number 274, July 2002. Nonsurgical diagnosis and management of vaginal agenesis. Obstet Gynecol. 2002 Jul;100(1):213-6. doi: 10.1016/s0029-7844(02)02158-0
- ACOG Committee on Adolescent Health Care. ACOG Committee Opinion No. 355: Vaginal agenesis: diagnosis, management, and routine care. Obstet Gynecol. 2006 Dec;108(6):1605-9. doi: 10.1097/00006250-200612000-00059
- Committee opinion: no. 562: müllerian agenesis: diagnosis, management, and treatment. Obstet Gynecol. 2013 May;121(5):1134-1137. doi: 10.1097/01.AOG.0000429659.93470.ed
- Dear J, Creighton SM, Conway GS, Williams L, Liao LM. Sexual Experience before Treatment for Vaginal Agenesis: A Retrospective Review of 137 Women. J Pediatr Adolesc Gynecol. 2019 Jun;32(3):300-304. doi: 10.1016/j.jpag.2018.12.005
- Callens N, De Cuypere G, Wolffenbuttel KP, Beerendonk CC, van der Zwan YG, van den Berg M, Monstrey S, Van Kuyk ME, De Sutter P; Belgian-Dutch Study Group on DSD; Dessens AB, Cools M. Long-term psychosexual and anatomical outcome after vaginal dilation or vaginoplasty: a comparative study. J Sex Med. 2012 Jul;9(7):1842-51. doi: 10.1111/j.1743-6109.2012.02747.x
- Hayashida SA, Soares JM Jr, Costa EM, da Fonseca AM, Maciel GA, Mendonça BB, Baracat EC. The clinical, structural, and biological features of neovaginas: a comparison of the Frank and the McIndoe techniques. Eur J Obstet Gynecol Reprod Biol. 2015 Mar;186:12-6. doi: 10.1016/j.ejogrb.2014.12.025
- Kang J, Chen N, Song S, Zhang Y, Ma C, Ma Y, Zhu L. Sexual function and quality of life after the creation of a neovagina in women with Mayer-Rokitansky-Küster-Hauser syndrome: comparison of vaginal dilation and surgical procedures. Fertil Steril. 2020 May;113(5):1024-1031. doi: 10.1016/j.fertnstert.2020.01.017
- Lappöhn RE. Congenital absence of the vagina–results of conservative treatment. Eur J Obstet Gynecol Reprod Biol. 1995 Apr;59(2):183-6. doi: 10.1016/0028-2243(94)02037-f
- Willemsen WN, Kluivers KB. Long-term results of vaginal construction with the use of Frank dilation and a peritoneal graft (Davydov procedure) in patients with Mayer-Rokitansky-Küster syndrome. Fertil Steril. 2015 Jan;103(1):220-7.e1. doi: 10.1016/j.fertnstert.2014.10.014
- Nakhal RS, Creighton SM. Management of vaginal agenesis. J Pediatr Adolesc Gynecol. 2012 Dec;25(6):352-7. doi: 10.1016/j.jpag.2011.06.003
- White PM. Commercialization, Altruism, Clinical Practice: Seeking Explanation for Similarities and Differences in Californian and Canadian Gestational Surrogacy Outcomes. Womens Health Issues. 2018 May-Jun;28(3):239-250. doi: 10.1016/j.whi.2018.01.004
- Hodson N, Townley L, Earp BD. Removing Harmful Options: The Law and Ethics of International Commercial Surrogacy. Med Law Rev. 2019 Nov 1;27(4):597-622. doi: 10.1093/medlaw/fwz025
- Saran J, Padubidri JR. New laws ban commercial surrogacy in India. Med Leg J. 2020 Sep;88(3):148-150. doi: 10.1177/0025817219891881
- Davydov S. Colpopoiesis from the peritoneum of the ueterorectal space. Obstet Gynecol. 1969;12:255–257.
- Banister JB, McIndoe AH. Congenital Absence of the Vagina, treated by Means of an Indwelling Skin-Graft. Proc R Soc Med. 1938 Jul;31(9):1055-6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2076989/pdf/procrsmed00453-0047.pdf
- Baldwin JF. XIV. The Formation of an Artificial Vagina by Intestinal Trransplantation. Ann Surg. 1904 Sep;40(3):398-403. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1425761/pdf/annsurg01000-0140.pdf
- WILLIAMS EA. CONGENITAL ABSENCE OF THE VAGINA: A SIMPLE OPERATION FOR ITS RELIEF. J Obstet Gynaecol Br Commonw. 1964 Aug;71:511-2. doi: 10.1111/j.1471-0528.1964.tb04315.x
- Raya-Rivera AM, Esquiliano D, Fierro-Pastrana R, López-Bayghen E, Valencia P, Ordorica-Flores R, Soker S, Yoo JJ, Atala A. Tissue-engineered autologous vaginal organs in patients: a pilot cohort study. Lancet. 2014 Jul 26;384(9940):329-36. doi: 10.1016/S0140-6736(14)60542-0
- Sabatucci I, Palaia I, Marchese C, Muzii L, Morte CD, Giorgini M, Musella A, Ceccarelli S, Vescarelli E, Panici PB. Treatment of the Mayer-Rokitansky-Küster-Hauser syndrome with autologous in vitro cultured vaginal tissue: descriptive study of long-term results and patient outcomes. BJOG. 2019 Jan;126(1):123-127. doi: 10.1111/1471-0528.15477
- Zhang X, Liu Z, Yang Y, Yao Y, Tao Y. The clinical outcomes of vaginoplasty using tissue-engineered biomaterial mesh in patients with Mayer-Rokitansky-Küster-Hauser syndrome. Int J Surg. 2017 Aug;44:9-14. doi: 10.1016/j.ijsu.2017.06.026
- Vecchietti G. Neovagina nella sindrome di Rokitansky-Küster-Hauser [Creation of an artificial vagina in Rokitansky-Küster-Hauser syndrome]. Attual Ostet Ginecol. 1965 Mar-Apr;11(2):131-47. Italian.
- Rall K, Schickner MC, Barresi G, Schönfisch B, Wallwiener M, Wallwiener CW, Wallwiener D, Brucker SY. Laparoscopically assisted neovaginoplasty in vaginal agenesis: a long-term outcome study in 240 patients. J Pediatr Adolesc Gynecol. 2014 Dec;27(6):379-85. doi: 10.1016/j.jpag.2014.02.002
- Brännström M. The Swedish uterus transplantation project: the story behind the Swedish uterus transplantation project. Acta Obstet Gynecol Scand. 2015 Jul;94(7):675-679. doi: 10.1111/aogs.12661
- Brännström M, Diaz-Garcia C, Hanafy A, Olausson M, Tzakis A. Uterus transplantation: animal research and human possibilities. Fertil Steril. 2012 Jun;97(6):1269-76. doi: 10.1016/j.fertnstert.2012.04.001
- Olausson M, Johannesson L, Brattgård D, Diaz-Garcia C, Lundmark C, Groth K, Marcickiewizc J, Enskog A, Akouri R, Tzakis A, Rogiers X, Janson PO, Brännström M. Ethics of uterus transplantation with live donors. Fertil Steril. 2014 Jul;102(1):40-3. doi: 10.1016/j.fertnstert.2014.03.048
- Díaz-García C, Johannesson L, Shao R, Bilig H, Brännström M. Pregnancy after allogeneic uterus transplantation in the rat: perinatal outcome and growth trajectory. Fertil Steril. 2014 Dec;102(6):1545-52.e1. doi: 10.1016/j.fertnstert.2014.09.010
- Brännström M, Johannesson L, Dahm-Kähler P, Enskog A, Mölne J, Kvarnström N, Diaz-Garcia C, Hanafy A, Lundmark C, Marcickiewicz J, Gäbel M, Groth K, Akouri R, Eklind S, Holgersson J, Tzakis A, Olausson M. First clinical uterus transplantation trial: a six-month report. Fertil Steril. 2014 May;101(5):1228-36. doi: 10.1016/j.fertnstert.2014.02.024
- Brännström M, Johannesson L, Bokström H, Kvarnström N, Mölne J, Dahm-Kähler P, Enskog A, Milenkovic M, Ekberg J, Diaz-Garcia C, Gäbel M, Hanafy A, Hagberg H, Olausson M, Nilsson L. Livebirth after uterus transplantation. Lancet. 2015 Feb 14;385(9968):607-616. doi: 10.1016/S0140-6736(14)61728-1
- Brännström M, Bokström H, Dahm-Kähler P, Diaz-Garcia C, Ekberg J, Enskog A, Hagberg H, Johannesson L, Kvarnström N, Mölne J, Olausson M, Olofsson JI, Rodriguez-Wallberg K. One uterus bridging three generations: first live birth after mother-to-daughter uterus transplantation. Fertil Steril. 2016 Aug;106(2):261-6. doi: 10.1016/j.fertnstert.2016.04.001
- Mölne J, Broecker V, Ekberg J, Nilsson O, Dahm-Kähler P, Brännström M. Monitoring of Human Uterus Transplantation With Cervical Biopsies: A Provisional Scoring System for Rejection. Am J Transplant. 2017 Jun;17(6):1628-1636. doi: 10.1111/ajt.14135
- Testa G, McKenna GJ, Gunby RT Jr, Anthony T, Koon EC, Warren AM, Putman JM, Zhang L, dePrisco G, Mitchell JM, Wallis K, Klintmalm GB, Olausson M, Johannesson L. First live birth after uterus transplantation in the United States. Am J Transplant. 2018 May;18(5):1270-1274. doi: 10.1111/ajt.14737
- Ejzenberg D, Andraus W, Baratelli Carelli Mendes LR, Ducatti L, Song A, Tanigawa R, Rocha-Santos V, Macedo Arantes R, Soares JM Jr, Serafini PC, Bertocco de Paiva Haddad L, Pulcinelli Francisco R, Carneiro D’Albuquerque LA, Chada Baracat E. Livebirth after uterus transplantation from a deceased donor in a recipient with uterine infertility. Lancet. 2019 Dec 22;392(10165):2697-2704. doi: 10.1016/S0140-6736(18)31766-5
- Ayoubi JM, Carbonnel M, Pirtea P, Kvarnström N, Brännström M, Dahm-Kähler P. Laparotomy or minimal invasive surgery in uterus transplantation: a comparison. Fertil Steril. 2019 Jul;112(1):11-18. doi: 10.1016/j.fertnstert.2019.05.038





{kind=link}