What is myeloproliferative disorder
Myeloproliferative disorders is now called myeloproliferative neoplasms, which are a rare group of diseases in which the bone marrow stem cells grow and reproduce abnormally. In 1951, William Dameshek coined the term myeloproliferative disorders which are now reformed to myeloproliferative neoplasms by the World Health Organization (WHO) 1. In myeloproliferative disorder (myeloproliferative neoplasm) abnormal stem cells produce excess numbers of one or more types of blood cells (red blood cells, white blood cells and/or platelets). These abnormal blood cells cannot function properly and can cause serious health problems unless properly treated and controlled. Myeloproliferative disorders (myeloproliferative neoplasms) are chronic diseases that, in most cases, remain stable for many years and progress gradually over time.
Main types of myeloproliferative disorder (myeloproliferative neoplasm) are chronic myeloid leukemia (CML), polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF) 1. WHO classification also included chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia (CEL), and myeloproliferative neoplasm unclassifiable 2. Out of the classic types of myeloproliferative neoplasms, chronic myeloid leukemia is BCR-ABL1 positive, but polycythemia vera, essential thrombocythemia, and primary myelofibrosis are BCR-ABL1 negative. Besides, the fourth edition of WHO classification for myeloid and acute leukemia was revised in 2016 due to recent advances in hematology with the identification of molecular markers and prognostic markers, giving a better understanding of the molecular pathogenesis and genetics of the hematological malignancies 3.
Chronic myeloid leukemia: It comprises 0.5% of all new cancer cases in the United States. As per recent SEER (Surveillance, Epidemiology, and End Results) national cancer database from the US, chronic myeloid leukemia is commonly diagnosed in older age groups, ranging from 65 to 74 years, and the median age of diagnosis is 65 years. It is more common in males, with an incidence rate of about 2.4 new cases per 100,000 versus 1.4 new cases per 100,000 in females. About 67.6% of patients have a 5-year survival rate, and the median age of death is about 77 years.
Polycythemia vera: The median age of diagnosis of polycythemia vera is 60 years. It is more prevalent in males as compared to females, with a male to female ratio of 1.8: 1. The estimated incidence of polycythemia vera ranges from 0.4 to 2.8 per 100,000 per year.
Essential thrombocythemia: The median age for diagnosis of essential thrombocythemia is 60 years. It is more common in females, with a male to female ratio of 1:2. The estimated incidence of essential thrombocythemia ranges from 1 to 2.5/100,000/year, and incidence increases with increasing age 4.
Primary myelofibrosis: The median age at the diagnosis is 67 years. The incidence rate varies from 0.8 to 2.1/100,000/year.
Other myeloproliferative neoplasms such as chronic neutrophilic leukemia, chronic eosinophilic leukemia, and unclassified myeloproliferative neoplasm are rare; exact incidence and prevalence are not known due to the rarity of the disorders.
Myeloproliferative neoplasms are sometimes described as being clonal blood stem cell disorders. This means that they result from a change, or mutation, in the DNA of a single blood stem cell. This change (or changes) results in abnormal blood cell development and in this case the overproduction of blood cells.
In myeloproliferative neoplasm the original mutation is preserved when the affected stem cell divides (proliferates) and produces a ‘clone’: a group of identical stem cells all with the same defect. Mutations in dividing cells occur all the time and healthy cells have sophisticated mechanisms within them to stop these abnormalities persisting. But the longer you live, the more chance you have of acquiring mutations that manage to escape these safeguards.
Myeloproliferative neoplasms are usually described according to the type of blood cell which is most affected. Myeloproliferative neoplasms are closely related diseases, so it’s not uncommon for people to have features of more than one myeloproliferative neoplasm when they are first diagnosed, or during the course of their illness. In some cases, one myeloproliferative disease may transform over time to another, or to a type of leukemia called acute myeloid leukemia.
Most people with an myeloproliferative neoplasm have no family history of the myeloproliferative disease. Myeloproliferative neoplasm is more commonly diagnosed in people over the age of 50 although it can rarely occur in younger people, even very rarely in children.
Normally, the bone marrow makes blood stem cells (immature cells) that become mature blood cells over time.
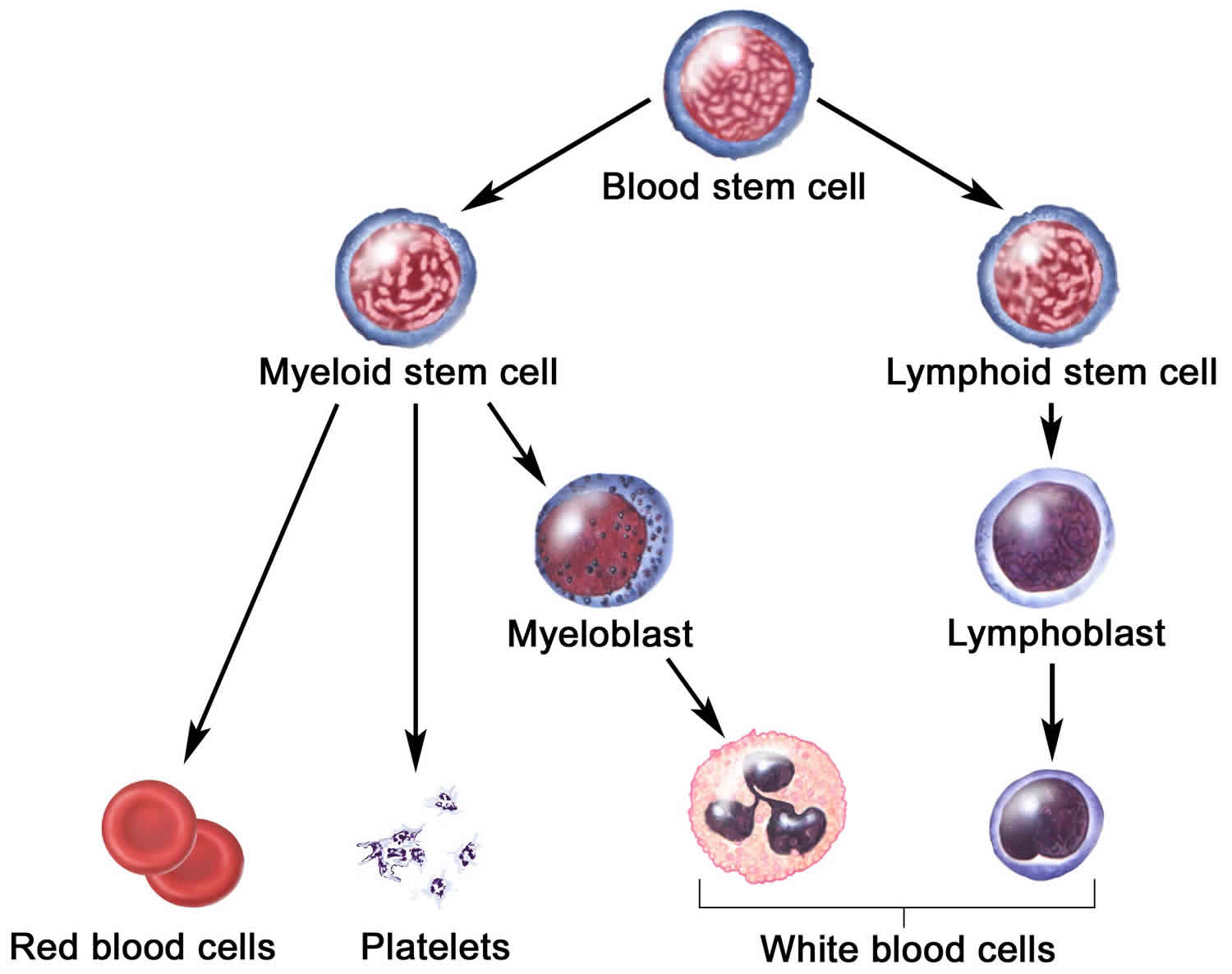
A blood stem cell may become a myeloid stem cell or a lymphoid stem cell. A lymphoid stem cell becomes a white blood cell. A myeloid stem cell becomes one of three types of mature blood cells:
- Red blood cells that carry oxygen and other substances to all tissues of the body.
- White blood cells that fight infection and disease.
- Platelets that form blood clots to stop bleeding.
In myeloproliferative neoplasms, too many blood stem cells become one or more types of blood cells. The neoplasms usually get worse slowly as the number of extra blood cells increases.
Figure 1. Blood cells production in bone marrow
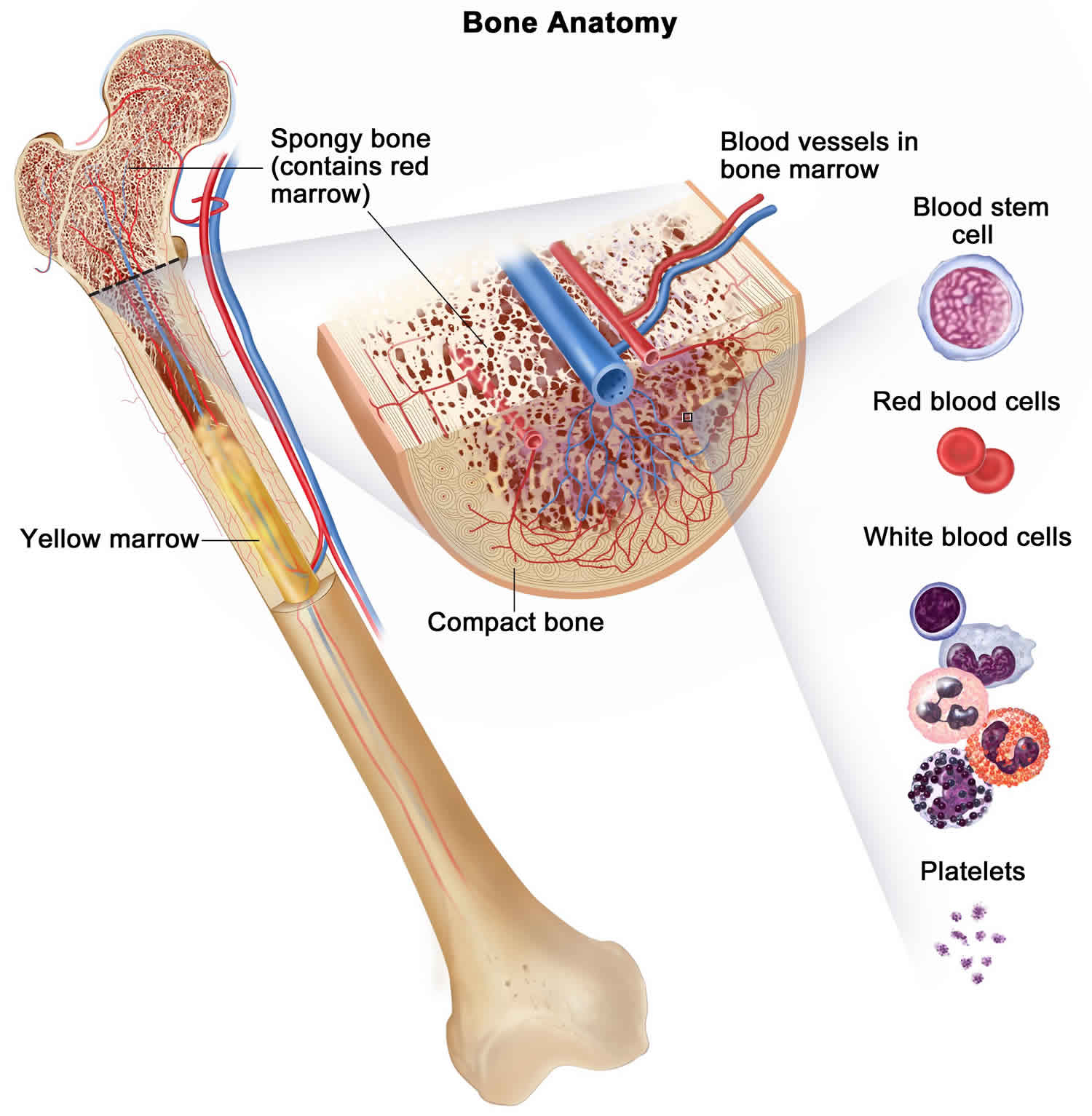
Figure 2. Blood cells development
Chronic myeloproliferative neoplasms types
The type of myeloproliferative neoplasm is based on whether too many red blood cells, white blood cells, or platelets are being made. Sometimes the body will make too many of more than one type of blood cell, but usually one type of blood cell is affected more than the others are. Chronic myeloproliferative neoplasms include the following 6 types:
- Chronic myelogenous leukemia.
- Polycythemia vera.
- Primary myelofibrosis (also called chronic idiopathic myelofibrosis).
- Essential thrombocythemia.
- Chronic neutrophilic leukemia.
- Chronic eosinophilic leukemia.
Chronic myeloproliferative neoplasms sometimes become acute leukemia, in which too many abnormal white blood cells are made.
Myeloproliferative neoplasms causes
The exact cause of myeloproliferative neoplasms remain unknown but there are likely to be a number of factors involved. That’s why myeloproliferative neoplasms, like most leukemias and other cancers, become more common as you get older. A mutation of a particular gene (a segment of DNA that makes proteins) known as Janus kinase 2 (JAK2) is found in a large proportion of people with myeloproliferative neoplasms. The exact meaning of this mutation remains unclear but it appears to play a role in the overproduction of blood cells seen in myeloproliferative disorders. The discovery of a mutation in the JAK2 gene is important because it is likely to have a significant impact on the way myeloproliferative neoplasms are diagnosed and treated.
Long-term exposure to high levels of benzene or very high doses of ionizing radiation may increase the risk of myelofibrosis in a small number of cases. Around one third of people with myelofibrosis have been previously diagnosed with polycythemia vera or essential thrombocythemia.
Myeloproliferative neoplasms pathophysiology
In the last decade, the treatment paradigm has changed with the detection of new driver mutations and a better understanding of the molecular pathogenesis of the myeloproliferative neoplasms. Mutations are divided into “restricted” which are found only in myeloproliferative neoplasms and “unrestricted” which are also detected in other myeloid malignancies; these mutations in hematopoietic stem cells result in the clonal expansion of all myeloid cells, B cell, and natural killer cells, leading to myeloproliferative neoplasms 5.
Cytogenetics of chronic myeloid leukemia is characterized by a reciprocal translocation between long arms of chromosomes 22 and 9, leading to the shortening of the length of the chromosome 22, also known as Philadelphia chromosomes, which is diagnostic of chronic myeloid leukemia. The translocation involves the fusion of breakpoint cluster gene (BCR) on chromosome 22 with oncogene Abelson murine leukemia virus (ABL) on the long arm of chromosome 9 resulting in BCR-ABL fusion gene. The tyrosine kinase activity of the ABL oncogene is further enhanced with the fusion of BCR gene due to an encoding of a chimeric protein from BCR-ABL fusion gene 6.
In BCR-ABL1 negative myeloproliferative neoplasms, the restricted driver mutations are JAK2, myeloproliferative leukemia virus proto-oncogene (MPL), CALR, and CSF3R, which encodes for various tyrosine kinases.
Janus kinase (JAK) 2 gene mutation
Janus kinase gene is a type of protein kinase which phosphorylates signal transducer and activator of transcription (STAT) in the JAK-STAT pathway, and activation of this pathway results in the pathogenesis of the myeloproliferative neoplasms 7. The JAK2 mutation is found in about 70% of myeloproliferative neoplasms. It is a somatic mutation which involves substitution of valine to phenylalanine at codon 617 (JAK2 V617F) in the pseudokinase domain. The frequency of JAK2 V617F mutation is 95% in patients with PV, about 50% to 70% in ET, and 40% to 50% in PMF, respectively.
In 5% of polycythemia vera with JAK2 V617F negative individuals, JAK2 exon 12 mutation is found; however, no such mutation was found in essential thrombocythemia and primary myelofibrosis 8.
MPL proto-oncogene mutation
The MPL gene codes the thrombopoietin (TPO) receptor which regulates megakaryopoiesis through a JAK-STAT pathway. The most common somatic MPL mutations are MPL W515L and MPL W515K which cause spontaneous activation of the JAK-STAT pathway, resulting in abnormal hematopoietic cells proliferation 9. These mutations are found in up to 6% of essential thrombocythemia and up to 10% of primary myelofibrosis as per literature 5. Furthermore, this mutation will help categorize patients diagnosed with negative JAK2 V617F mutation and Philadelphia chromosome myeloproliferative neoplasm. MPL mutations increase the risk of thrombotic complications as compared to JAK2 V617F mutation patients and are associated with low hemoglobin, low bone marrow cellularity, high platelets, and high serum erythropoietin levels 10.
Calreticulin (CALR) gene mutation
The CALR gene is located in chromosome 19 (exon 9) which encodes calreticulin, a calcium-binding endoplasmic reticulum chaperone protein that regulates cellular proliferation, differentiation, and apoptosis; this protein also plays a role in immune system function and wound healing. In 2013, the somatic mutation (usually frameshift mutation) of the CALR gene was discovered in patients with essential thrombocythemia and primary myelofibrosis who were negative for JAK2 and MPL mutation 11. The CALR exon 9 mutations are found in approximately 50% to 75% of patients with essential thrombocythemia and primary myelofibrosis 12.
About 10% to 15% of essential thrombocythemia and primary myelofibrosis patients lack all three driver mutations (JAK2 V617F, MPL W515L/K, and CALR exon 9), which are also known as triple negative myeloproliferative neoplasm.
Chronic neutrophilic leukemia is one of the myeloproliferative neoplasms which is characterized by peripheral leukocytosis with neutrophilia, hypercellularity of the bone marrow with less than 5% myeloblasts, and normal neutrophil maturation. The JAK2 V617F mutation is common in chronic neutrophilic leukemia; however, detection of mutation in colony-stimulating factor 3 receptor (CSF3R) gene is one of the WHO diagnostic criteria for chronic neutrophilic leukemia and commonly occurs at a CSF3R T618I location in the gene 13.
Myeloproliferative neoplasms symptoms
Many people have no symptoms when they are first diagnosed with an myeloproliferative neoplasm and the disease is picked up accidentally during a routine blood test or physical examination. In other cases, people go to see their doctor because they have some troubling symptoms of their disease. When symptoms do occur, they develop gradually over time. Common symptoms include:
- headaches
- blurred vision
- fatigue
- weakness
- dizziness
- itchiness (pruritus)
- night sweats
- raised blood pressure (hypertension).
Other symptoms experienced in myeloproliferative neoplasm are a result of the affected cell involved with the myeloproliferative neoplasm.
Polycythemia vera
Enlargement of the spleen (splenomegaly) is common and occurs in around 75% of cases. In some cases the liver may also be enlarged: this is called hepatomegaly. Some people experience gout, which usually presents as a painful inflammation of the big toe or foot. Some individuals may develop erythromelalgia, a rare condition characterised by intense burning pain of affected extremities and an increased skin temperature. In many cases, people with polycythemia vera have a ruddy (red) complexion, and a reddening of the palms of their hands, the soles of their feet, ear lobes, mucous membranes and their eyes. This is due to the high numbers of red cells in the circulation.
As the blood is thicker than normal it cannot flow as easily, especially through the smaller blood vessels. If left untreated, this increases the risk of the formation of a blood clot within a blood vessel. Blood clots are a common complication of polycythemia vera and occur in around 30% of people, even before they are diagnosed. Bleeding and easy bruising can also occur. This is usually minor and occurs in around one quarter of all patients.
Essential thrombocythemia
Similar to polycythemia vera many people have no symptoms when they are first diagnosed with essential thrombocythemia. Thrombosis (blood clot) is a major complication of essential thrombocythemia. Blood clots can occur in large or small arteries, interfering with the blood and oxygen supply to various organs or tissues. Older patients and those with a high platelet count, or a prior history of thrombosis, may be at increased risk.
A major aim of treatment in essential thrombocythemia is to reduce your platelet count, and therefore your risk of thrombosis. Less commonly, people experience symptoms of abnormal bleeding including bruising for no apparent reason, or exaggerated or prolonged bleeding following minor cuts or injury. In pregnancy, uncontrolled essential thrombocythemia can reduce the blood supply to the placenta or fetus. This can cause problems with fetal growth and may in some cases lead to miscarriage. An enlarged spleen is common and occurs in around 30% of cases of essential thrombocythemia. In some cases the liver may also be enlarged (hepatomegaly).
Primary myelofibrosis
Virtually all patients with myelofibrosis have an enlarged spleen (splenomegaly) when they are first diagnosed, although around 20% of people will have no symptoms. In around a third of cases the spleen is very enlarged. Abdominal discomfort can also result from an enlarged liver (hepatomegaly), which occurs in around two-thirds of cases. Other less common symptoms include bone and joint pain, and bleeding problems. Primary myelofibrosis is characterized by extramedullary hematopoiesis and bone marrow fibrosis. Physical examination findings include pallor, petechiae, ecchymoses, splenomegaly, hepatomegaly, lymphadenopathy, pleural effusion, pericardial effusion, ascites, pulmonary edema, seizure, altered mental status, and spinal cord compression.
Chronic neutrophilic leukemia
Most patients are asymptomatic, but common symptoms on presentation include fatigue, night sweats, loss of appetite, weight loss, easy bruising, and bone pain. A patient may have splenomegaly at diagnosis on physical examination.
Myeloproliferative neoplasms complications
Thrombosis and hemorrhagic events are the most common complications in myeloproliferative neoplasms. Other complications based on diseases are as follows:
Chronic myeloid leukemia (CML): Infection is the most common complication due to the immunocompromised state and blast transformation.
Polycythemia vera (PV): Arterial and venous thrombosis and thromboembolic complications may manifest as transient ischemic stroke (TIA)/stroke and pulmonary embolism, myocardial infarction, and hemorrhage. Some patients with polycythemia vera may present as acquired von Willebrand disease, and they have an increased tendency for bleeding, particularly if they are taking aspirin. Post-polycythemia vera myelofibrosis and transformation to myelodysplastic syndromes or acute myeloid leukemia are other complications of polycythemia vera. Erythromelalgia is the microvascular complication of polycythemia vera which manifest as pallor, erythema, or cyanosis of the hands and feet.
Essential thrombocythemia (ET): Small and large vessel thrombosis, hemorrhage, thromboembolic complications such as stroke, pulmonary embolism, myocardial infarction, pulmonary hypertension, and priapism.
Primary myelofibrosis (PMF): Portal hypertension, gastrointestinal bleeding, spinal cord compression, respiratory distress, transformation to acute leukemia, thrombotic events, immune deficiency leading to infections, and pulmonary hypertension.
Myeloproliferative neoplasms diagnosis
Myeloproliferative neoplasms are diagnosed by examining samples of your blood and bone marrow. Sometimes the diagnosis of myeloproliferative neoplasm is only made when other causes of altered blood count have been excluded.
The laboratory workup for the evaluation of myeloproliferative neoplasms includes the following:
- Complete blood count with differential
- Peripheral blood with microscopic examination
- Comprehensive metabolic panel
- Electrolytes
- Peripheral blood fluorescence in situ hybridization (FISH) or Reverse transcription polymerase chain reaction (RT-PCR) for BCR-ABL1
- Leukocyte alkaline phosphatase (LAP) score which helps to differentiate between reactive leukocytosis from chronic myeloid leukemia. The LAP score is low in patients with chronic myeloid leukemia and paroxysmal nocturnal hemoglobinuria, however, the score is high in leukemoid reaction.
- Red blood cell mass study
- Serum uric acid
- Lactate dehydrogenase (LDH)
- Erythropoietin (EPO) level – normal or high in secondary polycythemia, but low or absent in polycythemia vera.
- Von Willebrand factor – polycythemia vera patients may develop acquired von Willebrand disease with platelet counts greater than 1 million/microL, and these patients are at increased risk of bleeding, particularly if they are on aspirin.
- Bone marrow aspiration and biopsy
- Cytogenetic analysis of the bone marrow biopsy or aspirate for BCR-ABL1, JAK2, MPL, CALR, and CSF3R mutations.
Evaluation of myeloproliferative neoplasms involves comprehensive hematological/laboratory workup, bone marrow biopsy, cytogenetic analysis, and clinical assessment.
Complete blood count
The first step in the diagnosis is a simple blood test called a complete blood count (CBC) or full blood count (FBC). This involves a sample of blood from a vein in your arm being sent to the laboratory for investigation. People with polycythemia vera have a high red cell count, hemoglobin level and hematocrit due to the excessive production of red cells. A raised white cell count (especially a raised neutrophil count) and a raised platelet count are also common findings. Other findings that help confirm the diagnosis of polycythemia vera is the presence of the JAK2 mutation or other cytogenetic abnormalities in your blood or bone marrow cells.
A persistently raised platelet count is the most common sign of essential thrombocythemia. Around a third of people with essential thrombocythemia will also have a mildly raised red cell and/or white cell count.
People with primary myelofibrosis commonly present with varying degrees of anemia. When examined under the microscope the red cells are often described as being ‘teardrop-shaped’. Higher than normal numbers of white cells and platelets may be found in the early stages of this disorder, but low white cell and platelet counts are common in more advanced disease.
Bone marrow examination
If the results of your blood tests suggest that you might have myeloproliferative neoplasm, a bone marrow biopsy may be required to help confirm the diagnosis. A bone marrow biopsy involves taking a sample of bone marrow, usually from the back of the hip bone and sending it to the laboratory for examination under the microscope. The bone marrow biopsy may be done in the haematologist’s rooms, clinic or day procedure centre and is usually performed under local anaesthesia with sedation given either by tablet or through a small drip in your arm. The sample of bone marrow is examined in the laboratory to determine the number and type of cells present and the amount of haemopoiesis (blood forming) activity taking place there.
Other tests
Once the diagnosis of myeloproliferative neoplasm is made, blood and bone marrow cells are examined further using special laboratory tests. These include immunophenotyping and cytogenetic tests. These tests provide more information about the exact type of disease you have, the likely course of your disease and the best way to treat it. Other tests may be conducted to provide information on your general health and how your vital organs are functioning. These include a combination of further blood tests and imaging tests (x-rays, scans and EKG).
These results will provide a baseline of your disease and general health, which will be compared with later results to assess how well you are progressing and responding to treatment.
WHO classification of myeloproliferative neoplasm
Owing to recent advances in molecular markers or driver mutations, 2008 WHO classification of myeloid neoplasm was updated in 2016 to incorporate new molecular biomarkers. Diagnostic criteria for myeloproliferative neoplasms based on revised and updated WHO classification are as follows 3:
WHO diagnostic criteria for chronic myeloid leukemia (Accelerated and Blast Phase)
The diagnosis of the chronic myeloid leukemia requires any one or more of the following for accelerated phase:
- Persistent or increasing white blood cell (greater than 10 X 109/L), Unresponsive to therapy
- Persistent thrombocytosis (greater than 1000 X 109/L), Unresponsive to therapy
- Persistent thrombocytopenia (greater than 100 X 109/L), Unrelated to therapy
- Persistent or increasing splenomegaly, Unresponsive to therapy
- Basophils greater than or equal to 20% in peripheral blood
- Blast cells 10% to 19% in peripheral blood or bone marrow
- Additional clonal chromosomal abnormality in Philadelphia positive cells at diagnosis
- Any new clonal chromosomal abnormality in Philadelphia positive cells during therapy
The diagnosis criteria for chronic myeloid leukemia (blast phase) includes any one of the above criteria and blast cells greater than or equal to 20% in peripheral blood or bone marrow or extramedullary involvement of lymph nodes, skin, lung, central nervous system, and bone.
Revised WHO Diagnostic Criteria for Polycythemia Vera
Diagnosis of polycythemia vera necessitates the presence of either all three major criteria or the first two major criteria and one minor criterion.
Major criteria
- Hemoglobin greater than 16.5 g/dL in male and hemoglobin greater than 16.0 g/dL in female or hematocrit greater than 49% in male and hematocrit greater than 48% in female or increased red cell mass greater than 25% above mean normal predicted value
- Bone marrow biopsy with trilineage myeloproliferation (panmyelosis) and hypercellularity for age
- Presence of JAK2 V617F or JAK exon 12 mutation
Minor criterion
- Serum erythropoietin below normal
Of note, patients with polycythemia vera who fulfill the diagnostic criteria should also be evaluated for secondary causes of polycythemia.
Revised WHO Diagnostic Criteria for Essential Thrombocythemia
Diagnosis of essential thrombocythemia requires either the presence of all four major criteria or the first three major criteria and the minor criterion.
Major criteria
- Platelet count greater than or equal to 450 X 109/L
- Bone marrow biopsy with a proliferation of megakaryocyte lineage with increased numbers of large, mature megakaryocytes. No increase in neutrophil granulopoiesis or erythropoiesis.
- Not meeting WHO criteria for BCR-ABL1 positive chronic myeloid leukemia, primary myelofibrosis, polycythemia vera, myelodysplastic syndromes, or other myeloid neoplasms
- Detection of JAK2, CALR, or MPL mutation
Minor criterion
- Presence of clonal marker or absence of evidence for reactive thrombocytosis
Revised WHO Diagnostic Criteria for Primary Myelofibrosis
The diagnosis of primary myelofibrosis requires the presence of all three major criteria and at least one of the minor criteria. Further division of primary myelofibrosis to overt primary myelofibrosis and prefibrotic primary myelofibrosis is based on a degree of fibrosis.
Major criteria
- Bone marrow findings of megakaryocyte proliferation with atypia, usually accompanied by reticulin and/or collagen fibrosis
- Not meeting the WHO criteria for BCR-ABL1 positive chronic myeloid leukemia, essential thrombocythemia, polycythemia vera, myelodysplastic syndromes, or other myeloid neoplasms
- Detection of JAK2, CALR, or MPL mutation or presence of clonal markers in the absence of these mutations
Minor criteria
- Anemia not attributed to another comorbid condition
- Leukocytosis greater than or equal to 11 X 109/L
- Palpable splenomegaly
- Elevated LDH
- Leukoerythroblastosis
Revised WHO diagnostic criteria for Chronic Neutrophilic Leukemia
- Peripheral blood with WBC greater than or equal to 25 X 109/L
- , segmented neutrophils plus band forms greater than or equal to 80% of white blood cells, neutrophil precursors less than 10%, rare myeloblasts, monocyte count less than 1 X 109/L, and no dysgranulopoiesis
- Bone marrow hypercellularity with elevated neutrophil granulocytes, normal neutrophil maturation, and myeloblast less than 5% of nucleated cells
- Not meeting WHO criteria for BCR-ABL1 positive chronic myeloid leukemia, essential thrombocythemia, polycythemia vera, or primary myelofibrosis
- No rearrangement of PDGFRA, PDGFRB, or FGFR1, or PCM1-JAK2
- Detection of CSF3R T618I or other activating CSF3R mutation or in the absence of a CSF3R mutation sustained neutrophilia for at least 3 months, splenomegaly and no identifiable cause of reactive neutrophilia including an absence of a plasma cell neoplasm
Myeloproliferative neoplasms treatment
Management or treatment of the different myeloproliferative neoplasms are as follows:
Chronic myeloid leukemia
There are different types of treatment for patients with chronic myelogenous leukemia.
Different types of treatment are available for patients with chronic myelogenous leukemia (chronic myeloid leukemia). Some treatments are standard (the currently used treatment), and some are being tested in clinical trials. A treatment clinical trial is a research study meant to help improve current treatments or obtain information about new treatments for patients with cancer. When clinical trials show that a new treatment is better than the standard treatment, the new treatment may become the standard treatment. Patients may want to think about taking part in a clinical trial. Some clinical trials are open only to patients who have not started treatment.
Six types of standard treatment are used:
Targeted therapy
Targeted therapy is a type of treatment that uses drugs or other substances to identify and attack specific cancer cells without harming normal cells. Tyrosine kinase inhibitors are targeted therapy drugs used to treat chronic myelogenous leukemia.
Imatinib mesylate, nilotinib, dasatinib, and ponatinib are tyrosine kinase inhibitors that are used to treat chronic myeloid leukemia.
Chemotherapy
Chemotherapy is a cancer treatment that uses drugs to stop the growth of cancer cells, either by killing the cells or by stopping them from dividing. When chemotherapy is taken by mouth or injected into a vein or muscle, the drugs enter the bloodstream and can reach cancer cells throughout the body (systemic chemotherapy). When chemotherapy is placed directly into the cerebrospinal fluid, an organ, or a body cavity such as the abdomen, the drugs mainly affect cancer cells in those areas (regional chemotherapy). The way the chemotherapy is given depends on the type and stage of the cancer being treated.
Biologic therapy
Biologic therapy is a treatment that uses the patient’s immune system to fight cancer. Substances made by the body or made in a laboratory are used to boost, direct, or restore the body’s natural defenses against cancer. This type of cancer treatment is also called biotherapy or immunotherapy.
High-dose chemotherapy with stem cell transplant
High doses of chemotherapy are given to kill cancer cells. Healthy cells, including blood -forming cells, are also destroyed by the cancer treatment. Stem cell transplant is a treatment to replace the blood-forming cells. Stem cells (immature blood cells) are removed from the blood or bone marrow of the patient or a donor and are frozen and stored. After the patient completes chemotherapy, the stored stem cells are thawed and given back to the patient through an infusion. These reinfused stem cells grow into (and restore) the body’s blood cells.
Donor lymphocyte infusion
Donor lymphocyte infusion (DLI) is a cancer treatment that may be used after stem cell transplant. Lymphocytes (a type of white blood cell) from the stem cell transplant donor are removed from the donor’s blood and may be frozen for storage. The donor’s lymphocytes are thawed if they were frozen and then given to the patient through one or more infusions. The lymphocytes see the patient’s cancer cells as not belonging to the body and attack them.
Surgery
Splenectomy is surgery to remove the spleen.
Polycythemia vera
The goal of treatment for polycythemia vera is to reduce the number of cells in your blood and help you to maintain a normal blood count. This helps control any symptoms of your disease and reduces the risk of complications due to blood clotting, or bleeding. The treatment or combination of treatments chosen for you will depend on several factors including the duration and severity of your disorder, whether or not you have a history of blood clots, your age and your general health. In addition to treatment, your doctor will advise you on ways to stay healthy and reduce any lifestyle factors that might increase your risk of thrombosis. For example you may be advised to stop smoking, and/or take a series of steps to maintain a healthy weight range and blood pressure. Your doctor or nurse will explain any side effects you might experience while you are having these treatments and how they can be managed.
Venesection/phlebotomy
Venesection (or phlebotomy) is a procedure in which a controlled amount of blood is removed from your bloodstream. This procedure is commonly used when people are first diagnosed with polycythemia vera because it can help to rapidly reduce a high red cell count. It is a process similar to a blood donation. This is usually done in the outpatient’s department of the hospital. This procedure may need to be repeated frequently at first, usually every few days, until your blood count is reduced to the desired level.After this, you may need to have the procedure repeated periodically. For many people, particularly younger patients and those with mild disease, regular venesection (every few months) may be all that is needed to control their disease for many years.
Chemotherapy
These drugs are commonly used for people with an extremely high platelet count, complications due to blood clotting or bleeding, or symptoms of an enlarged spleen or for some people who are unable to tolerate venesection. The most commonly used myelosuppressive agent is an oral capsule chemotherapy drug called hydroxyurea. It is particularly useful in controlling a high platelet count and therefore reducing the risk of thrombosis. Another less commonly used chemotherapy drug is busulfan. This drug is also given in tablet form. Chemotherapy taken in capsule form is tolerated well by most people and side effects tend to be few and mild.
Interferon
Interferon is a substance produced naturally by the body’s immune system. It plays an important role in fighting disease. In polycythemia vera, interferon is sometimes prescribed for younger patients to help control the production of blood cells. Interferon is usually given as an injection under the skin (subcutaneous injection) using a very small needle. You or a family member (or friend) will be taught how to do this at home. The main side effects are flu-like symptoms such as chills, fevers, aches and pains and weakness.
Aspirin
Many people are prescribed small daily doses of aspirin, which have been shown to significantly reduce the risk of thrombosis in people with polycythemia vera. Aspirin works by preventing your platelets from clumping together to form harmful blood clots in different parts of your body.
Anagrelide hydrochloride
Anagrelide hydrochloride (Agrylin) is a drug used to reduce high platelet counts in people with polycythemia vera and essential thrombocythaemia. Anagrelide affects platelet-producing cells in the bone marrow called megakaryocytes, slowing down platelet production and therefore reducing the number of platelets in the circulating blood. Anagrelide is taken in capsule form by mouth. The most commonly reported side effects include headaches, fast or forceful heart beat (palpitations), diarrhoea, weakness, fluid retention, nausea, dizziness, abdominal pain and shortness of breath. You should report any side effects you are experiencing to your doctor. You need to contact your doctor immediately if you experience the following symptoms: shortness of breath or difficulty breathing, swollen ankles, fast or irregular heartbeat, and/or chest pain. You should not stop taking this or any other medication for polycythemia vera unless instructed by your doctor. Stopping these medications suddenly can be harmful.
Essential thrombocythemia
The goal of treatment for people with essential thrombocythemia is to prevent complications like abnormal bleeding and bruising and in some cases reducing the number of platelets in the blood. You may not have any symptoms of essential thrombocythemia when you are first diagnosed and therefore may not require any treatment for some time. Instead your doctor may recommend a ‘watch and wait’ strategy which involves regular check-ups and blood counts to carefully monitor your health. In addition they will advise you on the steps you can take to stay healthy and reduce any lifestyle-related risk factors you may have that increase your chances of developing a blood clot. You may be advised, for example, on ways to help you stop smoking and maintain a healthy weight range and blood pressure.
Chemotherapy
For people at high risk of thrombosis, a chemotherapy drug called hydroxyurea with or without low-dose aspirin is often used as a first-line treatment. Hydroxyurea works by suppressing the function of your bone marrow and thereby controlling platelet production, while aspirin prevents your platelets from aggregating and forming harmful clots in your body.
Anagrelide hydrochloride and Interferon
Anagrelide hydrochloride (Agrylin) and interferon may also be used. Interferon works by suppressing the abnormal megakaryocyte clone in your bone marrow thereby reducing the overproduction of platelets. Those at low risk may be simply treated using low-dose aspirin or an equivalent drug alone. They usually have a very good outlook with no difference to the general population.
Primary myelofibrosis
Some people have no symptoms when they are first diagnosed with primary myelofibrosis and do not require treatment straight away, apart from regular check-ups with their doctor to carefully monitor their disease. For others treatment is largely supportive and is aimed at preventing complications due to low blood counts and an enlarged spleen. This may include periodic blood transfusions and taking antibiotics to prevent and treat any infections. A chemotherapy drug such as hydroxyurea, or low-doses of a drug called thalidomide may be used to reduce an enlarged spleen. In some cases, the surgical removal of the spleen (splenectomy) may be considered. Small doses of radiation to the spleen can also be given to reduce its size. This usually provides temporary relief for about three to six months.
Hematopoietic stem cell transplant
Some younger patients who have a suitably matched donor may be offered an allogeneic (donor) stem cell transplant. This is a medical procedure that offers the only chance of cure or long-term survival in patients with myelofibrosis. Stem cell transplants carry significant risks and are only suitable for a small minority of younger patients (usually under 60 years).
JAK2 Inhibitors
JAK2 inhibitors work by blocking the activity of the JAK2 protein, which may lead to a reduction in splenomegaly and decreased symptoms. They also work in patients with myelofibrosis without the JAK2 mutation. Side effects may include worsening anaemia or a low platelet count. Ruxolitinib is the only JAK2 inhibitor currently licenced for use in Australia. A number of JAK2 inhibitors may be available in clinical trials or may become available in the near future.
Chronic neutrophilic leukemia
Because of the rarity of the disease, there is no specific treatment. Most patients are managed with a cytoreductive agent (hydroxyurea).
Treatment of chronic neutrophilic leukemia may include the following:
- Donor bone marrow transplant.
- Chemotherapy.
- Biologic therapy using interferon alfa.
- A clinical trial of a new treatment.
Chronic eosinophilic leukemia
Treatment of chronic eosinophilic leukemia may include the following:
- Bone marrow transplant.
- Biologic therapy using interferon alfa.
- A clinical trial of a new treatment.
Myeloproliferative neoplasms survival rate
With the advent of the tyrosine kinase inhibitors, the prognosis of the chronic myeloid leukemia has significantly improved.
Patients with essential thrombocythemia usually have a good prognosis with normal life expectancy.
Triple negative primary myelofibrosis patients have been observed as a worse prognosis. Furthermore, Kampfl et al. 11 reported better survival benefit in patients with CALR exon 9 mutations as compared to JAK2 or MPL mutations.
Patients with polycythemia vera who transform to myelodysplastic syndromes or acute myeloid leukemia have a very poor prognosis. As per one of the retrospective studies, older age (greater than 70 years), leukocytosis, and thrombosis are prognostic factors for worse survival in patients with polycythemia vera 14.
References- Thapa B, Rogers HJ. Cancer, Myeloproliferative Neoplasms. [Updated 2018 Oct 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531464
- Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer. 2009 Sep 01;115(17):3842-7
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May 19;127(20):2391-405
- Srour SA, Devesa SS, Morton LM, Check DP, Curtis RE, Linet MS, Dores GM. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br. J. Haematol. 2016 Aug;174(3):382-96
- Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017 Feb 09;129(6):667-679
- Faderl S, Talpaz M, Estrov Z, O’Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N. Engl. J. Med. 1999 Jul 15;341(3):164-72
- Saeidi K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit. Rev. Oncol. Hematol. 2016 Feb;98:375-89
- Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007 Feb 01;356(5):459-68
- Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, Cuker A, Wernig G, Moore S, Galinsky I, DeAngelo DJ, Clark JJ, Lee SJ, Golub TR, Wadleigh M, Gilliland DG, Levine RL. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006 Jul;3(7):e270
- Akpınar TS, Hançer VS, Nalçacı M, Diz-Küçükkaya R. MPL W515L/K Mutations in Chronic Myeloproliferative Neoplasms. Turk J Haematol. 2013 Mar;30(1):8-12
- Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant’Antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schönegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013 Dec 19;369(25):2379-90
- Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, Avezov E, Li J, Kollmann K, Kent DG, Aziz A, Godfrey AL, Hinton J, Martincorena I, Van Loo P, Jones AV, Guglielmelli P, Tarpey P, Harding HP, Fitzpatrick JD, Goudie CT, Ortmann CA, Loughran SJ, Raine K, Jones DR, Butler AP, Teague JW, O’Meara S, McLaren S, Bianchi M, Silber Y, Dimitropoulou D, Bloxham D, Mudie L, Maddison M, Robinson B, Keohane C, Maclean C, Hill K, Orchard K, Tauro S, Du MQ, Greaves M, Bowen D, Huntly BJP, Harrison CN, Cross NCP, Ron D, Vannucchi AM, Papaemmanuil E, Campbell PJ, Green AR. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013 Dec 19;369(25):2391-2405
- Elliott MA, Tefferi A. Chronic neutrophilic leukemia 2016: Update on diagnosis, molecular genetics, prognosis, and management. Am. J. Hematol. 2016 Mar;91(3):341-9
- Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, Randi ML, Vaidya R, Cazzola M, Rambaldi A, Gisslinger B, Pieri L, Ruggeri M, Bertozzi I, Sulai NH, Casetti I, Carobbio A, Jeryczynski G, Larson DR, Müllauer L, Pardanani A, Thiele J, Passamonti F, Barbui T. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013 Sep;27(9):1874-81





{kind=link}