Epidermolytic hyperkeratosis
Epidermolytic hyperkeratosis also called epidermolytic ichthyosis or bullous congenital ichthyosiform erythroderm, is a skin disorder that is present at birth. Epidermolytic hyperkeratosis is part of a group of conditions called ichthyosis, which refers to the scaly skin seen in individuals with related disorders. However, in epidermolytic hyperkeratosis, the skin is thick but not scaly as in some of the other conditions in the group. Epidermolytic hyperkeratosis presents at birth with affected babies may have very red skin (erythroderma), severe blisters, and erosions and evolves over time into varying degrees of skin thickening (hyperkeratosis) 1. Because newborns with this disorder are missing the protection provided by normal skin, they are at risk of becoming dehydrated and developing infections in the skin or throughout the body (sepsis) 2.
As affected individuals get older, blistering is less frequent, erythroderma becomes less evident, and the skin becomes thick (hyperkeratotic), especially over joints, on areas of skin that come into contact with each other, or on the scalp or neck. This thickened skin is usually darker than normal. Bacteria can grow in the thick skin, often causing a distinct odor.
Epidermolytic hyperkeratosis can be categorized into two types. People with palmoplantar-type epidermolytic hyperkeratosis have thick skin on the palms of their hands and soles of their feet (palmoplantar or palm/sole hyperkeratosis) in addition to other areas of the body. People with the other type, non-palmoplantar-type, do not have extensive palmoplantar hyperkeratosis but do have hyperkeratosis on other areas of the body.
Epidermolytic hyperkeratosis affects approximately 1 in 200,000 to 300,000 people worldwide 2.
At this time, there is no cure for epidermolytic hyperkeratosis or epidermolytic ichthyosis and treatment is a challenge 3. The main goal of therapy is to ease the symptoms. This may be achieved with the following, sometimes in combination:
- Topical keratolytics (medications which help shed outer layers of skin) – examples include lactic acid, alpha-hydroxy acid, or urea.
- Topical emollients (products that soften the skin)
- Topical retinoids or oral retinoids – these can significantly improve symptoms, but care must be taken to avoid causing increased skin fragility 1.
Antiseptic washes can reduce the risk for bacterial infections so as to avoid frequent antibiotic therapy 4. When blistering is severe, treatment is focused on wound healing and preventing infection.
Affected newborns with open skin lesions should be transferred to the neonatal ICU to be monitored and treated for infections as needed. They should be handled gently to avoid further trauma to the skin
- 1.
Figure 1. Epidermolytic hyperkeratosis
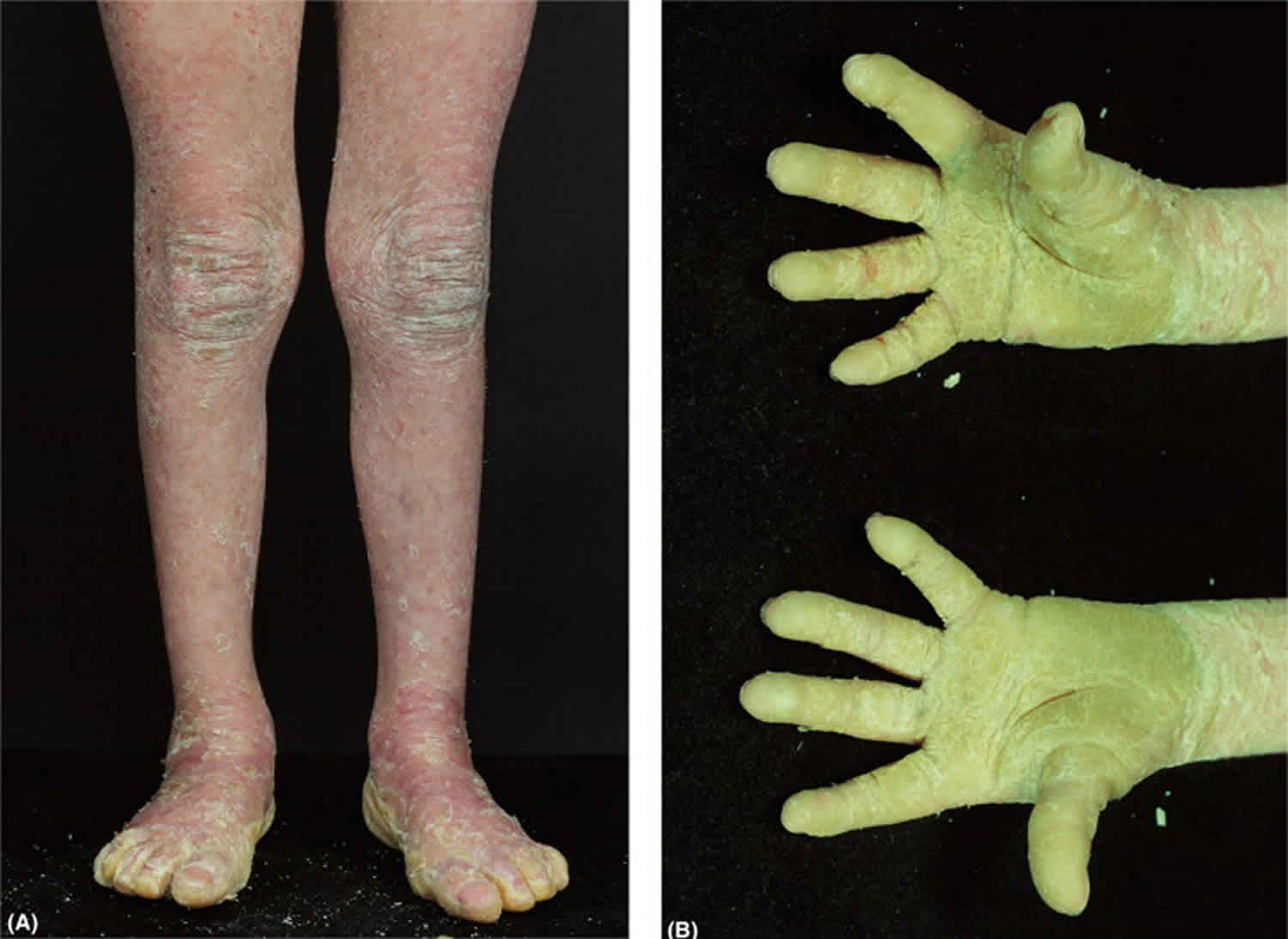
Footnote: (A) Generalized hyperkeratosis with background erythema of the lower limbs; (B) palmoplantar hyperkeratosis extending proximally, typical of epidermolytic hyperkeratosis.
Epidermolytic hyperkeratosis causes
Mutations in the keratin 1 (KRT1) or keratin 10 (KRT10) genes are responsible for epidermolytic hyperkeratosis. These genes provide instructions for making proteins called keratin 1 and keratin 10, which are found in cells called keratinocytes in the outer layer of the skin (the epidermis). The tough, fibrous keratin proteins attach to each other and form fibers called intermediate filaments, which form networks and provide strength and resiliency to the epidermis.
Mutations in the KRT1 or KRT10 genes lead to changes in the keratin proteins, preventing them from forming strong, stable intermediate filament networks within cells. Without a strong network, keratinocytes become fragile and are easily damaged, which can lead to blistering in response to friction or mild trauma. It is unclear how these mutations cause the overgrowth of epidermal cells that results in hyperkeratotic skin.
KRT1 gene mutations are associated with palmoplantar-type epidermal hyperkeratosis, and KRT10 gene mutations are usually associated with non-palmoplantar-type. The keratin 1 protein is present in the keratinocytes of the skin on the palms of the hands and the soles of the feet as well as other parts of the body, so mutations in the KRT1 gene lead to skin problems in these areas. The keratin 10 protein is not found in the skin of the palms and soles, so these areas are unaffected by mutations in the KRT10 gene.
Epidermolytic hyperkeratosis inheritance pattern
Epidermolytic hyperkeratosis can have different inheritance patterns. About half of the cases of epidermolytic hyperkeratosis result from new mutations in the keratin 1 (KRT1) or keratin 10 (KRT10) gene and occur in people with no history of the disorder in their family. However, while people with sporadic epidermolytic hyperkeratosis did not inherit the condition from a parent, they may still pass the condition on to their children.
When epidermolytic hyperkeratosis is inherited, it is usually in an autosomal dominant pattern, which means one copy of the altered KRT1 or KRT10 gene in each cell is sufficient to cause the disorder. When a person with a mutation that causes an autosomal dominant condition has children, each child has a 50% (1 in 2) chance to inherit that mutation. Typically, EI due to a new mutation will follow autosomal dominant inheritance in subsequent generations.
Very rarely, epidermolytic hyperkeratosis caused by mutations in the KRT10 gene can be inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Affected people inherit one mutated copy of the gene from each parent, who is referred to as a carrier. Carriers of an autosomal recessive condition typically do not have any signs or symptoms (they are unaffected). When 2 carriers of an autosomal recessive condition have children, each child has a:
- 25% (1 in 4) chance to be affected
- 50% (1 in 2) chance to be an unaffected carrier like each parent
- 25% chance to be unaffected and not be a carrier.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Epidermolytic hyperkeratosis symptoms
Epidermolytic hyperkeratosis or epidermolytic ichthyosis presents at birth or shortly thereafter with red, blistering, and/or denuded skin with visible areas of skin thickening. Phenotype evolves in early infancy to varying degrees of generalized skin thickening (hyperkeratosis). Over time, there is a gradual decrease in blistering, but an increase in the severity of the scaling and skin thickening. Scales tend to form in parallel rows of spines or ridges. A generalized erythroderma (redness of the skin) may be present in some individuals. Patients with epidermolytic hyperkeratosis are at an increased risk for recurrent infections. Skin infections with common bacteria can be a problem. Heat intolerance is common. A palmoplantar keratoderma may be present and can be so severe as to limit ambulation and hand function. Surgical intervention may then be required. On the other end of the scale, there are individuals who have only minimal blistering in areas subject to friction, or have only a palmoplantar keratoderma. Rarely patients are covered in brown-grey hyperkeratotic spines. This is called ichthyosis hystrix (Curth-Macklin).
Epidermolytic hyperkeratosis or epidermolytic ichthyosis presents in neonates as widespread superficial blisters, which, when ruptured, leave raw, denuded areas. Within the first few months of life, hyperkeratosis begins to predominate, and while skin fragility persists over time, it becomes far less severe, with most patients experiencing infrequent blisters and erosions. Hyperkeratosis can vary from mild to severe and is typically more prominent over joints and in flexural sites. The scale is classically described as corrugated or cardboardlike. Palmoplantar keratoderma is seen primarily in patients with KRT1 mutations and can be severe and disabling at times. Some patients also experience joint contractures. Hair, nails, and teeth are normal and ectropion is generally absent. Superficial bacterial infections are common and are often associated with a characteristic odor of the skin.
Epidermolytic hyperkeratosis has been infrequently found to be associated with other clinical findings. Rare cases of patients with epidermolytic ichthyosis and hypocalcemic rickets, with or without vitamin D resistance, have been reported 5. A report also describes epidermolytic ichthyosis and congenital platelike osteoma cutis in a child 6, as well as epidermolytic ichthyosis localized to the vulva 7.
Epidermolytic hyperkeratosis diagnosis
Epidermolytic hyperkeratosis is diagnosed by physical signs and symptoms. Molecular genetic testing for mutations in the KRT1 and KRT10 genes is available to confirm the diagnosis. Genetic studies can be performed on buccal swabs or blood. Once a mutation is identified in an affected individual, mutation-specific testing for relatives and prenatal diagnosis is available. Prenatal diagnosis can be made through chorionic villus sampling, analysis of amniotic cells, or fetal skin biopsies.
Along with clinical presentation and history, skin biopsy can be helpful, with the histologic findings confirming a diagnosis of epidermolytic ichthyosis.
Epidermolytic hyperkeratosis treatment
Treating epidermolytic hyperkeratosis is a challenge. The medications that help to remove the excess thickened skin layers (topical keratolytics or oral retinoids) often remove too much scale, leaving a very fragile epidermis (underlying living cell layers) exposed. Severe palmoplantar keratoderma is very difficult to treat. A combination of therapies may help, including: application of a barrier repair formula containing ceramides or cholesterol; application of a barrier repair formula containing petrolatum or lanolin; topical or systemic anti-bacterial agents; and cautious use of keratolytics (lotions containing alpha-hydroxy acids, propylene glycol, lactic acid or urea). Since bacterial colonization is almost always present due to the scaling, it is recommended that patients wash with antiseptic soap 2-3 times per week. Many patients find baths with salt or sodium bicarbonate beneficial (helps to descale). Also, some bleach added to the bath may assist in combating bacterial overgrowth.
Epidermolytic hyperkeratosis prognosis
Epidermolytic hyperkeratosis is a lifelong condition. Some patients may experience amelioration of symptoms as they age. Risk for morbidity and mortality is highest in the neonatal period, where infants are at increased risk for complications such as sepsis and dehydration because of impaired barrier function. Later in life, affected patients may experience recurrent skin infections.
References- Epidermolytic Ichthyosis (Epidermolytic Hyperkeratosis or Bullous Congenital Ichthyosiform Erythroderma). https://emedicine.medscape.com/article/1112403-overview
- Epidermolytic hyperkeratosis. https://ghr.nlm.nih.gov/condition/epidermolytic-hyperkeratosis
- Epidermolytic Ichthyosis: A Patient’s Perspective. http://www.firstskinfoundation.org/types-of-ichthyosis/epidermolytic-ichthyosis
- Autosomal dominant epidermolytic ichthyosis. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=312
- Bhat YJ, Baba AN, Manzoor S, Qayoom S, Ahmed SM. Bullous icthyosiform erythroderma with rickets in child of a parent with naevus unius lateralis. Indian J Dermatol Venereol Leprol. 2010 Mar-Apr. 76(2):192-4.
- Blalock TW, Teague D, Sheehan DJ. Epidermolytic hyperkeratosis and congenital platelike osteoma cutis in a child. Cutis. 2011 Jun. 87(6):278-80.
- Russell P, Valmadre S, Howard V. Localised epidermolytic hyperkeratosis of the vulva: a case of mistaken identity. Pathology. 2010. 42(5):483-5.





{kind=link}