Rhizomelic chondrodysplasia punctata
Rhizomelic chondrodysplasia punctata is a class of peroxisomal disorders characterized by defective plasmalogen biosynthesis that impairs the normal development of many parts of the body 1. The major features of rhizomelic chondrodysplasia punctata include skeletal abnormalities, distinctive facial features, intellectual disability, and respiratory problems.
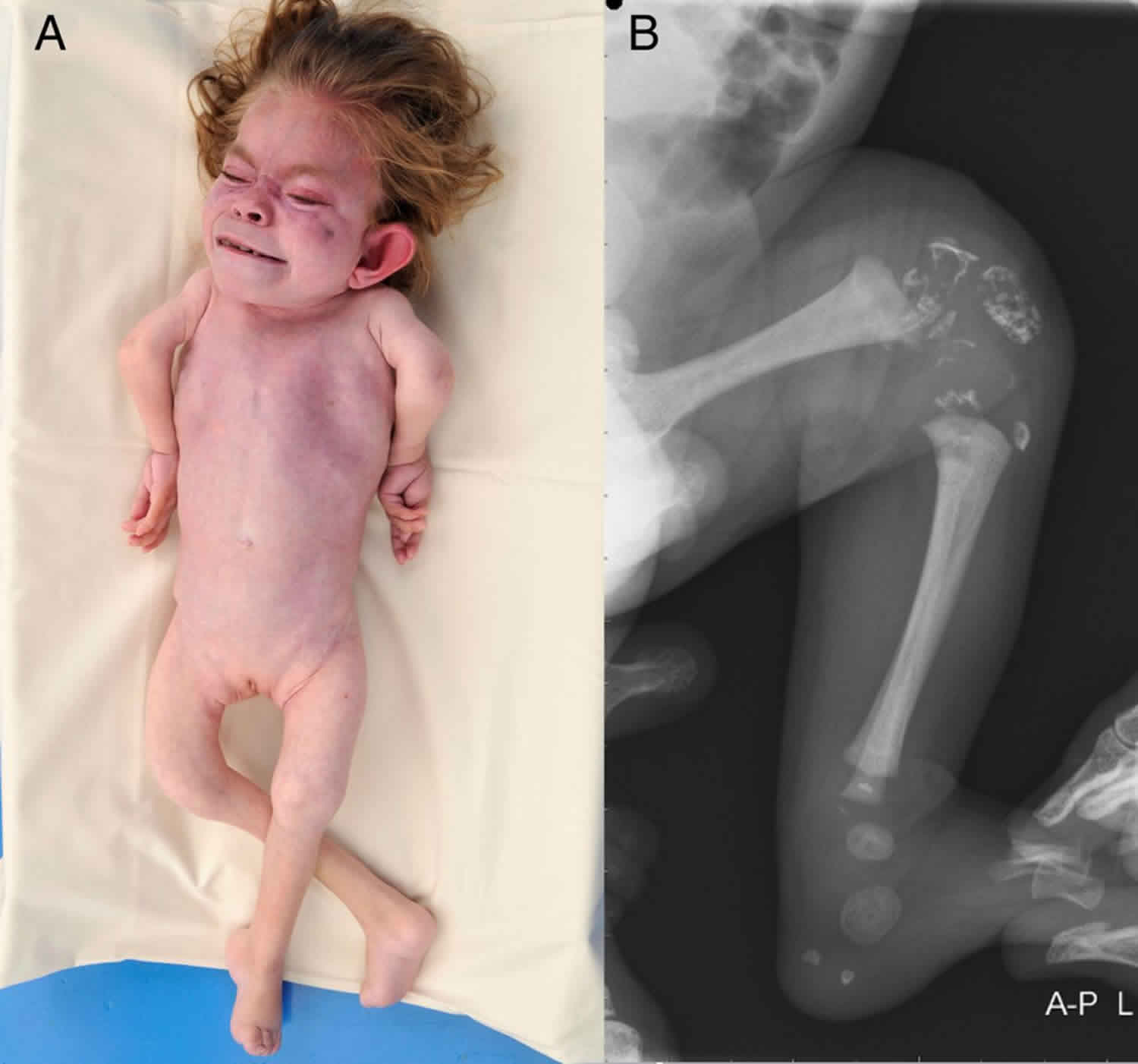
Rhizomelic chondrodysplasia punctata is characterized by shortening of the bones in the upper arms and thighs (rhizomelia). People with rhizomelic chondrodysplasia punctata also have a specific bone abnormality called chondrodysplasia punctata, which affects the growth of the long bones and can be seen on x-rays. People with rhizomelic chondrodysplasia punctata often develop joint deformities (contractures) that make the joints stiff and painful.
Distinctive facial features are also seen with rhizomelic chondrodysplasia punctata. These include a prominent forehead, widely set eyes (hypertelorism), a sunken appearance of the middle of the face (midface hypoplasia), a small nose with upturned nostrils, and full cheeks. Additionally, almost all affected individuals have clouding of the lenses of the eyes (cataracts). The cataracts are apparent at birth (congenital) or develop in early infancy.
Rhizomelic chondrodysplasia punctata is associated with significantly delayed development and severe intellectual disability. Most children with this condition do not achieve developmental milestones such as sitting without support, feeding themselves, or speaking in phrases. Affected infants grow much more slowly than other children their age, and many also have seizures. Recurrent respiratory infections and life-threatening breathing problems are common. Because of their severe health problems, most people with rhizomelic chondrodysplasia punctata survive only into childhood. It is rare for affected children to live past age 10. However, a few individuals with milder features of the condition have lived into early adulthood.
Researchers have described 5 types of rhizomelic chondrodysplasia punctata, classified according to the associated gene mutations 2:
- Rhizomelic chondrodysplasia punctata type 1 with PEX7 gene
- Rhizomelic chondrodysplasia punctata type 2 with GNPAT gene
- Rhizomelic chondrodysplasia punctata type 3 with AGPS gene
- Rhizomelic chondrodysplasia punctata type 4 (peroxisomal fatty acyl-CoA reductase 1 disorder) with FAR1 gene
- Rhizomelic chondrodysplasia punctata type 5 with PEX5 gene
All these genes are involved in the formation and function of sac-like cell structures called peroxisomes that contain enzymes needed to break down many substances, including fatty acids known as plasmalogens 3. Deficiency of plasmalogen affects bone growth 2.
Rhizomelic chondrodysplasia punctata affects fewer than 1 in 100,000 people worldwide. Rhizomelic chondrodysplasia punctata type 1 is more common than rhizomelic chondrodysplasia punctata type 2 or rhizomelic chondrodysplasia punctata type 3.
Inheritance is autosomal recessive. There is no cure for rhizomelic chondrodysplasia punctata. Treatment is symptomatic and may include physiotherapy and orthopedic procedures, eye surgery, and nutritional plans. For example, rhizomelic chondrodysplasia punctata1 patients may need diet restriction of phytanic acid 4.
Figure 1. Rhizomelic chondrodysplasia punctata
Footnote: X-ray of a newborn with classic (severe) rhizomelic chondrodysplasia punctata showing rhizomelia and chondrodysplasia punctata (arrows).
[Source 4 ]Rhizomelic chondrodysplasia punctata causes
Rhizomelic chondrodysplasia punctata results from mutations in one of three genes. Mutations in the PEX7 gene, which are most common, cause RCDP1. Changes in the GNPAT gene lead to RCDP2, while AGPS gene mutations result in RCDP3.
The genes associated with rhizomelic chondrodysplasia punctata are involved in the formation and function of structures called peroxisomes. Peroxisomes are sac-like compartments within cells that contain enzymes needed to break down many different substances, including fatty acids and certain toxic compounds. They are also important for the production of fats (lipids) used in digestion and in the nervous system.
Within peroxisomes, the proteins produced from the PEX7, GNPAT, and AGPS genes play roles in the formation (synthesis) of lipid molecules called plasmalogens. Plasmalogens are found in cell membranes throughout the body, although little is known about their function. Mutations in the PEX7, GNPAT, or AGPS genes prevent peroxisomes from making plasmalogens. Researchers are working to determine how problems with plasmalogen synthesis lead to the specific signs and symptoms of rhizomelic chondrodysplasia punctata.
Rhizomelic chondrodysplasia punctata inheritance pattern
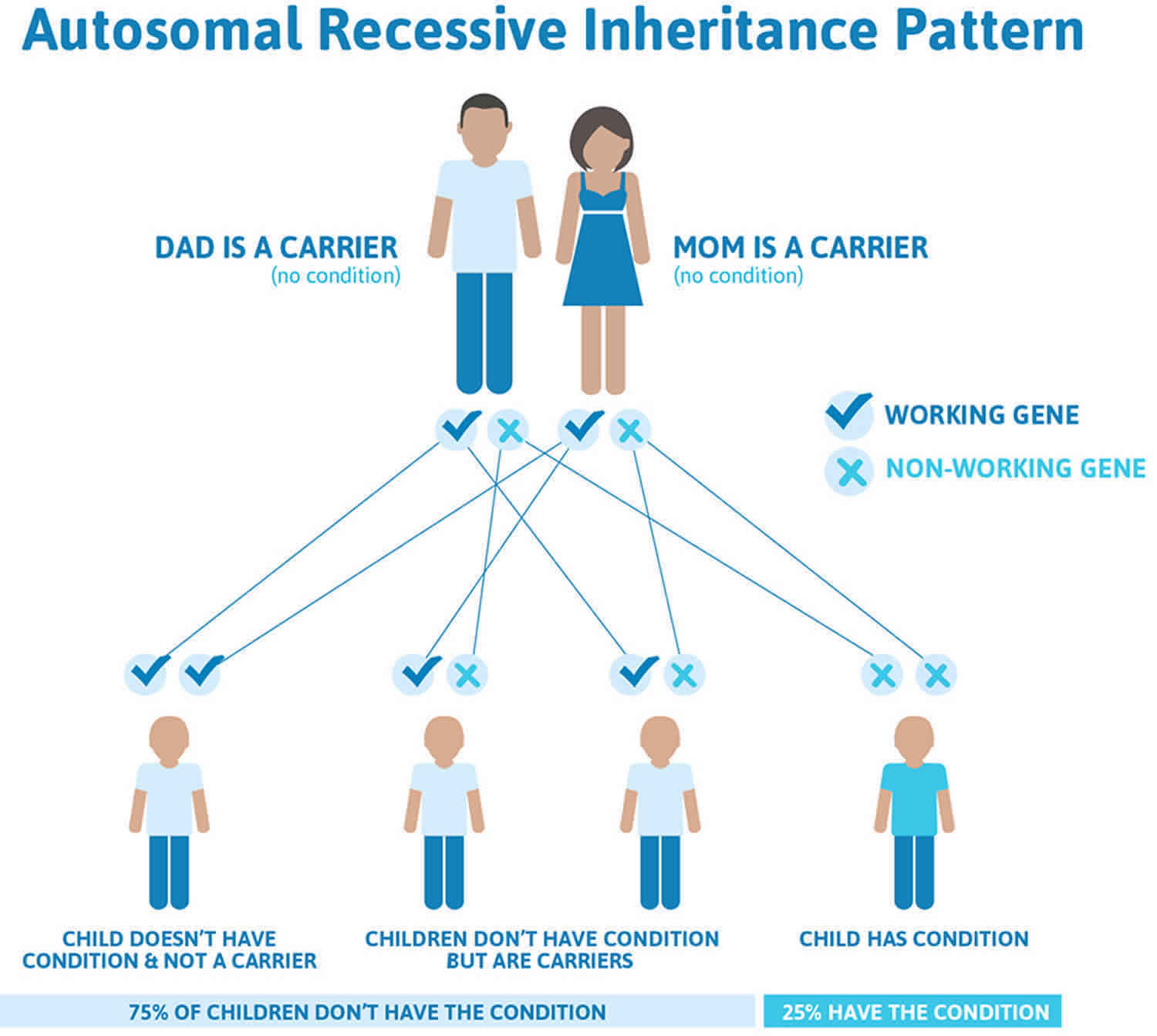
Rhizomelic chondrodysplasia punctata is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Rhizomelic chondrodysplasia punctata autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Rhizomelic chondrodysplasia punctata symptoms
Classic (severe) rhizomelic chondrodysplasia punctata type 1
The characteristic clinical features of classic rhizomelic chondrodysplasia punctata type 1 are skeletal abnormalities, cataracts, growth restriction, and intellectual disability.
Rhizomelic chondrodysplasia punctata life expectancy is shortened 4. The majority of children do not survive beyond the first decade of life and a proportion die in the neonatal period. Of 35 affected children older than age one month, 90% survived to age one year, 55% to age five years, and approximately 20% to age 12 years 5. In a separate review of 66 patients with rhizomelic chondrodysplasia punctata type 1, 80% survived to age five years, 45% to age 12 years, and 35% to adulthood 6. Most deaths in these cohorts were secondary to respiratory complications. Some infants may die in the neonatal period; this number is not known. Clinical experience suggests that neonatal deaths were associated with congenital heart disease or lung hypoplasia 7.
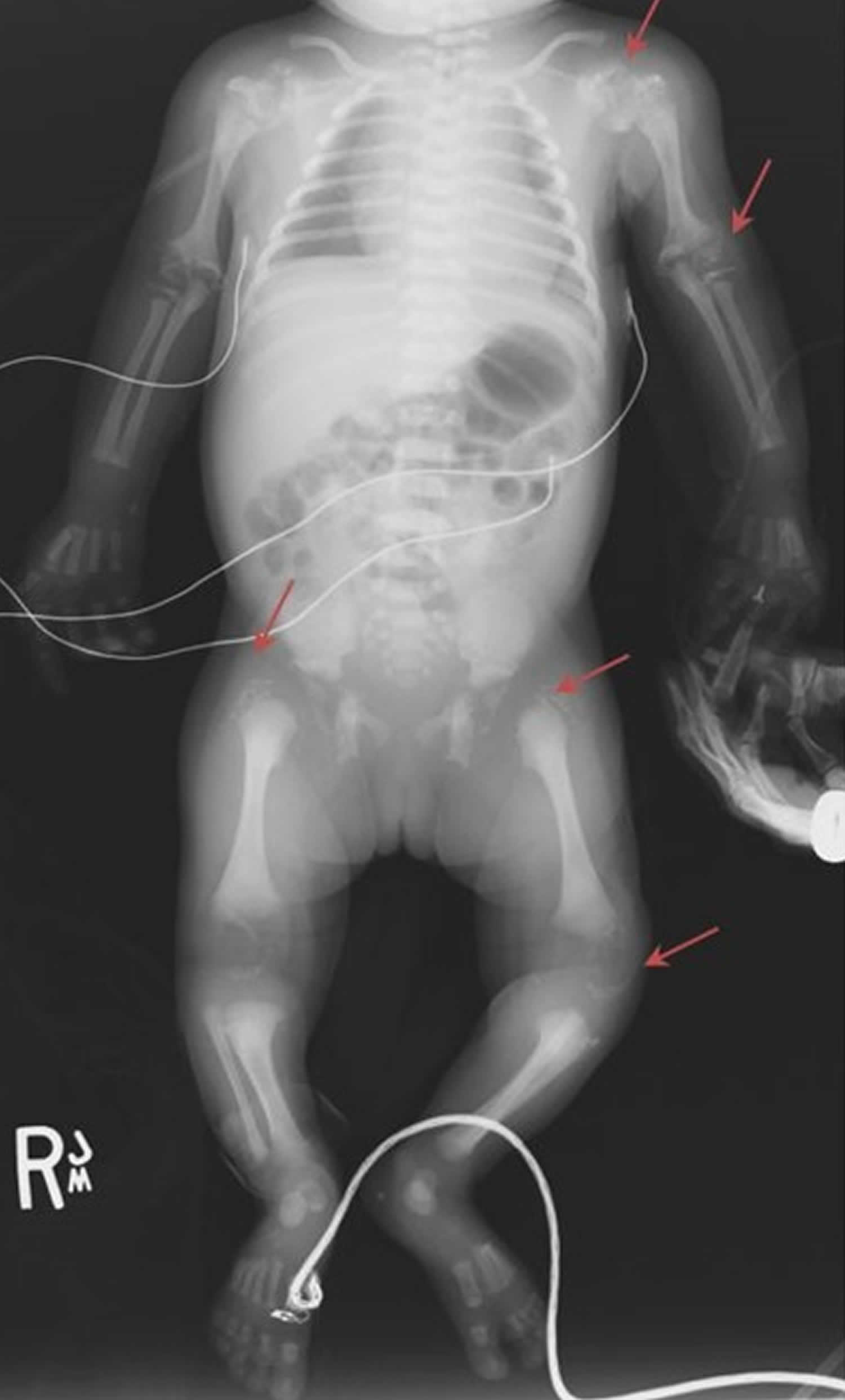
Skeletal findings. Infants with rhizomelic chondrodysplasia punctata type1 have bilateral shortening of the humerus and to a lesser degree the femur. They typically have contractures and stiff, painful joints, causing irritability in infancy.
In a study of the MRIs of children with classic rhizomelic chondrodysplasia punctata type1, all patients had cervical stenosis. More global spinal stenosis, cervical kyphosis, and thoracolumbar kyphosis were seen, but with less frequency. Tethered cord was also identified 8.
Cataracts. Bilateral cortical cataracts develop in virtually all affected individuals. They are usually present at birth or appear in the first few months of life and are progressive.
Growth restriction. Whereas birth weight, length, and head circumference are often at the lower range of normal, profound postnatal growth deficiency is evident throughout the life span 9. At age three years, height, weight and head circumference are around the 50th percentile for a child age 4-6 months. Rate of weight gain is slow, dropping to 5 g/day at age six months, and <2 g/day expected after age three years. rhizomelic chondrodysplasia punctata type1 height, weight, and head circumference growth charts are available as well as height-for-weight charts and charts showing rate of weight gain over time 9.
Intellectual disability. Developmental quotients are below 30. Early developmental skills such as smiling and recognizing voices are achieved by most children with rhizomelic chondrodysplasia punctata type1, but at delayed ages. Skills achieved in typically developing children after age six months are never seen in children with rhizomelic chondrodysplasia punctata type1 10.
Seizures. The majority of children develop seizures 10. Myoclonic jerks are the most frequent type of seizure reported, but seizure frequency and types are variable. The median age at seizure onset was 2.5 years 10.
Recurrent respiratory tract infections. Most children with rhizomelic chondrodysplasia punctata type 1 have recurrent respiratory tract infections caused by a combination of neurologic compromise, aspiration, immobility, and a small chest with restricted expansion. Plasmalogen deficiency may also play a role in the chronic respiratory disease as these lipids are enriched in lung tissues and an integral component of surfactant 6.
Congenital heart disease. Cardiac malformations have been identified in 52% and 64% of individuals with rhizomelic chondrodysplasia punctata type1 in the Dutch and North American cohorts respectively 11. Septal defects, tetralogy of Fallot, and peripheral pulmonary stenosis were most commonly reported. Mitral valve prolapse was also noted in several patients.
Eczema, mild ichthyosis, and skin rashes were noted in around 50% of individuals in the cohort studied by White et al 5.
Other malformations observed in one affected individual include: ureteropelvic junction obstruction 12, cleft palate, diaphragmatic hernia, hypospadias, and cryptorchidism 5.
Routine brain imaging is normal or shows cerebral and cerebellar atrophy with enlargement of the ventricles and CSF spaces 13. Cerebellar atrophy is progressive 14. MRI and MR spectroscopy have shown delayed myelinization, signal abnormalities in supratentorial white matter, decreased choline-to-creatine ratios, and increased levels of mobile lipids, thought to reflect the deficiency of plasmalogens, which are substantial components of myelin 10.
Nonclassic (mild) rhizomelic chondrodysplasia punctata type 1
This group is defined clinically by the ability to walk with or without support and the ability to use verbal or nonverbal types of communication 10. The majority of individuals with nonclassic (mild) rhizomelic chondrodysplasia punctata type 1 have presented in early childhood with bilateral cataracts, multiple joint contractures, and developmental delays 14. A few individuals manifest cataracts within the first two years, no skeletal findings, and behavioral disorders that develop at school age 15. Overall life expectancy is considerably longer than that of classic rhizomelic chondrodysplasia punctata type 1, with survival to adulthood 16; in a recent study 11 of 12 affected individuals survived to adulthood 6.
Skeletal. Most individuals with mild rhizomelic chondrodysplasia punctata type1 have limited joint mobility due to flexion contractures of the elbows, knees, and hips. While the radiographic finding of chondrodysplasia punctata is commonly noted at the time of rhizomelic chondrodysplasia punctata type 1 diagnosis, rhizomelic limb shortening is uncommon in this group. Deformities in hip joints including coxa vara and small femoral heads have been reported 10. Many of these individuals required orthopedic surgeries over time to improve mobility and activities of daily living 17.
Cataract. Most individuals with mild rhizomelic chondrodysplasia punctata type 1 have bilateral cataracts diagnosed in the first two years of life.
Growth of individuals with mild rhizomelic chondrodysplasia punctata type 1 can be within normal ranges at birth. Postnatal growth rates are also better than those in classic rhizomelic chondrodysplasia punctata type 1. Growth curves based on four individuals with nonclassic rhizomelic chondrodysplasia punctata type 1 have been published 9.
Intellect. Most individuals with mild rhizomelic chondrodysplasia punctata type 1 have had developmental delays and learning disabilities. However, they were able to achieve gross and fine motor skills never achieved in individuals with classic rhizomelic chondrodysplasia punctata type1. All individuals with mild rhizomelic chondrodysplasia punctata type1 were able to walk and most can communicate verbally; all required some degree of special education. Brain MRI was normal in all three individuals with nonclassic rhizomelic chondrodysplasia punctata type 1 reported by Bams-Mengerink et al 14.
Seizures. In one cohort, three of four individuals with mild rhizomelic chondrodysplasia punctata type 1 developed seizures in late childhood (age range 7-21 years). The type of seizures more commonly seen in this group were absence and tonic-clonic seizures 10.
Congenital heart disease. In six individuals with mild rhizomelic chondrodysplasia punctata type1, cardiac defects including atrial septal defects were reported in two individuals, one of whom also had first-degree heart block. Two individuals developed mitral valve prolapse, possibly indicating degenerative cardiac changes 16.
Behavior disorders, usually identified at school age in the limited number of individuals described with mild rhizomelic chondrodysplasia punctata type1, included attention-deficit/hyperactivity disorder and autism spectrum disorder 15.
In one family, two siblings with the PEX7 variant p.Ser25Phe had congenital cataract. One sibling had autism spectrum disorder, intellectual disability, and epilepsy; the other had normal intellect with attention-deficit disorder. Elevated blood phytanic acid was observed on an unrestricted diet.
In another family, two siblings had the PEX7 variant p.Trp75Cys and autism spectrum disorder, intellectual disability, epilepsy, and cataracts.
Retinitis pigmentosa and peripheral neuropathy. An individual with the PEX7 variant c.-45C>T had developmental delays and poor growth in childhood; retinitis pigmentosa and peripheral neuropathy developed in adolescence 18. Cataracts were not reported.
Rhizomelic chondrodysplasia punctata diagnosis
The diagnosis of rhizomelic chondrodysplasia punctata is established with suggestive clinical, radiographic, and laboratory findings and molecular genetic testing.
Biochemical testing
The finding of deficiency of plasmalogens in red blood cells, increased plasma concentration of phytanic acid (when diet includes phytanic acid sources), and normal plasma concentration of very long chain fatty acids (VLCFA) has consistently predicted the PEX7 receptor defect in rhizomelic chondrodysplasia punctata type 1 19.. These assays are extremely specialized and are reliably performed in a limited number of laboratories worldwide.
Plasmalogen levels are valuable in distinguishing between classic and milder rhizomelic chondrodysplasia punctata type 1: erythrocyte plasmalogen levels are around 10- to 30-fold higher in milder rhizomelic chondrodysplasia punctata type1 than in classic rhizomelic chondrodysplasia punctata type 1 11.
Rhizomelic chondrodysplasia punctata treatment
Management is supportive and limited by the multiple handicaps present at birth and poor outcome. Poor feeding and recurrent aspiration may necessitate placement of a gastrostomy tube; attention to respiratory function and good pulmonary toilet. Cataract extraction may restore some vision. Physical therapy to improve contractures; orthopedic procedures may improve function in some individuals. Management of developmental delay/intellectual disability as per standard of care.
In classic (severe) rhizomelic chondrodysplasia punctata type 1, management is supportive because of the multiple handicaps present at birth and the poor outcome.
In children with nonclassic (mild) rhizomelic chondrodysplasia punctata type 1, orthopedic surgeries have been performed to maintain gait and joint mobility. Individual education plans have enabled these children to maximize benefit from the school system.
Prevention of primary manifestations
Dietary restriction of phytanic acid to avoid the consequences of phytanic acid accumulation over time may benefit individuals with mild rhizomelic chondrodysplasia punctata type 1.
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., Augmentative and Alternative Communication) for individuals who have expressive language difficulties. An Augmentative and Alternative Communication evaluation can be completed by a speech language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. Augmentative and Alternative Communication devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice generating devices. Contrary to popular belief, Augmentative and Alternative Communication devices to not hinder verbal development of speech and in many cases, can improve it.
Social and behavioral concerns
Children with nonclassic (mild) rhizomelic chondrodysplasia punctata type 1 may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). Applied behavior analysis therapy is targeted to the individual child’s behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Frequent monitoring of growth, nutritional status, and developmental and educational needs; regular assessments for evidence of aspiration, respiratory insufficiency, seizure control, vision, hearing, contractures, and orthopedic complications.
Rhizomelic chondrodysplasia punctata life expectancy
Classic (severe) rhizomelic chondrodysplasia punctata type 1 life expectancy is shortened 4. The majority of children do not survive beyond the first decade of life and a proportion die in the neonatal period. Of 35 affected children older than age one month, 90% survived to age one year, 55% to age five years, and approximately 20% to age 12 years 5. In a separate review of 66 patients with rhizomelic chondrodysplasia punctata type 1, 80% survived to age five years, 45% to age 12 years, and 35% to adulthood 6. Most deaths in these cohorts were secondary to respiratory complications. Some infants may die in the neonatal period; this number is not known. Clinical experience suggests that neonatal deaths were associated with congenital heart disease or lung hypoplasia 7.
Nonclassic (mild) rhizomelic chondrodysplasia punctata type 1 overall life expectancy is considerably longer than that of classic rhizomelic chondrodysplasia punctata type 1, with survival to adulthood 16; in a recent study 11 of 12 affected individuals survived to adulthood 6.
References- Duker AL, Niiler T, Eldridge G, Brereton NH, Braverman NE, Bober MB. Growth charts for individuals with rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2017;173(1):108–113. doi:10.1002/ajmg.a.37961
- Duker AL, Niiler T, Eldridge G, Brereton NH, Braverman NE & Bober MB. Growth charts for individuals with rhizomelic chondrodysplasia punctata. Am J Med Genet Part A. 2017; 173A:108–113. https://www.ncbi.nlm.nih.gov/pubmed/27616591
- Rhizomelic chondrodysplasia punctata. https://ghr.nlm.nih.gov/condition/rhizomelic-chondrodysplasia-punctata
- Braverman NE, Steinberg SJ, Fallatah W, et al. Rhizomelic Chondrodysplasia Punctata Type 1. 2001 Nov 16 [Updated 2020 Jan 30]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1270
- White AL, Modaff P, Holland-Morris F, Pauli RM. Natural history of rhizomelic chondrodysplasia punctata. Am J Med Genet. 2003;118A:332–42.
- Duker AL, Niiler T, Schouten M, Poll-The BT, Braverman NE, Bober MB. Rhizomelic chondrodysplasia punctata morbidity and mortality, an update. Am J Med Genet A. 2019. Epub ahead of print.
- Oswald G, Lawson C, Raymond G, Golden WC, Braverman N. Rhizomelic chondrodysplasia punctata type 1 and fulminant neonatal respiratory failure, a case report and discussion of pathophysiology. Am J Med Genet A. 2011;155A:3160–3.
- Abousamra O, Kandula V, Duker AL, Rogers KJ, Bober MB, Mackenzie WG. Cervical spine deformities in children with rhizomelic chondrodysplasia punctata. J Pediatr Orthop. 2017. Epub ahead of print.
- Duker AL, Niiler T, Eldridge G, Brereton NH, Braverman NE, Bober MB. Growth charts for individuals with rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2017;173:108–13.
- Bams-Mengerink AM, Koelman JH, Waterham H, Barth PG, Poll-The BT. The neurology of rhizomelic chondrodysplasia punctata. Orphanet J Rare Dis. 2013;8:174.
- Duker AL, Eldridge G, Braverman NE, Bober MB. Congenital heart defects common in rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2016;170A:270–2.
- Khanna AJ, Braverman NE, Valle D, Sponseller PD. Cervical stenosis secondary to rhizomelic chondrodysplasia punctata. Am J Med Genet. 2001;99:63–6.
- Powers JM, Kenjarski TP, Moser AB, Moser HW. Cerebellar atrophy in chronic rhizomelic chondrodysplasia punctata: a potential role for phytanic acid and calcium in the death of its Purkinje cells. Acta Neuropathol (Berl). 1999;98:129–34.
- Bams-Mengerink AM, Majoie CBLM, Duran M, Wanders RJA, Van Hove J, Scheurer CD, Barth PG, Poll-The BT. MRI of the brain and certical spinal cord in rhizomelic chondrodysplasia punctata. Neurology. 2006;66:798–803.
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, Schmitz-Abe K, Harmin DA, Adli M, Malik AN, D’Gama AM, Lim ET, Sanders SJ, Mochida GH, Partlow JN, Sunu CM, Felie JM, Rodriguez J, Nasir RH, Ware J, Joseph RM, Hill RS, Kwan BY, Al-Saffar M, Mukaddes NM, Hashmi A, Balkhy S, Gascon GG, Hisama FM, LeClair E, Poduri A, Oner O, Al-Saad S, Al-Awadi SA, Bastaki L, Ben-Omran T, Teebi AS, Al-Gazali L, Eapen V, Stevens CR, Rappaport L, Gabriel SB, Markianos K, State MW, Greenberg ME, Taniguchi H, Braverman NE, Morrow EM, Walsh CA. Using whole exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–73.
- Huffnagel IC, Clur SA, Bams-Mengerink AM, Blom NA, Wanders RJ, Waterham HR, Poll-The BT. Rhizomelic chondrodysplasia punctata and cardiac pathology. J Med Genet. 2013;50:419–24.
- Barth PG, Wanders RJ, Schutgens RB, Staalman CR. Variant rhizomelic chondrodysplasia punctata (RCDP) with normal plasma phytanic acid: clinico-biochemical delineation of a subtype and complementation studies. Am J Med Genet. 1996;62:164–8.
- Braverman N, Chen L, Lin P, Obie C, Steel G, Douglas P, Chakraborty PK, Clarke JT, Boneh A, Moser A, Moser H, Valle D. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum Mutat. 2002;20:284–97.
- Motley AM, Brites P, Gerez L, Hogenhout E, Haasjes J, Benne R, Tabak HF, Wanders RJ, Waterham HR. Mutational spectrum in the PEX7 gene and functional analysis of mutant alleles in 78 patients with rhizomelic chondrodysplasia punctata type 1. Am J Hum Genet. 2002;70:612–24.




{kind=link}