What is oligodendroglioma
Oligodendroglioma is a rare type of glioma brain tumor that develop from cells called oligodendrocytes. About 4 out of every 100 (4%) brain tumors are oligodendrogliomas, representing about 10-15% of the gliomas. Oligodendrocytes (cells that cover and protect nerve cells in the brain and spinal cord) (Figure 3) line up in small groups and wrap their cell processes around the thicker axons in the CNS (central nervous system= brain+spinal cord), producing fatty white substance that covers nerves, called myelin. The myelin sheath is a multilayered lipid and protein covering around some axons that insulates them and increases the speed of nerve impulse conduction.
Oligodendrogliomas are most likely to be diagnosed in adults, although they do occur in young children. Only 6% of these oligodendrogliomas are found in infants and children. Most oligodendrogliomas occur in adults ages 50-60, and are found in men more often than women. Rarely oligodendroglioma can spread within the central nervous system, in the fluid that circulates around the brain and spinal cord.
Oligodendrogliomas can be found anywhere within the cerebral hemisphere of the brain including the spinal cord, although the frontal and temporal lobes are the most common locations. The majority of oligodendroglioma are located supratentorially (85%), with the frontal lobe being the most common location (50–65%) 1. Infratentorial location is rare. Extremely rare primary locations are within the ventricular system, brain stem, spinal cord, retina and leptomeninges 2.
Oligodendroglioma tumors can be low grade (grade 2, slow growing) or high grade (grade 3, fast growing also called anaplastic).
Oligodendrogliomas are generally soft, grayish-pink tumors that often contain mineral deposits (calcifications), areas of hemorrhage, and/or cysts. Oligodendrogliomas tend to grow slowly and may be present for many years before they are diagnosed.
Common symptoms include seizures, headaches and changes in personality. Other symptoms vary by the size and location of the tumor. The exact cause of opigodendrogliomas is unknown. Some appear to have a chromosome abnormality involving loss of chromosomes 1p (short arm of chromosome 1) and 19q (long arm of chromosome 19), which is not only of diagnostic but also of prognostic and predictive relevance 1.
Treatment of oligodendroglioma generally involves surgical removal of the tumor followed by radiation therapy and/or chemotherapy. Recurrent tumors may need additional surgery, radiation and chemotherapy.
Neuroglia
Neuroglia or glia make up about half the volume of the CNS (central nervous system= brain+spinal cord). There are 6 types of neuroglia—4 in the CNS and 2 in the PNS (peripheral nervous system). Their name derives from the idea of early histologists that they were the “glue” that held nervous tissue together. Scientists now know that neuroglia are not merely passive bystanders but rather actively participate in the activities of nervous tissue. Generally, neuroglia are smaller than neurons, and they are 5 to 25 times more numerous. In contrast to neurons, glial cells do not generate or propagate action potentials, and they can multiply and divide in the mature nervous system. In cases of injury or disease, neuroglia multiply to fill in the spaces formerly occupied by neurons. Brain tumors derived from glia, called gliomas, tend to be highly malignant and to grow rapidly. Of the six types of neuroglia, four—astrocytes, oligodendrocytes, microglia, and ependymal cells—are found only in the CNS. The remaining two types—Schwann cells and satellite cells—are present in the PNS (peripheral nervous system).
Neuroglia of the CNS (central nervous system= brain+spinal cord) can be classified on the basis of size, cytoplasmic processes, and intracellular organization into four types (Figure 3):
- Astrocytes,
- Oligodendrocytes,
- Microglial cells, and
- Ependymal cells.
Figure 1. Human brain
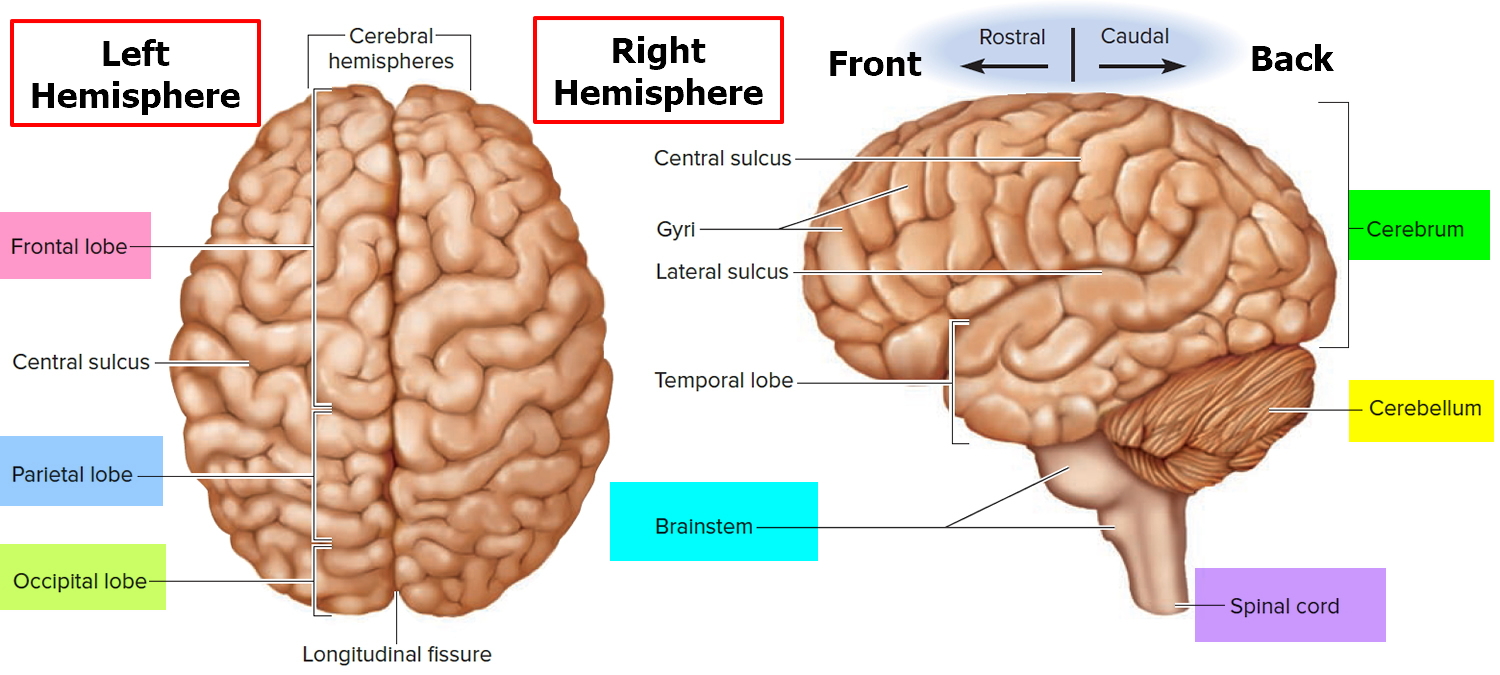
Figure 2. Cerebrum of the brain
Figure 3. Glia cells (Neuroglia) of the CNS
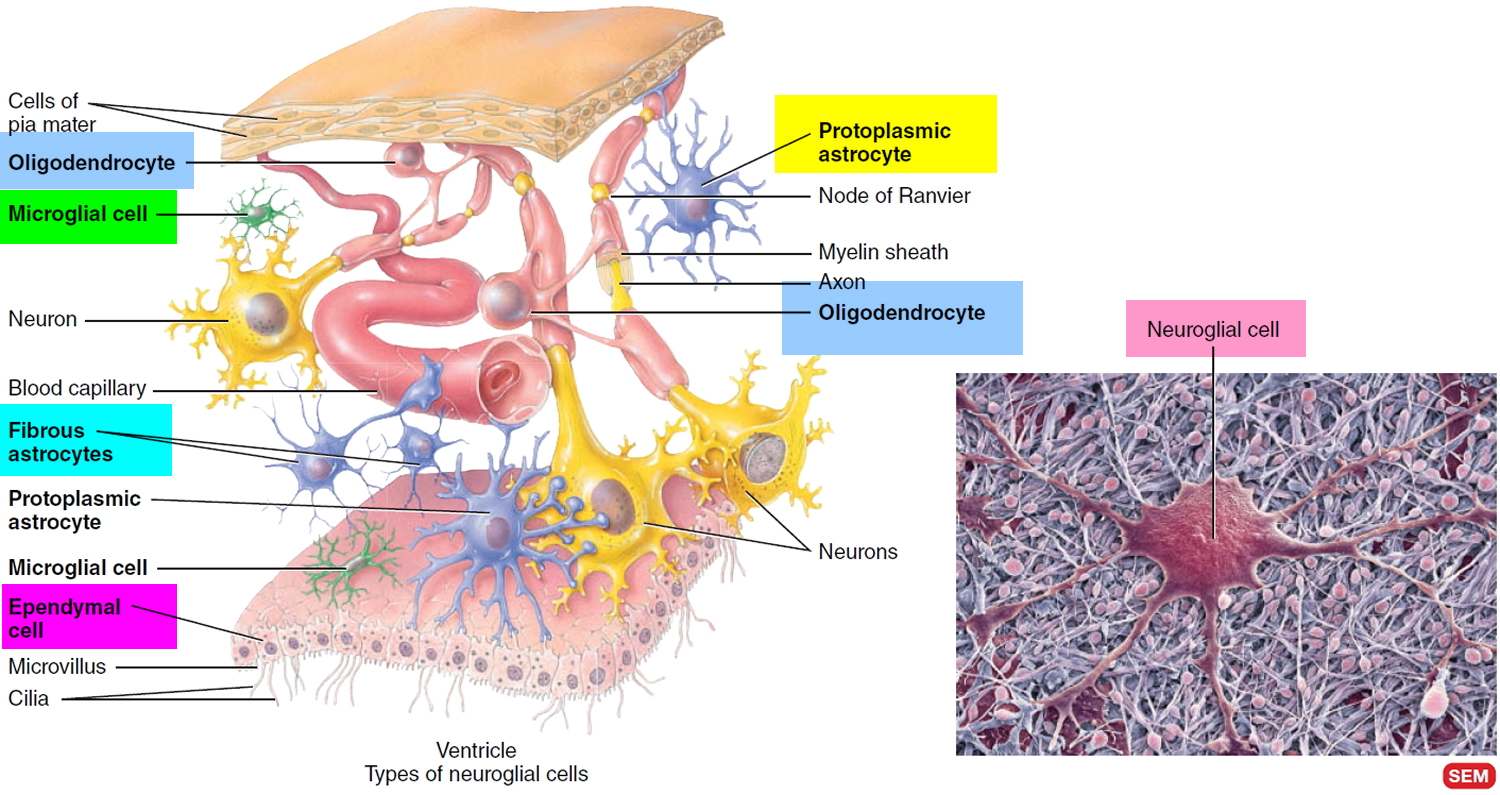
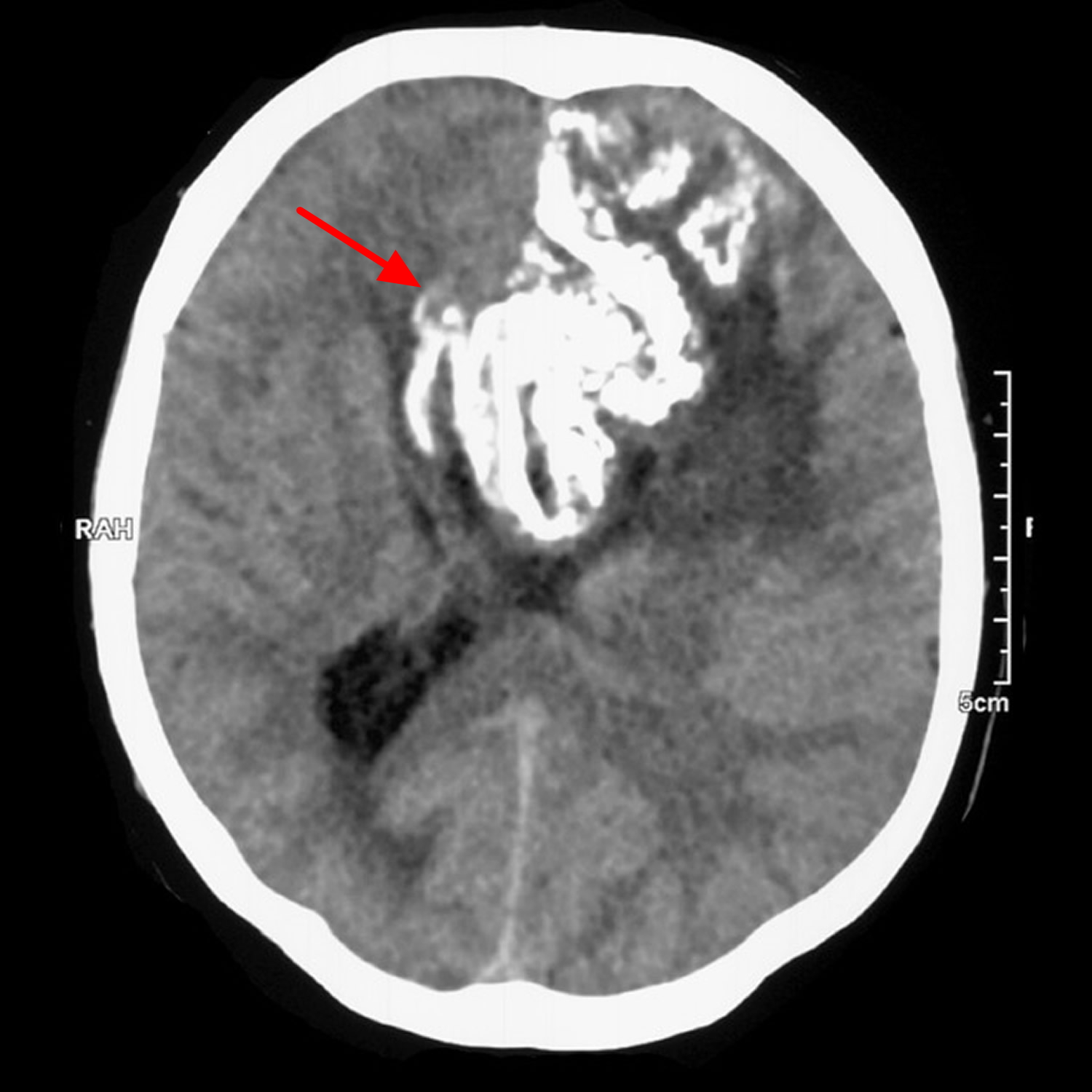
Figure 4. Oligodendroglioma (CT scan demonstrating a large left frontal lobe mass with prominent gyriform calcification. It appears to extend across the corpus callosum)
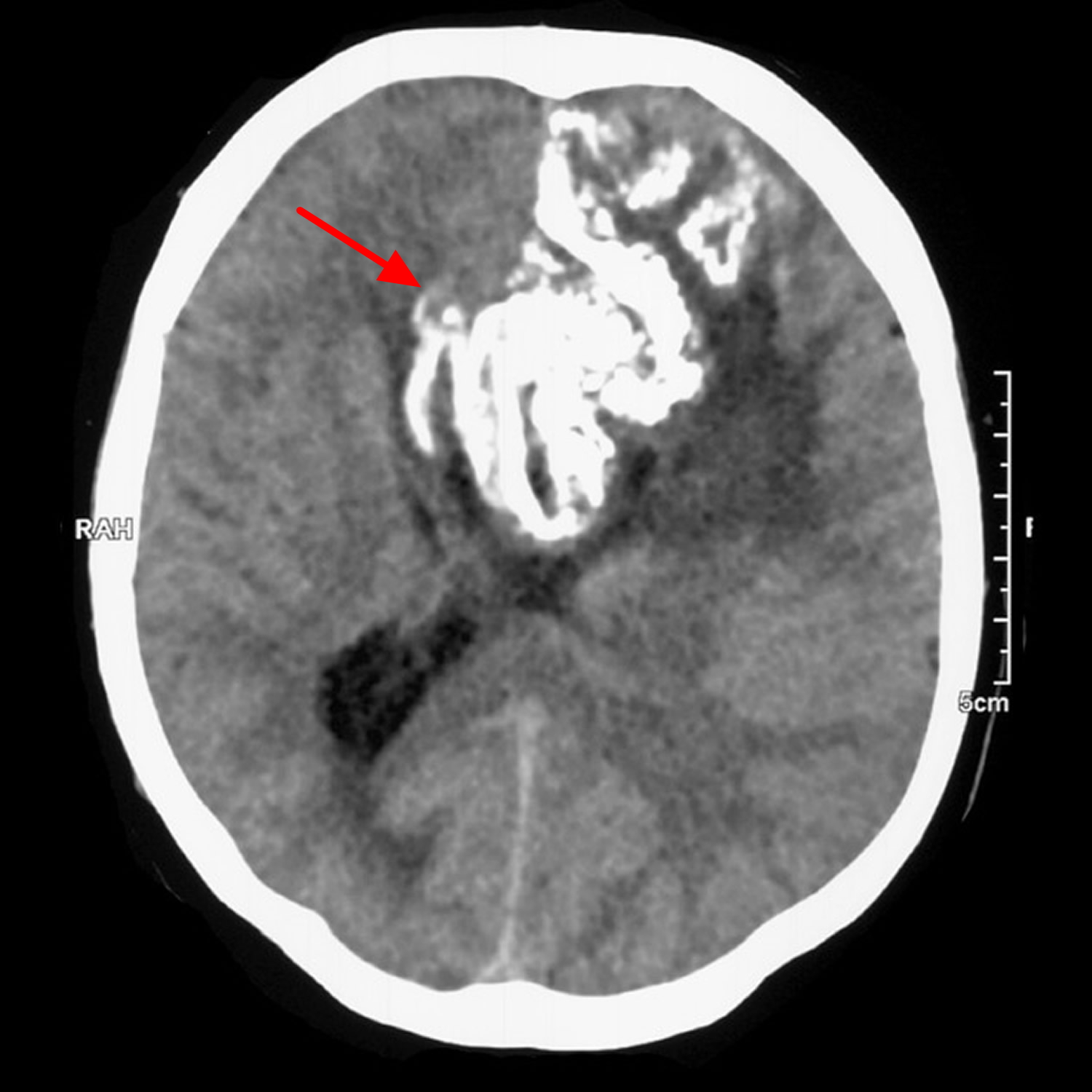
Figure 5. Oligodendroglioma
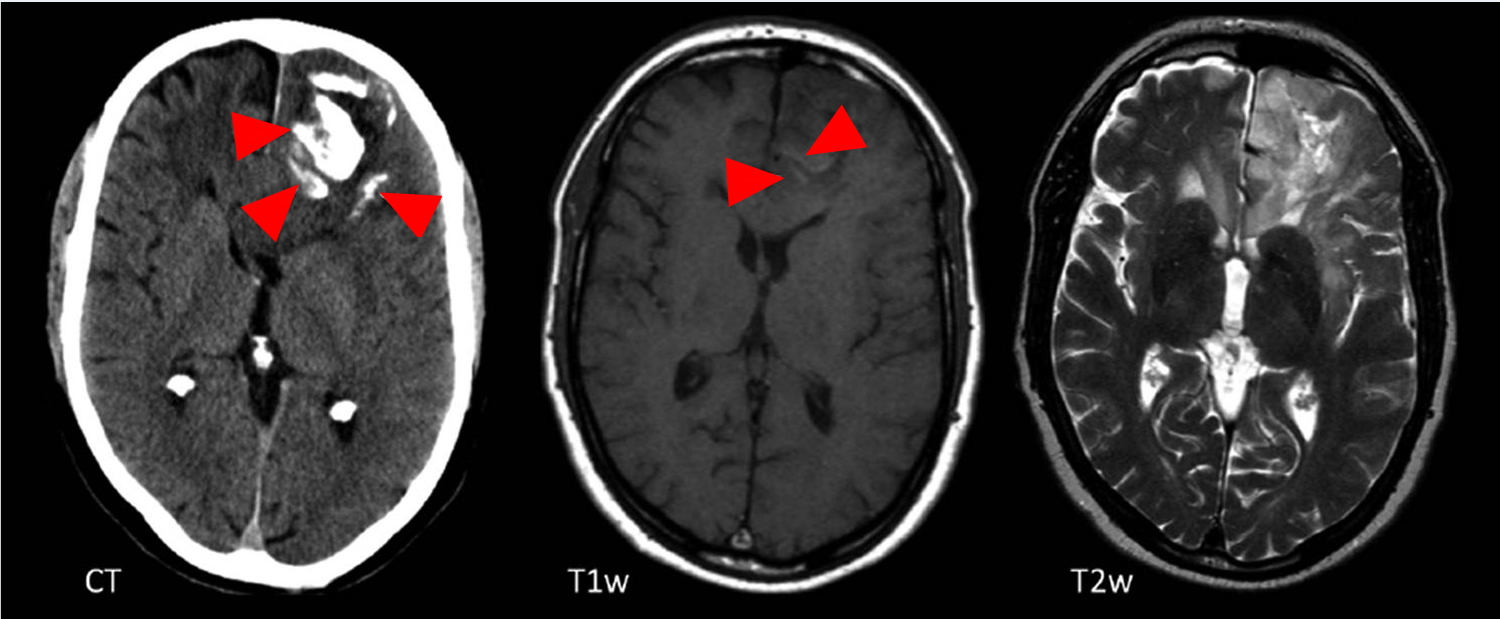
Note: Transverse non-enhanced CT, T1 weighted (T1w) and T2 weighted (T2w) MRI sections, showing extensive, gyriform calcifications (arrows) on CT in the left frontal lobe. Note that the calcifications are only barely visible on T1w images as discrete linear hyperintensities (arrows) and are not at all visible on T2w imaging.
Oligodendroglioma Terminology
Historically, oligodendrogliomas have been defined on histological grounds. As of 2016, however, with the update to the WHO classification of CNS tumors the diagnosis of oligodendroglioma is made by identifying a diffuse infiltrating glioma with isocitrate dehydrogenase (IDH) gene mutation and 1p19q codeletion 3. This is a very significant change, as the tumors previously diagnosed as oligodendrogliomas on the grounds of histology, and those currently diagnosed on the basis of the molecular marker are not identical, with different imaging features. As such it is essential when reading about gliomas to ascertain which definition is being used. As a general rule, anything written before 2016 will use histology alone.
The reliance on molecular markers is, however, not foolproof. Sometimes molecular markers are unavailable (in many countries or regional centres) or sometimes equivocal. In such instances, the diagnosis reverts to histological features alone, and are termed Not-Otherwise-Specified (NOS). Clearly, as the two groups are not the same, this is creating problems when interpreting the literature.
The WHO classification of CNS tumors, therefore, recognizes four diagnoses related to oligodendrogliomas 3:
- oligodendroglioma (WHO grade II): IDH-mt, 1p19q co-deleted
- anaplastic oligodendroglioma (WHO grade III): IDH-mt, 1p19q co-deleted
- oligodendroglioma NOS (WHO grade II): IDH and 1p19q indeterminate
- anaplastic oligodendroglioma NOS (WHO grade III): IDH and 1p19q indeterminate
NOS: not otherwise specified
1p19q codeletion stands for the combined loss of the short arm chromosome 1 (i.e. 1p) and the long arm of chromosome 19 (19q) and is recognized as a genetic marker predictive of therapeutic response to both chemotherapy and combined chemoradiotherapy and overall longer survival in patients with diffuse gliomas, especially those with oligodendroglial components 4.
Historically, the 1p19q codeletion was present in up to 70-85% of oligodendrogliomas and 50% of oligoastrocytomas 4. The recent (2016) revision to the WHO classification of CNS tumors has highlighted the importance of 1p19q codeletion by making it essential for the diagnosis of oligodendroglioma (along with the IDH mutation) 5.
Tumors who have mutations of IDH genes are referred to as “IDH-mutant” or in older literature “IDH positive”. The majority of low-grade diffuse gliomas (astrocytomas and oligodendrogliomas) are IDH-mutant. A minority of glioblastomas are also IDH-mutant, and it is believed that these usually represent secondary glioblastomas (i.e. GBMs that have arisen from pre-existing low-grade tumours) 6. It is important to note that most IDH mutant tumors are also MGMT-methylated (>80%) 9,11.
In other words:
- IDH positive + 1p19q codeletion = oligodendroglioma = better prognosis
- IDH positive + no 1p19q codeletion = diffuse astrocytoma
When the IDH is negative, it corresponds to the ‘wild type’ and the tumor behaves far more aggressively, with a poor prognosis, similar to that of primary glioblastoma multiforme 7. This is discussed further in the article on IDH.
In addition, oligoastrocytomas are also recognized but discussed separately.
It is essential to appreciate that as of the 2016 update to the WHO classification of CNS tumors, to formally make the diagnosis of oligodendroglioma, a tumor must be shown to have IDH-mutation and 1p19q co-deletion.
Oligodendroglioma grade 2 (WHO grade II)
Neoplastic oligodendrocytes appear as regular cells with spherical nuclei containing finely granular chromatin surrounded by a halo of cytoplasm “fried egg” appearance under the light microscope. Typically contains a delicate network of anastomosing capillaries giving it a so-called “chicken wire” appearance 8. These tumors are slowly growing.
Oligodendroglioma grade 3 (WHO grade III)
Anaplastic oligodendroglioma is a WHO grade III diffuse infiltrating glioma that has histological features of anaplasia, and molecular markers consistent with an oligodendroglioma (1p19q co-deletion and IDH mutation) as per the current (2016) WHO classification of CNS tumours 5. They make up 20-50% of all oligodendrogliomas. If the molecular status is unavailable then the diagnosis relies on histology alone and is given the designation anaplastic oligodendroglioma not otherwise specified.
Anaplastic oligodendrogliomas tend to present in a slightly older age group than oligodendroglioma grade 2, presumably on account of them having been undiagnosed for some time.
Anaplastic oligodendrogliomas demonstrate increased cellular density, increased mitotic activity, microvascular proliferation and necrosis. Nuclear anaplasia is also common.
Importantly, and unlike astrocytomas, oligodendrogliomas with necrosis and microvascular proliferation are considered only WHO grade III anaplastic oligodendrogliomas and not WHO grade IV glioblastomas 5. In other words, if you have a tumour that is both IDH mutation and 1p19q co-deleted you can never call it a glioblastoma. Having said that occasionally IDH wild-type glioblastomas may have 1p19q deletions.
Clinical presentation is non-specific with symptoms related to increased intracranial pressure and focal neurological deficits being common. Seizures are also fairly common, but not as common as with oligodendroglioma grade 2.
Oligoastrocytoma
Oligoastrocytomas are tumors of mixed oligodendroglioma and astrocytoma cell populations, that have historically been variably reported depending on local practice. As of the 2016 update to the WHO classification of CNS tumors, to make the diagnosis genomic evidence of both astrocytic and oligodendroglial components will be required to make the diagnosis, and as such, they are likely to become rare 5.
Figure 6. Grading of selected CNS tumors according to the 2016 CNS WHO

Oligodendroglioma survival rate
Response to radiochemotherapy and survival rate depends significantly on presence or absence of 1p19q gene deletion 9.
Survival statistics are primarily available for histologically defined oligodendrogliomas and tend to demonstrate 10 year-survival of approximately 50% 5.
Oligodendroglioma prognosis
Keep in mind that these predictions are estimates. When your doctor talks with you about prognosis, s/he will take into account your age, the location of the tumor, grade of the tumor cells, whether your tumor has deletions of 1p and 19q, and the amount of tumor removed during surgery. Low-grade oligodendrogliomas tend to be slow growing tumors. Anaplastic oligodendrogliomas are more aggressive tumors which grow more quickly. Oligoastrocytoma growth generally depends on the percent of astrocytoma in the tumor, as astrocytomas tend to grow more rapidly than oligodendrogliomas. Scientists continue to study the impact of natural biologic differences amongst all of these tumors and the role of various treatment plans.
Oligodendroglioma life expectancy
Analysis of clinical outcomes showed that persons who had lower-grade gliomas with an IDH mutation and no 1p/19q codeletion had shorter overall survival than did those who had lower-grade gliomas with an IDH mutation with codeletion, yet both of these groups had substantially longer overall survival than did persons who had lower-grade gliomas with wild-type IDH 10.
Patients who had lower-grade gliomas with wild-type IDH were older than those who had lower-grade gliomas with mutated IDH and were more likely to have a family history of cancer.The anatomical locations of the tumors also differed; lower-grade gliomas with mutated IDH arose in frontal lobes more often than did those with wild-type IDH. Among the patients for whom clinical follow-up data were available, 77 of 250 (31%) had tumor recurrence, and 60 of 289 (21%) were deceased at the time of analysis. Patients who had lower-grade gliomas with wild-type IDH had substantially shorter overall survival than did those with lower-grade gliomas with mutated IDH. Their prognosis (median survival, 1.7 years) was intermediate between those of persons who had glioblastomas with wild-type IDH (median survival, 1.1 years) and persons who had glioblastomas with mutated IDH (median survival, 2.1 years) (Figure 7B). In comparison, persons who had lower-grade gliomas with an IDH mutation and 1p/19q codeletion had a median survival of 8.0 years, and those with an IDH mutation and no codeletion had a median survival of 6.3 years 11.
Figure 7. Oligodendroglioma life expectancy
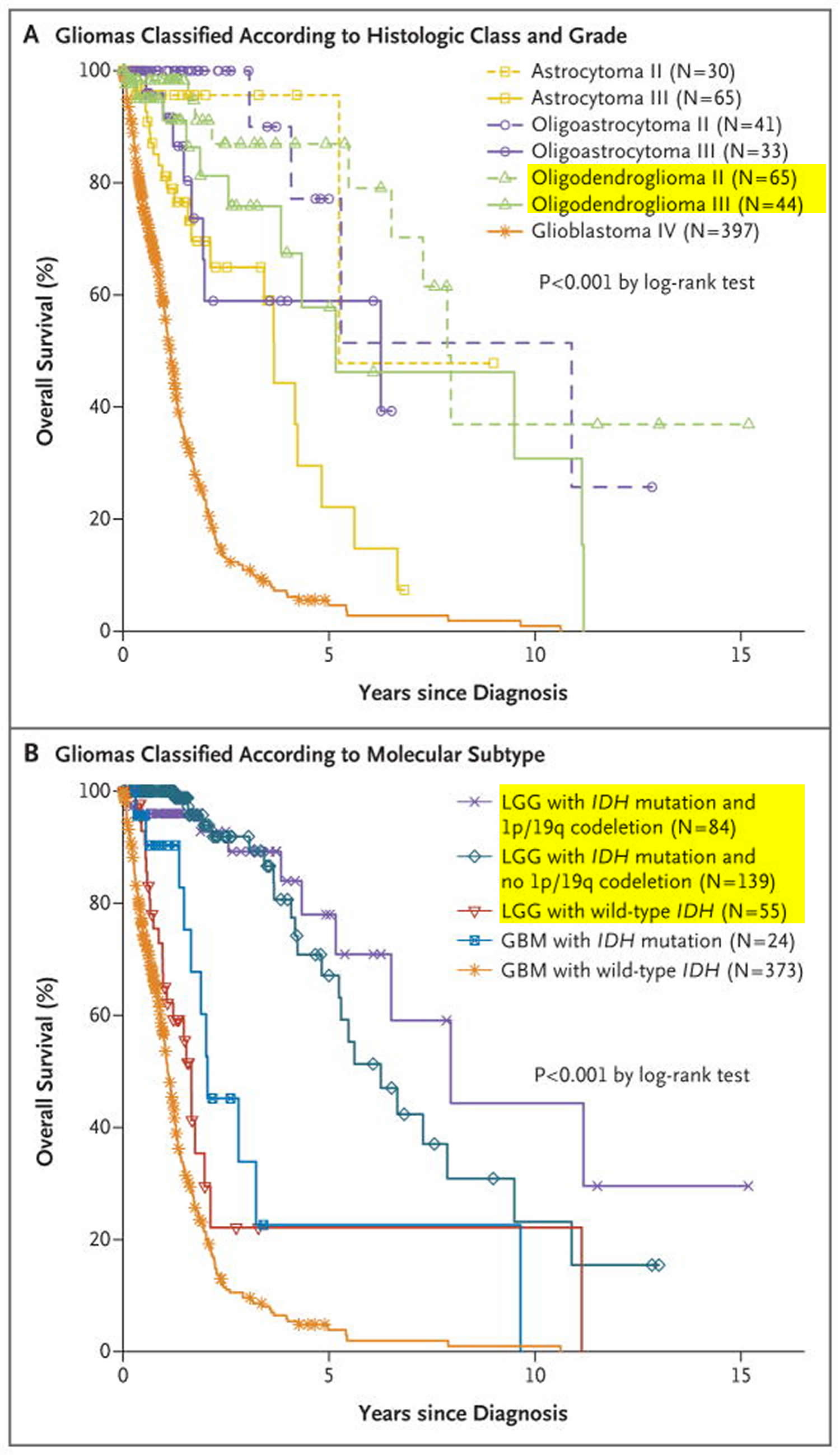
Note: LGG = Low-Grade Gliomas; GBM = Glioblastoma Multiforme
[Source 11]Oligodendroglioma causes
The exact cause of oligodendroglioma is not known. However, scientists have identified chromosomal abnormalities which may play a role in the development of these tumors.
Oligodendroglioma symptoms
Because of their generally slow growth, oligodendrogliomas are often present for years before they are diagnosed. The most common symptoms are seizures, headaches, and personality changes. Other symptoms vary by location and size of the tumor.
Oligodendrogliomas of the frontal lobe may cause weakness on one side of the body, personality or behavior changes, and difficulty with short-term memory. Temporal lobe tumors are usually “silent,” causing few symptoms other than perhaps seizures or language problems.
Oligodendroglioma diagnosis
Tests and procedures used to diagnose oligodendroglioma include:
- Neurological exam. During a neurological exam, your doctor will ask you about your signs and symptoms. He or she may check your vision, hearing, balance, coordination, strength and reflexes. Problems in one or more of these areas may provide clues about the part of your brain that could be affected by a brain tumor.
- Imaging tests. Imaging tests can help your doctor determine the location and size of your brain tumor. MRI is often used to diagnose brain tumors, and it may be used along with specialized MRI imaging, such as functional MRI and magnetic resonance spectroscopy. Other imaging tests may include CT and positron emission tomography (PET).
- Removing a sample of tissue for testing (biopsy). A biopsy can be done with a needle before surgery or during surgery to remove your oligodendroglioma, depending on your particular situation and the location of your tumor. The sample of suspicious tissue is analyzed in a laboratory to determine the types of cells and their level of aggressiveness. Specialized tests of the tumor cells can tell your doctor the types of mutations the cells have acquired. This gives your doctor clues about your prognosis and may guide your treatment options.
The diagnosis of oligodendroglioma and anaplastic oligodendroglioma requires the demonstration of both an IDH gene family mutation and combined whole-arm losses of 1p and 19q (1p/19q codeletion) 5.
In the absence of testing capabilities or in the setting of inconclusive genetic results, a histologically typical oligodendroglioma should be diagnosed as NOS [not otherwise specified] 5. In the setting of an anaplastic oligodendroglioma with non-diagnostic genetic results, careful evaluation for genetic features of glioblastoma may be undertaken 12. It is also recognized that tumors of childhood that histologically resemble oligodendroglioma often do not demonstrate IDH gene family mutation and 1p/19q codeletion; until such tumors are better understood at a molecular level, they should be included in the oligodendroglioma, NOS [not otherwise specified] category 5. However, care should be taken to exclude histological mimics like pilocytic astrocytoma, dysembryoplastic neuroepithelial tumor and clear cell ependymoma 5.
Oligodendroglioma treatment
Your treatment depends on whether your oligodendroglioma is a slow growing type (low grade or grade 2). Or a faster growing type (grade 3 or an anaplastic tumor).
Oligodendroglioma treatment options include:
- Surgery to remove the tumor. Your brain surgeon (neurosurgeon) will work to remove as much of the oligodendroglioma as possible without affecting healthy brain tissue. Specialized surgical techniques, such as awake brain surgery, can help ensure that sensitive brain tissue isn’t damaged during surgery. Additional treatments may be recommended after surgery if any tumor cells remain or if there’s an increased risk your tumor will recur.
- Chemotherapy. Chemotherapy uses drugs to kill cancer cells. Chemotherapy drugs can be taken in pill form or through a vein in your arm. Chemotherapy is often used after surgery to kill any cancer cells that might remain. It can be combined with radiation therapy for aggressive cancers. For people who can’t undergo surgery, radiation therapy and chemotherapy may be used as a primary treatment.
- Radiation therapy. Radiation therapy uses high-energy beams, such as X-rays or protons, to kill cancer cells. During radiation therapy, you lie on a table while a machine moves around you, directing beams to precise points in your brain. Radiation therapy is sometimes recommended after surgery and may be combined with chemotherapy.
- Clinical trials. Clinical trials are studies of new treatments. These studies give you a chance to try the latest treatment options, but the risk of side effects may not be known. Ask your doctor whether you might be eligible to participate in a clinical trial.
- Supportive (palliative) care. Palliative care is specialized medical care that focuses on providing relief from pain and other symptoms of a serious illness. Palliative care specialists work with you, your family and your other doctors to provide an extra layer of support that complements your ongoing care. Palliative care can be used while undergoing other aggressive treatments, such as surgery, chemotherapy or radiation therapy.
Oligodendroglioma grade 2
Monitoring
Some slow growing (low grade) oligodendrogliomas might be monitored with regular CT or MRI scans. This is called watchful waiting. The tumor might not change for many months or years.
Surgery
You might have surgery if your low grade oligodendroglioma is large, or is causing symptoms. Your surgeon will try and remove as much of the tumor as possible. Oligodendrogliomas tend to grow into the brain tissue surrounding the main tumor. This makes it very difficult to remove the tumor completely.
Radiotherapy or chemotherapy
After your operation, you might have radiotherapy or chemotherapy. Or your doctor might monitor you and delay treatment until your tumor is causing symptoms. If some of the tumor remains (also called “residual” tumor), radiation treatment is recommended following surgery. The best timing for radiation therapy (i.e., immediately or when the tumor appears to be growing again), is currently being studied in clinical trials.
You might have chemotherapy (instead of radiotherapy) if you have have a gene change called a 1p19q co-deletion.
Oligodendroglioma grade 3
High grade oligodendrogliomas tend to glow brightly (enhance) on a CT or MRI scan. You have an injection of a contrast drug into your bloodstream to show this.
Anaplastic oligodendroglioma is typically treated with a combination of radiation therapy and chemotherapy. Recurrent anaplastic oligodendroglioma may be treated with surgery and/or chemotherapy.
Surgery
You usually have an operation to remove your tumor. It is not usually possible to remove all of it.
Radiotherapy
After surgery you usually have radiotherapy. This aims to stop the tumor coming back. You usually have daily treatment from Monday to Friday for about 6 weeks.
Chemotherapy
Some research has shown that chemotherapy is useful for people who have a particular gene change called a 1p19q co-deletion. The trials found that some people who had chemotherapy before or after radiotherapy lived for longer.
You have chemotherapy as a drip into your bloodstream, or as a chemotherapy tablet called temozolamide.
If your tumor comes back
Your treatment depends on what treatment you have already had. You might one, or a combination of:
- surgery
- chemotherapy
- radiotherapy
Oligodendroglioma recurrence
Treatments for recurrent, low-grade or high-grade oligodendrogliomas are currently being studied in clinical trials.
References- Smits M. Imaging of oligodendroglioma. The British Journal of Radiology. 2016;89(1060):20150857. doi:10.1259/bjr.20150857. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4846213/
- Osborn A. Osborn’s brain imaging pathology anatomy. Salt Lake City, UT: Amirsys, Inc.; 2012.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK “WHO Classification of Tumours of the Central Nervous System. 4th Edition Revised” ISBN: 9789283244929
- Gadji M, Fortin D, Tsanaclis AM et-al. Is the 1p/19q deletion a diagnostic marker of oligodendrogliomas?. Cancer Genet. Cytogenet. 2009;194 (1): 12-22. doi:10.1016/j.cancergencyto.2009.05.004 https://www.ncbi.nlm.nih.gov/pubmed/19737649
- The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Louis, D.N., Perry, A., Reifenberger, G. et al. Acta Neuropathol (2016) 131: 803. https://doi.org/10.1007/s00401-016-1545-1
- Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13 (5): 345. doi:10.1007/s11910-013-0345-4 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4109985/
- Brat DJ, Verhaak RG et-al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015;372 (26): 2481-98. doi:10.1056/NEJMoa1402121 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4530011/
- Holodny AI. Functional Neuroimaging, A Clinical Approach. Informa HealthCare. (2008) ISBN:0849370566.
- Vogazianou AP, Chan R, Bäcklund LM et-al. Distinct patterns of 1p and 19q alterations identify subtypes of human gliomas that have different prognoses. Neuro-oncology. 2010;12 (7): 664-78. doi:10.1093/neuonc/nop075 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2940668/
- Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, Friedman H, Friedman AH, Riggins GJ, Holdhoff M, Burger P, McLendon R, Bigner DD, Vogelstein B, Meeker AK, Kinzler KW, Papadopoulos N, Diaz LA, Yan H. Oncotarget. 2012 Jul; 3(7):709-22. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3443254/
- The Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. The New England journal of medicine. 2015;372(26):2481-2498. doi:10.1056/NEJMoa1402121. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4530011/
- Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M et al (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372:2481–2498. doi: 10.1056/NEJMoa1402121 http://www.nejm.org/doi/10.1056/NEJMoa1402121




{kind=link}