Pendred syndrome
Pendred syndrome also known as goiter-deafness syndrome or nonsyndromic enlarged vestibular aqueduct, is a disorder typically associated with hearing loss and a thyroid condition called a goiter 1. A goiter is an enlargement of the thyroid gland, which is a butterfly-shaped organ at the base of the neck that produces hormones. If a goiter develops in a person with Pendred syndrome, it usually forms between late childhood and early adulthood. In most cases, this enlargement does not cause the thyroid to malfunction.
In most people with Pendred syndrome, severe to profound hearing loss (although mild-to-moderate progressive hearing impairment also occurs) caused by changes in the inner ear (sensorineural hearing loss) is evident at birth. Less commonly, hearing loss does not develop until later in infancy or early childhood. Some affected individuals also have problems with balance caused by dysfunction of the vestibular system, which is the part of the inner ear that helps maintain the body’s balance and orientation.
An inner ear abnormality called an enlarged vestibular aqueduct is a characteristic feature of Pendred syndrome. The vestibular aqueduct is a bony canal that connects the inner ear with the inside of the skull. Some affected individuals also have an abnormally shaped cochlea, which is a snail-shaped structure in the inner ear that helps process sound. The combination of an enlarged vestibular aqueduct and an abnormally shaped cochlea is known as Mondini malformation.
Pendred syndrome shares features with other hearing loss and thyroid conditions, and it is unclear whether they are best considered as separate disorders or as a spectrum of related signs and symptoms. These conditions include a form of nonsyndromic hearing loss (hearing loss that does not affect other parts of the body) called DFNB4, and, in a small number of people, a form of congenital hypothyroidism resulting from an abnormally small thyroid gland (thyroid hypoplasia). All of these conditions are caused by mutations in the same gene.
The prevalence of Pendred syndrome has been estimated to be between 7.5 and 10 per 100,000 2. However, researchers estimate that it accounts for to up to 10% of all hearing loss that is present from birth (congenital hearing loss) 3. Thus, Pendred syndrome may be the most frequent cause of syndromic deafness 4. Given its autosomal recessive mode of inheritance, the risk for inheritance from heterozygous parents is 25% 2.
Pendred syndrome treatment: Hearing habituation, hearing aids, and educational programs designed for the hearing impaired; consideration of cochlear implantation in individuals with severe-to-profound deafness; standard treatment of abnormal thyroid function.
Surveillance: Repeat audiometry every three to six months initially if hearing loss is progressive, then semiannually or annually. Baseline ultrasound examination of the thyroid with periodic ultrasound surveillance to monitor volumetric changes.
Agents/circumstances to avoid: Some evidence suggests that dramatic increases in intracranial pressure can be associated with a sudden drop in hearing. For this reason, advisability of weightlifting and/or contact sports should be discussed with a physician / health care provider prior to participation.
Figure 1. Pendred syndrome enlarged vestibular aqueduct
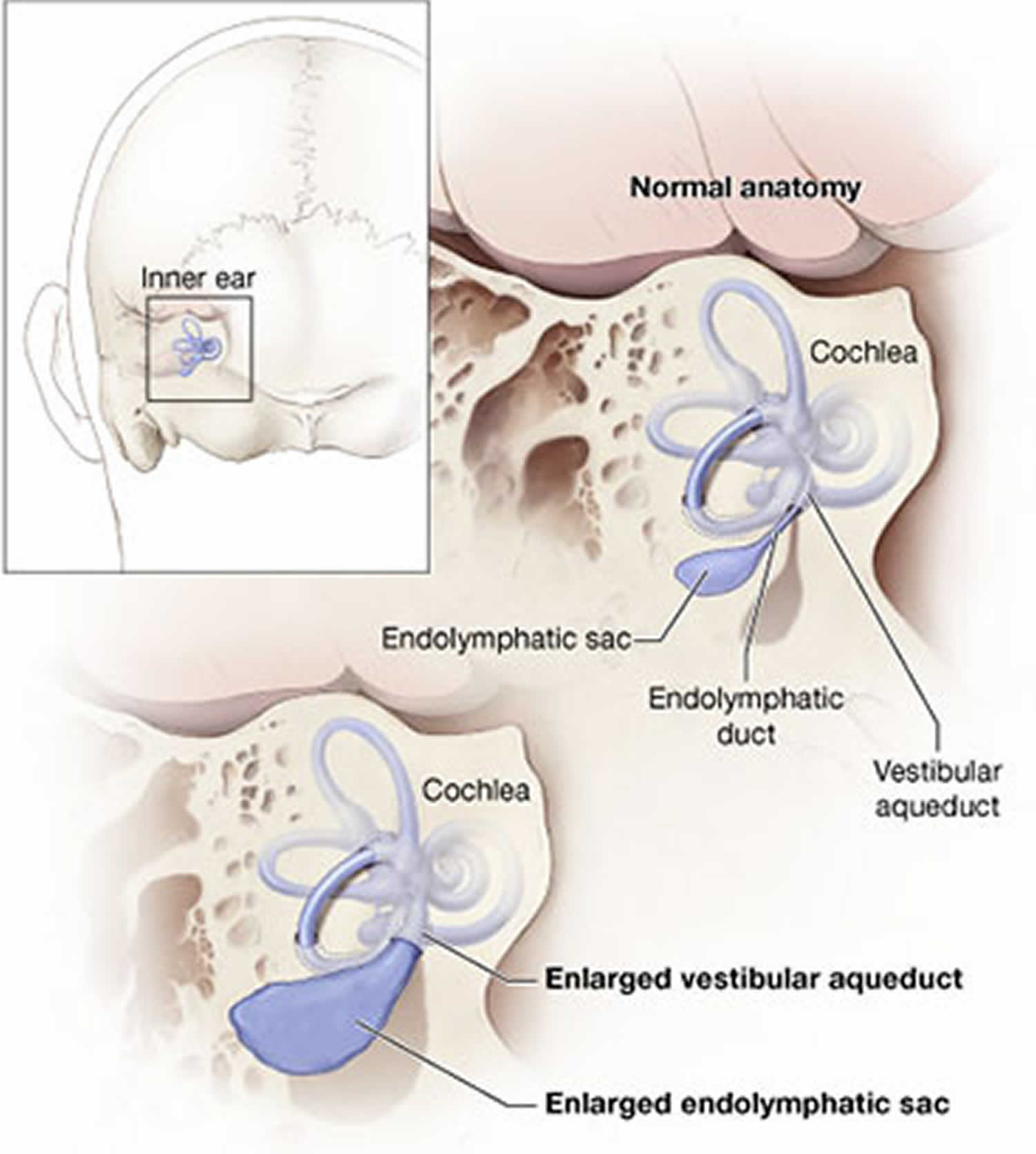
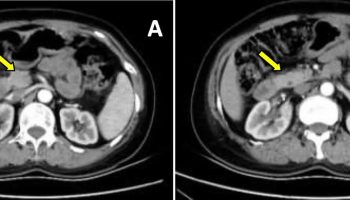
Figure 2. Pendred syndrome CT scan
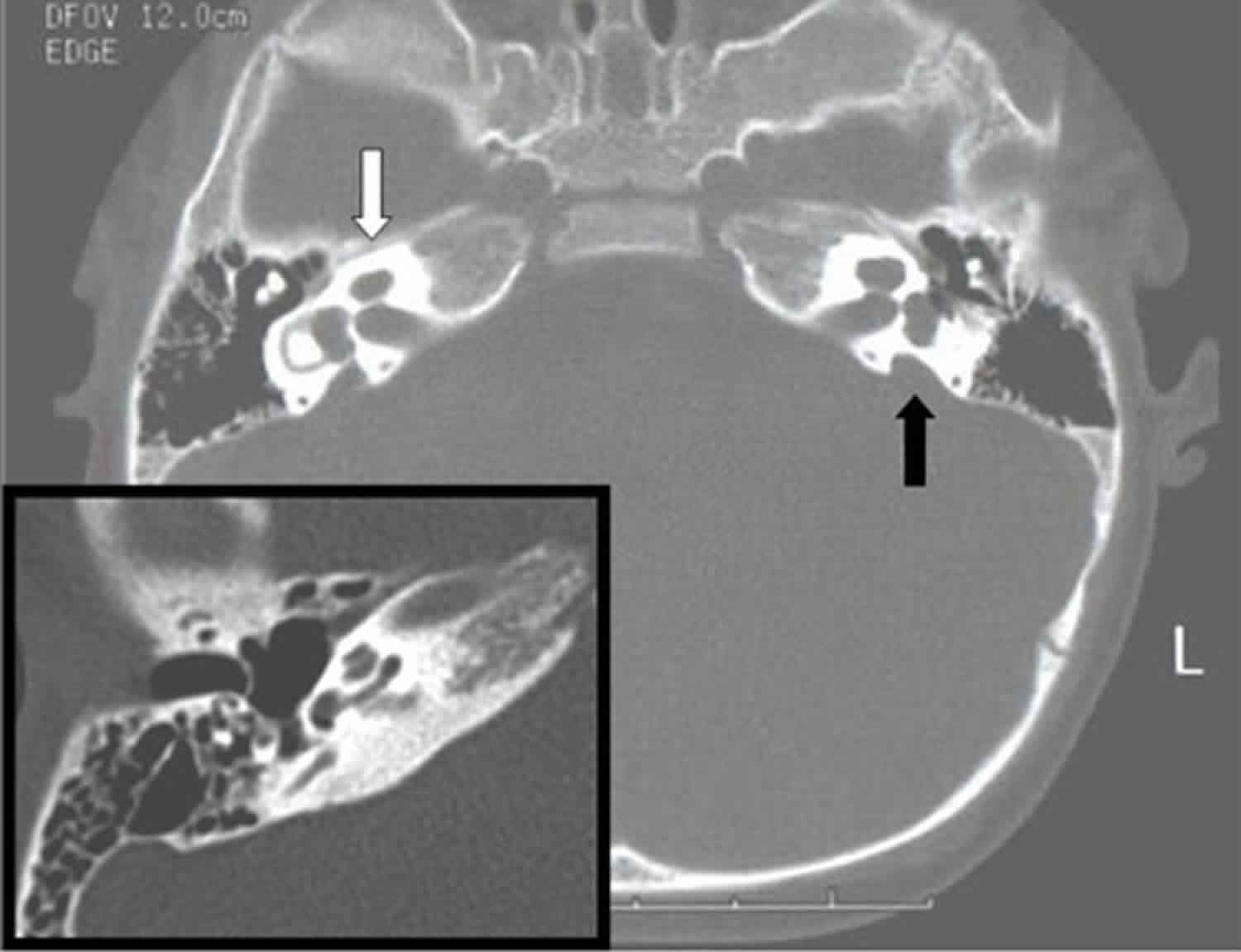
Footnote: Computed tomography in a proband with Pendred syndrome shows absence of the upper turn of the cochlea and deficiency of the modiolus (white arrow). Enlarged vestibular aqueduct is also present (black arrow). Inset shows a normal right cochlea and no enlargement of the vestibular aqueduct, which is not even apparent in this example.
[Source 1 ]How does Pendred syndrome affect other parts of the body?
Pendred syndrome can make the thyroid gland grow larger. An enlarged thyroid gland is called a goiter. The thyroid is a small, butterfly-shaped gland in the front of the neck, just above the collarbones. The thyroid plays a major role in how the body uses energy from food. In children, the thyroid is important for normal growth and development. Children with Pendred syndrome, however, rarely have problems growing and developing properly even if their thyroid is affected. Their levels of thyroid hormones are usually normal.
People with Pendred syndrome are significantly more likely than the general population to develop a goiter in their lifetime, although not everyone who has Pendred syndrome gets a goiter. The typical age for a goiter to develop is in adolescence or early adulthood. If a goiter becomes large, there may be problems with breathing and swallowing. In this case, a health professional should check the goiter and decide whether treatment is necessary. People with Pendred syndrome may need to visit an endocrinologist, who is a specially trained doctor familiar with diseases and disorders that involve the endocrine system, including the thyroid gland.
Pendred syndrome also can affect the vestibular system, which controls balance. Some people with Pendred syndrome will show vestibular weakness when their balance is tested. However, the brain is very good at making up for a weak vestibular system, and most children and adults with Pendred syndrome don’t have a problem with their balance or have difficulty doing routine tasks. Some babies with Pendred syndrome may start walking later than other babies, however.
Scientists don’t know why some people with Pendred syndrome develop a goiter or have balance problems and others don’t.
Will a person with Pendred syndrome have affected children?
Pendred syndrome is inherited in an autosomal recessive manner. The children of a person with Pendred syndrome will definitely be carriers of the condition. Carriers typically do not have any signs or symptoms. Children of a person with Pendred syndrome have a chance to have the condition if the other parent is a carrier.
People with specific questions about the genetics of Pendred syndrome and/or the probability of having a child with Pendred syndrome should speak with a genetics professional.
Pendred syndrome causes
Mutations in the SLC26A4 gene cause about half of all cases of Pendred syndrome. The SLC26A4 gene (formerly known as the PDS gene) provides instructions for making a protein called pendrin. This protein transports negatively charged particles (ions), including chloride, iodide, and bicarbonate, into and out of cells. Although the function of pendrin is not fully understood, this protein is important for maintaining the proper levels of ions in the thyroid and the inner ear. Mutations in the SLC26A4 gene alter the structure or function of pendrin, which disrupts ion transport. An imbalance of particular ions disrupts the development and function of the thyroid gland and structures in the inner ear, which leads to the characteristic features of Pendred syndrome.
In people with Pendred syndrome who do not have mutations in the SLC26A4 gene, the cause of the condition is unknown. Researchers suspect that other genetic and environmental factors may influence the condition.
Pendred syndrome gene
Pendred syndrome occurs due to mutation in The Pendrin Gene (SLC26A4), which encodes Pendrin/SLC26A4 Protein 3. SLC26A4 is on chromosome 7q22.3, and it expresses in multiple organs, including the inner ear, kidneys, thyroid, and bronchial epithelial cells 2. It a multifunctional anion exchanger that has an affinity to chloride, iodide, bicarbonate, and other anions 2.
In people with Pendred syndrome who do not have mutations in the SLC26A4 gene, the cause of the condition is unknown. Researchers suspect that other genetic and environmental factors may influence the condition.
Pendred syndrome inheritance pattern
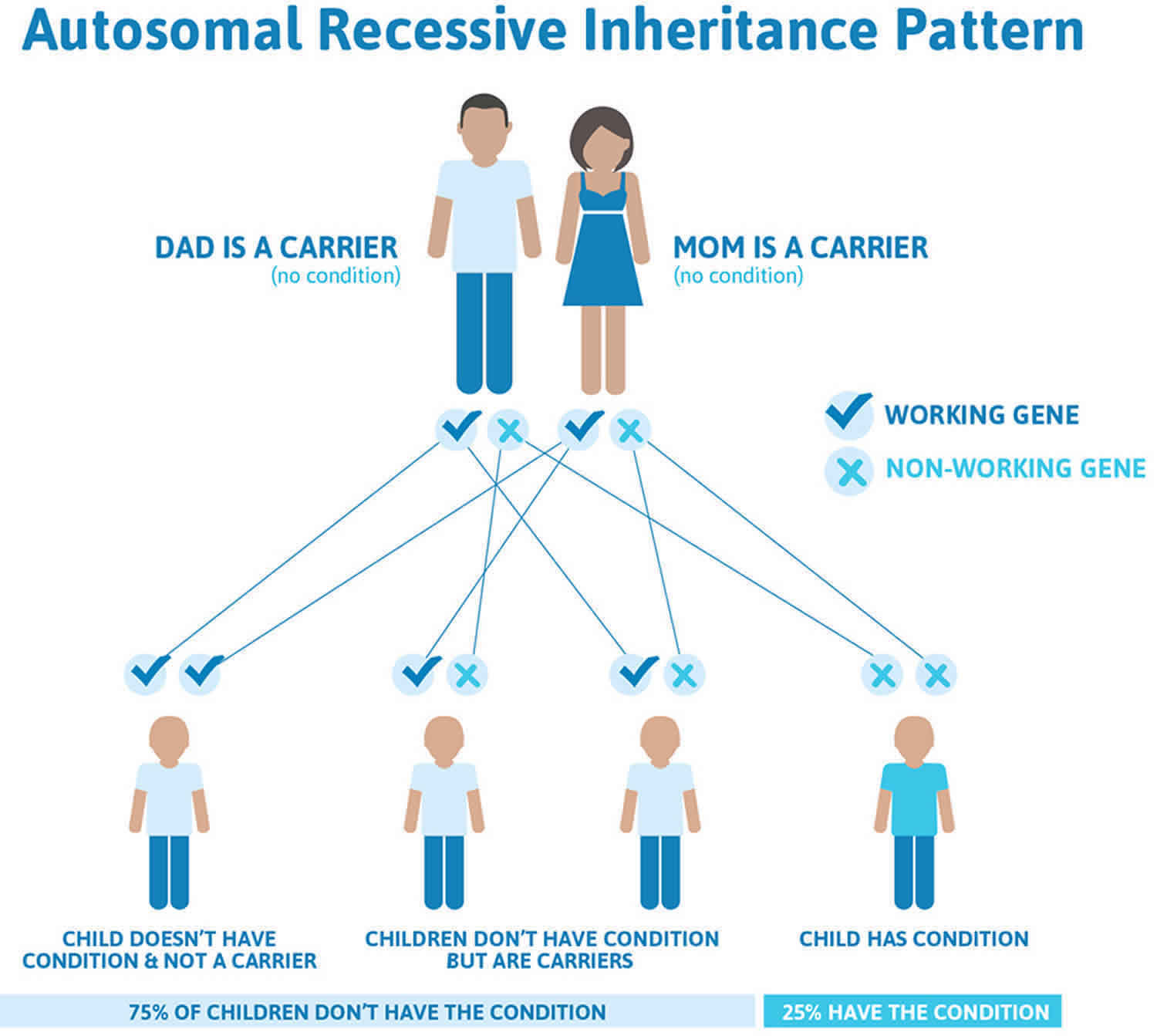
Pendred syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 3 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 3. Pendred syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Pendred syndrome pathophysiology
In the inner ear: Pendrin is expressed in the cochlea and vestibule of the inner ear, thus playing an important role in endolymphatic fluid resorption, acid-base balance, and proper function of the inner ear 3.
In the thyroid gland: Pendrin is expressed on the thyrocyte apical membrane, where its activity as a Cl-/I- exchanger is essential for cellular iodide efflux into the follicular lumen. A defect in the Pendrin gene usually leads to the partial impairment of thyroid organification. However, patients with Pendred syndrome usually have a euthyroid goiter 2.
In the Kidneys: Pendrin protein is located at or near the apical membrane of type B and non A non B intercalated cells of the cortical collecting ducts 3. Pendrin acts as chloride/anion exchanger, which leads to bicarbonate secretion to the tubular lumen and chloride reabsorption 2. Therefore, it plays an important role in the regulation of blood pressure 2 and fluid balance 5. Defect in Pendrin protein can cause metabolic alkalosis 6.
In Bronchial epithelium: Pendrin is also expressed in the airway epithelium at the apical membrane of bronchial epithelial cells. It helps to regulate airway surface liquid thickness via its function as a Chloride/bicarbonate exchanger. The defective pendrin gene affects mucus production and may play a vital role in patients with asthma and COPD. It also functions as SCN/Cl exchanger helps in the innate defense mechanism of mucosal surfaces by secreting SCN, which is an antioxidant to the lumen 3.
Pendred syndrome symptoms
- Hearing Impairment: Pendred syndrome patients have sensorineural hearing loss that is broad and can range from mild to profound 4. Hearing impairment typically is congenital or prelingual and profound. It can also develop later in infancy with progressive worsening, which may be aggravated by exposure to acoustic traumas, barotraumas, or head injury 2. Hearing impairment is usually bilateral, although asymmetry can be present. Early signs include the absence of a reaction to sound or a delay in language acquisition.
- Vestibular dysfunctions are usually not obvious but maybe noticed during progressive hearing loss period or in performances needing motor skills and balance 2. Some patients may show features of temporal bone abnormalities 7.
- Thyroid: Euthyroid goiter is the typical presentation of Pendred syndrome 8. Enlargement of the thyroid gland occurs because of iodide organification defects. It may also present with multinodular goiter in late childhood or early puberty 2. Some may develop thyroid goiter during their adult life. Approximately 75% of patients with Pendred syndrome have a goiter on physical examination 8. While the majority of patients present with congenital goiter 4, the rest can have normal size gland, especially if they have adequate iodine intake 2. Since Iodide organification is not solely dependent on pendrin, patients usually have partial iodide organification defects 8. While 50 % of patients will have normal thyroid function, others have subclinical hypothyroidism, which can be congenital 2.
- Renal System: Patients with Pendred syndrome might develop life-threatening metabolic alkalosis due to acid-base balance abnormalities 6.
Pendred syndrome complications
Pendred syndrome complications include:
- Progressive hearing impairment
- Hypothyroidism, euthyroid goiter or thyroid nodules
- Life-threatening metabolic alkalosis 6
- There is a report of a rare case of Hoffmann syndrome with this condition 9.
Pendred syndrome diagnosis
The diagnosis of Pendred syndrome is suggested by the following clinical, temporal bone imaging, and endocrine findings.
Clinical findings
Sensorineural hearing impairment is usually congenital (or prelingual), non-progressive, and severe to profound as measured by auditory brain stem response (ABR) testing or pure tone audiometry. For evaluation of hearing loss, see Hereditary Hearing Loss and Deafness Overview.
Temporal Bone imaging findings
Affected individuals have temporal bone abnormalities, which can be identified by thin-cut CT with detailed cochlear anatomy. The identification and interpretation of temporal bone defects require both the appropriate test (i.e., thin-cut CT as a routine CT of the temporal bones typically will not suffice) and detailed familiarity with cochlear anatomy.
The enlarged vestibular aqueduct is measurable by high-resolution CT of the temporal bone with coronal and axial sections. Confirmation of vestibular aqueduct enlargement is if the width of the middle portion of the descending limb of the vestibular aqueduct over 1.5 mm 10. Patients also can have cochlear hypoplasia when cochlea have 1.5 turn instead of normal 2.75 turns. If a patient has vestibular aqueduct enlargement associated with cochlear hypoplasia, it is referred to as Mondini malformation 3.
- Mondini malformation or dysplasia (bilateral enlarged vestibular aqueduct [EVA] with cochlear hypoplasia) is detected on thin-cut CT of the temporal bones. The cochlea is hypoplastic and has 1.5 cochlear turns instead of the expected 2.75 turns, and the vestibular aqueduct is enlarged, with a midpoint width exceeding 1.5 mm. The presence of both cochlear hypoplasia and enlarged vestibular aqueduct is known as a Mondini malformation or dysplasia. While the temporal bones are abnormal radiologically in all persons with Pendred syndrome 11, a range of findings can be present. In a study of individuals homozygous for the same SLC26A4 pathogenic variant, high-resolution CT revealed that 100% had deficiency of the modiolus (i.e., the bony polyhedral structure centered on the cochlea was not apparent on a mid-modiolar section); 80% had enlarged vestibular aqueduct (i.e., width in the middle portion of the descending limb of the vestibular aqueduct >1.5 mm); and 75% had absence of the upper turn of the cochlea (i.e., the interscalar septum was not seen between the upper and middle turns) (see Figure 2 above) 11.
Note: A radiologic diagnosis of EVA with or without cochlear hypoplasia does not equate to a clinical diagnosis of Pendred syndrome as there are other causes of these types of temporal bone malformations without associated thyroid abnormality (see Differential Diagnosis). - Nonsyndromic enlarged vestibular aqueduct (NSEVA) is detected on thin-cut CT of the temporal bones. The vestibular aqueduct is enlarged when its midpoint width exceeds 1.5 mm.
Thyroid Findings
Euthyroid goiter is the typical thyroid abnormality in Pendred syndrome, and it is detected by volumetric studies to assess the size of the gland, thyroid ultrasound helps to evaluate the volume of the thyroid gland and characterize the size and structure of the nodules 2. If Pendred syndrome diagnosis is confirmed, thyroid function tests should take place regularly. Pendred syndrome can also coexist with autoimmune thyroiditis 2.
Perchlorate test is a screening test for iodide organification defects done by administering radioactive iodine and measuring the intrathyroidal radioactive iodine content. If it is less than 10%, it suggests an organification defect. However, a negative test does not rule out the diagnosis of Pendred syndrome because multiple factors can affect the results. One of the factors is the previous use of high dose iodine intake, which can interfere with the testing 2.
Establishing the diagnosis
The clinical diagnosis of Pendred syndrome is established in a proband with sensorineural hearing loss, characteristic temporal bone abnormalities identified on thin-cut CT and euthyroid goiter.
Molecular genetic testing is the confirmatory test and should be performed to confirm the diagnosis 2. SLC26A4 gene is the most affected gene, and 50 % of Pendred syndrome patients have a mutation in this gene 7. Genetic testing is by direct sequencing of the coding region of the SLC26A4 gene 2. By identifying the biallelic pathogenic variant in the gene or the presence of double heterozygous for one pathogenic variant 8. FOXII and KCNJI0 gene mutations are affected in less than 2% of patients 7. Because of the diverse mutations in the SLC26A4 gene, researchers have identified approximately 200 sequence variants 2. Efforts should be made to recognize the full range of presentation of the syndrome to narrow down the suspected cases before performing the molecular studies, taking into account the more specific clinical criteria 10.
The molecular diagnosis of Pendred syndrome is established by identification of biallelic pathogenic variants in SLC26A4 or double heterozygosity for one pathogenic variant in SLC26A4 and one pathogenic variant in either FOXI1 or KCNJ10 (Table 1).
The outcome of testing varies by ethnicity and phenotype.
Ethnicity:
- In Korean and Japanese probands, more than 80% have two pathogenic variants in SLC26A4, slightly more than 10% have one pathogenic variant, and fewer than 10% have no pathogenic variants 12.
- In North American or European Caucasians with Pendred syndrome only about 25% have two pathogenic variants in SLC26A4, as would be expected for autosomal recessive inheritance 13. About half have no detectable SLC26A4 pathogenic variants, and in 25%, only one pathogenic variant is found 14.
Phenotype:
- The number of pathogenic variants in Caucasians is strongly correlated with the auditory and thyroid phenotypes: those with Pendred syndrome are more likely than those with Pendred syndrome to have biallelic pathogenic variants 15.
- The degree of hearing loss in persons with Pendred syndrome is greater if two (as opposed to 1 or 0) SCL26A4 pathogenic variants are identified 16.
An explanation for these molecular findings has been described by Chattaraj et al 17, who identified a haplotype Caucasian EVA (CEVA) – comprising 12 variants upstream of SLC26A4 – which is frequently found in persons with Pendred syndrome in trans with a coding or splice site variant.
Approach to molecular genetic testing. For all persons with hearing loss, the use of a multigene panel for hearing loss and deafness maximizes the diagnostic rate while minimizing the diagnostic expense.
Hearing loss and deafness multigene panels typically include SLC26A4, FOXI1, KCNJ10, and other genes of interest.
Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Note: Comprehensive genomic testing (i.e., exome sequencing and genome sequencing) is currently not justified as a primary screen for genetic causes of deafness 18.
Table 1. Molecular Genetic Testing Used in Pendred syndrome and nonsyndromic enlarged vestibular aqueduct
| Gene 1 | Proportion of Pendred syndrome & nonsyndromic enlarged vestibular aqueduct Attributed to Pathogenic Variants in Gene | Proportion of Pathogenic Variants Detectable by Method | ||
|---|---|---|---|---|
| Pendred syndrome | Nonsyndromic enlarged vestibular aqueduct | Sequence analysis 2 | Gene-targeted deletion/duplication analysis 3 | |
| FOXI1 | None described | <1% 4 | 2/2 4 | Unknown |
| KCNJ10 | None described | <1% 5 | 2/2 5 | Unknown |
| SLC26A4 | ~90% 8 | 50%-90% 6 | ~90% | ~10% 7 |
| Unknown | Unknown | ~50% | NA | |
Footnotes:
1. Genes are listed alphabetically.
2. Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
3. Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
4. In two families, persons with nonsyndromic enlarged vestibular aqueduct had a heterozygous pathogenic variant in both SLC26A4 and FOXI1 19.
5. In two families, persons with nonsyndromic enlarged vestibular aqueduct had a heterozygous pathogenic variant in both KCNJ10 and SLC26A4 20.
6. The proportion of Pendred syndrome and nonsyndromic enlarged vestibular aqueduct attributable to SLC26A4 varies by ascertainment, inheritance, and ethnicity. In patients ascertained for inner ear malformations (specifically enlarged vestibular aqueduct with or without cochlear hypoplasia), the proportion of cases attributable to SLC26A4 is ~40%-50% in the European-American population and higher in multiplex families and Asian populations 16.
7. Single-exon and multiexon SLC26A4 deletions have been reported 21.
Pendred syndrome treatment
There is no definite management for Pendred syndrome. The treatment basis is from clinical manifestations. Because Pendred syndrome is inherited and can involve thyroid and balance problems, many specialists may be involved in treatment, including a primary care physician, an audiologist, an endocrinologist, a clinical geneticist, a genetic counselor, an otolaryngologist, and a speech-language pathologist. Pendred syndrome isn’t curable, but a medical team will work together to encourage informed choices about treatment options. They also can help people prepare for increased hearing loss and other possible long-term consequences of the syndrome.
- Assessment of hearing impairment and treatment accordingly by providing hearing aids and cochlear implants 22.
- Monitoring thyroid function and thyroid gland size, which might need medical or surgical intervention 8.
- The importance of genetic counseling for the family and testing family members and discussion about offspring outcome 8.
To reduce the likelihood of hearing loss progression, children and adults with Pendred syndrome should avoid contact sports that might lead to head injury; wear head protection when engaged in activities such as bicycle riding and skiing that might lead to head injury; and avoid situations that can lead to barotrauma (extreme, rapid changes in pressure), such as scuba diving or hyperbaric oxygen treatment.
Children with Pendred syndrome should start early treatment to gain communication skills, such as learning sign language or cued speech or learning to use a hearing aid. Most people with Pendred syndrome will have hearing loss significant enough to be considered eligible for a cochlear implant. A cochlear implant is an electronic device that is surgically inserted into the cochlea. While a cochlear implant doesn’t restore or create normal hearing, it bypasses injured areas of the ear to provide a sense of hearing in the brain. Children as well as adults are eligible to receive an implant.
People with Pendred syndrome who develop a goiter need to have it checked regularly. The type of goiter found in Pendred syndrome is unusual because even though it grows in size, it still continues to make normal amounts of thyroid hormone. Such a goiter often is called a euthyroid goiter.
Pendred syndrome prognosis
- Pendred syndrome patients usually have a progressive hearing impairment. Patients with the syndrome can benefit from cochlear implantation, which provides optimal hearing rehabilitation, cochlear implantation in Pendred syndrome can have some surgical difficulties due to the inner ear malformations 23.
- Pendred syndrome also affects the thyroid gland and might lead to euthyroid goiter or clinical hypothyroidism 2.
- Smith RJH. Pendred Syndrome/Nonsyndromic Enlarged Vestibular Aqueduct. 1998 Sep 28 [Updated 2017 Oct 19]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1467
- Wémeau JL, Kopp P. Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2017 Mar;31(2):213-224.
- Rozenfeld J, Efrati E, Adler L, Tal O, Carrithers SL, Alper SL, Zelikovic I. Transcriptional regulation of the pendrin gene. Cell. Physiol. Biochem. 2011;28(3):385-96.
- Smith N, U-King-Im JM, Karalliedde J. Delayed diagnosis of Pendred syndrome. BMJ Case Rep. 2016 Sep 12;2016
- Rozenfeld J, Tal O, Kladnitsky O, Adler L, Efrati E, Carrithers SL, Alper SL, Zelikovic I. The pendrin anion exchanger gene is transcriptionally regulated by uroguanylin: a novel enterorenal link. Am. J. Physiol. Renal Physiol. 2012 Mar 01;302(5):F614-24.
- Kandasamy N, Fugazzola L, Evans M, Chatterjee K, Karet F. Life-threatening metabolic alkalosis in Pendred syndrome. Eur. J. Endocrinol. 2011 Jul;165(1):167-70.
- Koffler T, Ushakov K, Avraham KB. Genetics of Hearing Loss: Syndromic. Otolaryngol. Clin. North Am. 2015 Dec;48(6):1041-61.
- Smith RJH. Pendred Syndrome/Nonsyndromic Enlarged Vestibular Aqueduct. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. University of Washington, Seattle; Seattle (WA): Sep 28, 1998.
- Tahir F, Qadar LT, Khan M, Hussain H, Iqbal SU. Hoffmann’s Syndrome Secondary to Pendred Syndrome: A Rare Case. Cureus. 2019 Mar 06;11(3):e4195
- Fugazzola L, Cerutti N, Mannavola D, Crino A, Cassio A, Gasparoni P, Vannucchi G, Beck-Peccoz P. Differential diagnosis between Pendred and pseudo-Pendred syndromes: clinical, radiologic, and molecular studies. Pediatr. Res. 2002 Apr;51(4):479-84.
- Goldfeld M, Glaser B, Nassir E, Gomori JM, Hazani E, Bishara N. CT of the ear in Pendred syndrome. Radiology. 2005;235:537–40.
- Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, Jang HS, Moon SK, Lee SC, Chun YM, Lee HK, Choi JY, Jung SC, Griffith AJ, Koo SK. Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet. 2005;67:160–5.
- Ito T, Choi BY, King KA, Zalewski CK, Muskett J, Chattaraj P, Shawker T, Reynolds JC, Butman JA, Brewer CC, Wangemann P, Alper SL, Griffith AJ. SLC26A4 genotypes and phenotypes associated with enlargement of the vestibular aqueduct. Cell Physiol Biochem. 2011;28:545–52.
- Choi BY, Madeo AC, King KA, Zalewski CK, Pryor SP, Muskett JA, Nance WE, Butman JA, Brewer CC, Griffith AJ. Segregation of enlarged vestibular aqueducts in families with non-diagnostic SLC26A4 genotypes. J Med Genet. 2009a;46:856–61.
- Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith RJ. Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet. 2007;122:451–7.
- Rose J, Muskett JA, King KA, Zalewski CK, Chattaraj P, Butman JA, Kenna MA, Chien WW, Brewer CC, Griffith AJ. Hearing loss associated with enlarged vestibular aqueduct and zero or one mutant allele of SLC26A4. Laryngoscope. 2017;127:E238–43.
- Chattaraj P, Munjal T, Honda K, Rendtorff ND, Ratay JS, Muskett JA, Risso DS, Roux I, Gertz EM, Schäffer AA, Friedman TB, Morell RJ, Tranebjærg L, Griffith AJ. A common SLC26A4-linked haplotype underlying non-syndromic hearing loss with enlargement of the vestibular aqueduct. J Med Genet. 2017;54:665–73.
- Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJ. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441–50.
- Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, Smith RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet. 2007;80:1055–63.
- Yang T, Gurrola JG 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–7.
- Pera A, Villamar M, Viñuela A, Gandía M, Medà C, Moreno F, Hernández-Chico C. A mutational analysis of the SLC26A4 gene in Spanish hearing-impaired families provides new insights into the genetic causes of Pendred syndrome and DFNB4 hearing loss. Eur J Hum Genet. 2008;16:888–96.
- van Nierop JW, Huinck WJ, Pennings RJ, Admiraal RJ, Mylanus EA, Kunst HP. Patients with Pendred syndrome: is cochlear implantation beneficial? Clin Otolaryngol. 2016 Aug;41(4):386-94.
- Kontorinis G, Lenarz T, Lesinski-Schiedat A, Neuburger J. Cochlear implantation in Pendred syndrome. Cochlear Implants Int. 2011 Aug;12(3):157-63.




{kind=link}