
Spinal muscular atrophy
Spinal muscular atrophy also called SMA, is a group of rare genetic diseases with autosomal recessive inheritance that attacks nerve cells called motor neurons (alpha motor neurons (α-MNs) located in the anterior horn of the spinal cord), in the brain stem and spinal cord, that control essential skeletal muscle activity such as speaking, walking, breathing, feeding, coughing and swallowing, leading to muscle weakness and muscle atrophy and develop twitching called fasciculations 1, 2, 3, 4, 5, 6. Motor neurons cells communicate with your voluntary muscles – the ones you can control, like in your arms and legs. As the motor neurons die the messages that the brain tries to send along these motor neurons do not get through to the muscles. This causes the muscles to become weak and damaged. Over time they waste away (atrophy). This can affect walking, crawling, breathing, swallowing, and head and neck control. Spinal muscular atrophy is characterized by weakness and wasting (atrophy) in muscles used for movement (skeletal muscles) 7, 8, 9. The weakness tends to be more severe in the muscles that are close to the center of the body (proximal) compared to muscles away from the body’s center (distal) 10, 11. The muscle weakness usually worsens with age and ultimately result in paralysis and often death in severe cases 12. SMA is the leading genetic cause of infant mortality globally 13, 14 and the second most common fatal autosomal recessive disorder after cystic fibrosis 15, 16.
Spinal muscular atrophy (SMA) affects 1 per 8,000 to 10,000 people worldwide 17, 18, 19, 20, 21. SMA has generally been believed to affect as many as 10,000 to 25,000 children and adults in the United States 22. 1 in 6,000 to 10,000 children are born with spinal muscular atrophy (SMA) 22. One in 40 to 50 people (approximately 6 million Americans) are carriers of the SMN1 gene 22. These carriers have one healthy SMN1 gene and one missing or defective SMN1 gene. Carriers don’t develop SMA. There’s a 1 in 4 chance that two carriers will have a child with SMA.
There are many types of spinal muscular atrophy (SMA) that are caused by changes in the same genes. The types differ in age of onset and severity of muscle weakness; however, there is overlap between the types (see Figure 2 and Table 1 below). The most common form of spinal muscular atrophy (SMA) is caused by defects in both copies of the survival motor neuron 1 (SMN1) gene on chromosome 5q13.2 23, 24. The number of copies of the survival motor neuron 2 (SMN2) gene modifies the severity of spinal muscular atrophy and helps determine which type develops. Typically, people have two copies of the SMN1 gene and one to two copies of the SMN2 gene in each cell. However, the number of copies of the SMN2 gene varies, with some people having up to eight copies (see Figure 2 and Table 1 below). Extra copies of the SMN2 gene do not cause spinal muscular atrophy (SMA), but the number of copies of the SMN2 gene modify the severity of the disorder with a high number of SMN2 copies related to milder phenotypes and helps determine which type develops 25, 26, 27. In people with spinal muscular atrophy (SMA), having multiple copies of the SMN2 gene is usually associated with less severe features of the condition that develop later in life. Affected individuals with one or two functional copies of the SMN2 gene generally have severe muscle weakness that begins at birth or in infancy 27. Affected individuals with four or more copies of the SMN2 gene typically have mild muscle weakness that may not become noticeable until adulthood 27. For example, SMA type 1 patients generally have one or two SMN2 copies, while SMA type 3 or 4 patients have more than four copies 28, 29. However, this inverse relationship is not always true, as a few patients with two SMN2 copies showed milder SMA phenotypes, while there have also been patients with three SMN2 copies that have been defined as type 1 17, 30, 31, 32. Other factors, many unknown, also contribute to the variable severity of spinal muscular atrophy.
SMA type 1, 2, 3, and 4 are caused by changes (pathogenic variants, also know as genetic changes) in the SMN1 gene and are inherited in an autosomal recessive manner (a person must get the defective gene from both parents to be affected, meaning that a person must have two copies of a defective gene to have the disease). SMA carriers do not exhibit SMA symptoms, but do carry a defective copy of the SMN1 gene and one healthy SMN1 gene. If both parents are carriers of the defective SMN1 gene, then each of their children has a 1 in 4 chance of having spinal muscular atrophy.
The SMN1 and SMN2 genes both provide instructions for making a protein called the survival motor neuron (SMN) protein which maintains the health and normal function of motor neurons. Normally, most functional SMN protein is produced from the SMN1 gene, with a small amount produced from the SMN2 gene 21. Most people with spinal muscular atrophy (SMA) are missing a piece of the SMN1 gene, which impairs SMN protein production. The SMN protein is one of a group of proteins called the SMN complex, which is important for the maintenance of motor neurons. SMN complex is important for the development of specialized outgrowths from nerve cells called dendrites and axons. Dendrites and axons are required for the transmission of impulses between neurons and from neurons to muscles 27. Motor neurons transmit signals from the brain and spinal cord that tell skeletal muscles to tense (contract), which allows the body to move. A shortage of SMN protein leads to motor neuron death in the spinal cord, and as a result, signals are not transmitted between the brain and muscles. Muscles cannot contract without receiving signals from the brain, so many skeletal muscles become weak and waste away (atrophy), leading to the signs and symptoms of spinal muscular atrophy. This weakness is often more severe in the trunk and upper leg and arm muscles than in muscles of the hands and feet. The muscle damage gets worse over time and can affect speaking, walking, swallowing, and breathing.
However, the SMN protein produced by the SMN2 genes can help make up for the protein deficiency caused by SMN1 gene mutations. The number of copies of the SMN2 gene varies, with some people having up to eight copies. In people with spinal muscular atrophy (SMA), having multiple copies of the SMN2 gene is usually associated with less severe features of the condition that develop later in life. The SMN protein produced by the SMN2 genes can help make up for the SMN protein deficiency caused by SMN1 gene mutations. Several different versions of the SMN protein are produced from the SMN2 gene, but only one version is functional; the other versions are smaller and quickly broken down 21.
People with spinal muscular atrophy type 0 (also known as SMA type 1A) usually have one copy of the SMN2 gene in each cell, while those with SMA type 1 generally have one or two copies of the SMN2 gene in each cell, those with SMA type 2 usually have three copies of the SMN2 gene in each cell, those with SMA type 3 have three or four copies of the SMN2 gene in each cell, and those with SMA type 4 have four or more copies of the SMN2 gene in each cell 33, 34, 35, 36, 37, 6. Other factors, many unknown, also contribute to the variable severity of spinal muscular atrophy.
SMA type 0 is the rarest and most severe form of spinal muscular atrophy and begins before birth. SMA type 1 is the most common type accounting for about half of all cases and a severe form of the disorder with muscle weakness evident at birth or within the first few months of life, usually before 6 months of age and generally results in death before age two. SMA types 2 and 3 are the next most common and types 0 and 4 are rare. Patients with milder forms of SMA may not have symptoms of muscle weakness until much later in childhood or even as adults. Extra copies of the nearby related gene, SMN2, modify the severity of spinal muscular atrophy (SMA).
There are other rarer types of SMA caused by changes in different genes including the VAPB gene located on chromosome 20, the DYNC1H1 gene on chromosome 14, the BICD2 gene on chromosome 9, and the UBA1 gene on the X chromosome. Other autosomal recessive forms include SMA with progressive myoclonic epilepsy (SMA-PME) caused by changes in the ASAH1 gene and SMA with respiratory distress 1 (SMARD1) caused by changes in the IGHMBP2 gene. X-linked forms include X-linked infantile SMA caused by changes in UBA1. The types differ in age of onset and severity of muscle weakness; however, there is overlap between the types.
Diagnosis of SMA is suspected by symptoms and confirmed by genetic testing. Children with SMA generally appear normal at birth, with symptoms developing as early as a few months after they are born. Symptoms of SMA often overlap with other neuromuscular diseases, therefore diagnosis of SMA can be challenging for a non-specialist. Please consult your doctor if you suspect any symptoms of the disease in your child. A diagnosis of SMA is usually made by a pediatric neurologist and confirmed by a blood test, which is designed to identify genetic defects in the SMN1 gene.
The most severe forms of SMA are characterized by motor neuron degeneration and progressive loss of muscle function that culminate in death or permanent ventilation early in childhood 38. Other forms of spinal muscular atrophy and related motor neuron diseases, such as spinal muscular atrophy with progressive myoclonic epilepsy, spinal muscular atrophy with lower extremity predominance, X-linked infantile spinal muscular atrophy, and spinal muscular atrophy with respiratory distress type 1 are caused by mutations in other genes. Spinal muscular atrophy is the second leading cause of neuromuscular disease.
There is no cure for SMA. Treatments can help manage symptoms and prevent complications. They may include 39:
- Medicines to help the body make more of the proteins that the motor neurons need
- Gene therapy for children under 2 years of age
- Physical, occupational, and rehabilitation therapy to help to improve posture and the mobility of the joints. These therapies may also improve blood flow and slow muscle weakness and atrophy. Some people may also need therapy for trouble speaking, chewing, and swallowing.
- Assistive devices such as supports or braces, orthotics, speech synthesizers, and wheelchairs to help people stay more independent
- Good nutrition and a balanced diet to help maintain weight and strength. Some people might need a feeding tube in order to get the nutrition they need.
- Breathing support for people who have muscle weakness in the neck, throat, and chest. The support may include devices to help with breathing during the day and to prevent sleep apnea at night. Some people might need to be on a ventilator.
Several different compounds were investigated in randomized controlled trials, including treatments intended to increase muscle function and strength (e.g., hyperacetylation agents, anabolics, thyrotropin-releasing hormone, growth hormone, neuroprotective agents such as gabapentin, riluzole, and olesoxime), all produced negative results for their respective primary endpoints, and none were approved 40. However, with the advent of disease-modifying therapies (e.g., nusinersen, risdiplam, and onasemnogene abeparvovec), the prognosis for patients with SMA has significantly improved 4. To date, three medications have been approved by the U.S. Food and Drug Administration (FDA) for SMA, which are nusinersen (Spinraza®), onasemnogene abeparvovec-xioi (Zolgensma®) and recently approved risdiplam (Evrysdi®). However, the cost of nusinersen (Spinraza®) and onasemnogene abeparvovec-xioi (Zolgensma®) are astronomical in nature 41, 10, 42, while risdiplam (Evrysdi®) the cost has yet to be established.
- Nusinersen (Spinraza®) is an antisense oligonucleotide that modifies pre-messenger RNA splicing of the SMN2 gene to promote increased production of full-length, functional SMN protein. Treatment is initiated with four loading doses followed by maintenance dosage once every 4 months 43, 44. In a sham-controlled randomized controlled trial (ENDEAR; NCT02193074) 45, patients with nusinersen-treated SMA type 1 experienced a significantly greater likelihood of event-free survival and motor milestone response (51% vs. 0) compared with the control group 45. Overall survival was significantly greater in the nusinersen group versus the control group 45.
- Onasemnogene abeparvovec-xioi (Zolgensma®) is a gene replacement therapy that delivers a functional human SMN transgene to motor neurons via one-time intravenous infusion 46. In the Phase 1 START study (NCT02122952; n = 15), treatment of symptomatic SMA with onasemnogene abeparvovec resulted in significant improvements in survival, motor milestones, and function without the need for permanent ventilation 47. STR1VE-US (NCT03306277) 48, a completed Phase 3 study, demonstrated that the favorable risk-benefit profile first observed in START 47 was confirmed for a larger group of patients (n = 22 vs. n = 15). In STR1VE-US 48, 59% of patients receiving onasemnogene abeparvovec achieved functional independent sitting for 30 s or longer at the 18-months-of-age study visit (vs. 0 in the untreated cohort) and 91% survived free from permanent ventilation at age 14 months (vs. 26% in the untreated cohort).
- Risdiplam (Evrysdi®) is a SMN2 splicing modifier designed to treat patients with SMA that is caused by mutations in chromosome 5q13.2 that lead to SMN protein deficiency 49. In FIREFISH, 41% (7/17) of infants treated with the Risdiplam therapeutic dosage achieved the ability to sit without support for at least 5 seconds as measured by the Bayley Scales of Infant and Toddler Development Third Edition (BSID-III) 50. In addition, 90% (19/21) of infants were alive without permanent ventilation at 12 months of treatment and reached 15 months of age or older 51. In SUNFISH 52, children and adults treated with risdiplam experienced a clinically meaningful and statistically significant improvement in motor function at 12 months (1.55 point mean difference) compared with placebo (1.36 points; –0.19 points, respectively), as measured by a change from baseline in the Motor Function Measure-32 total score.
- Based upon these trial results, nusinersen has been approved for use in the United States, Europe, Canada, Japan, and several other countries in Asia and the Middle East. Onasemnogene abeparvovec has been approved for use in the United States, Europe, Japan, and many other countries in South America and Asia. Risdiplam has been approved in the United States and European Union 49, 53. Nusinersen and onasemnogene abeparvovec are also recommended by the National Institute for Health and Care Excellence (NICE) in the United Kingdom 54.
Due to advances in nutritional and respiratory care, physiotherapy and enablement strategies to maintain independent living put forth in the 2007 Consensus Statement for the Standard of Care in Spinal Muscular Atrophy, the prognosis of SMA has also been changing over the last few decades 55. The prognosis for even the most severely affected children with SMA has improved in terms of survival and health-related quality of life (HRQOL), albeit with severe disability 56, 37.
Figure 1. Spinal muscular atrophy causes
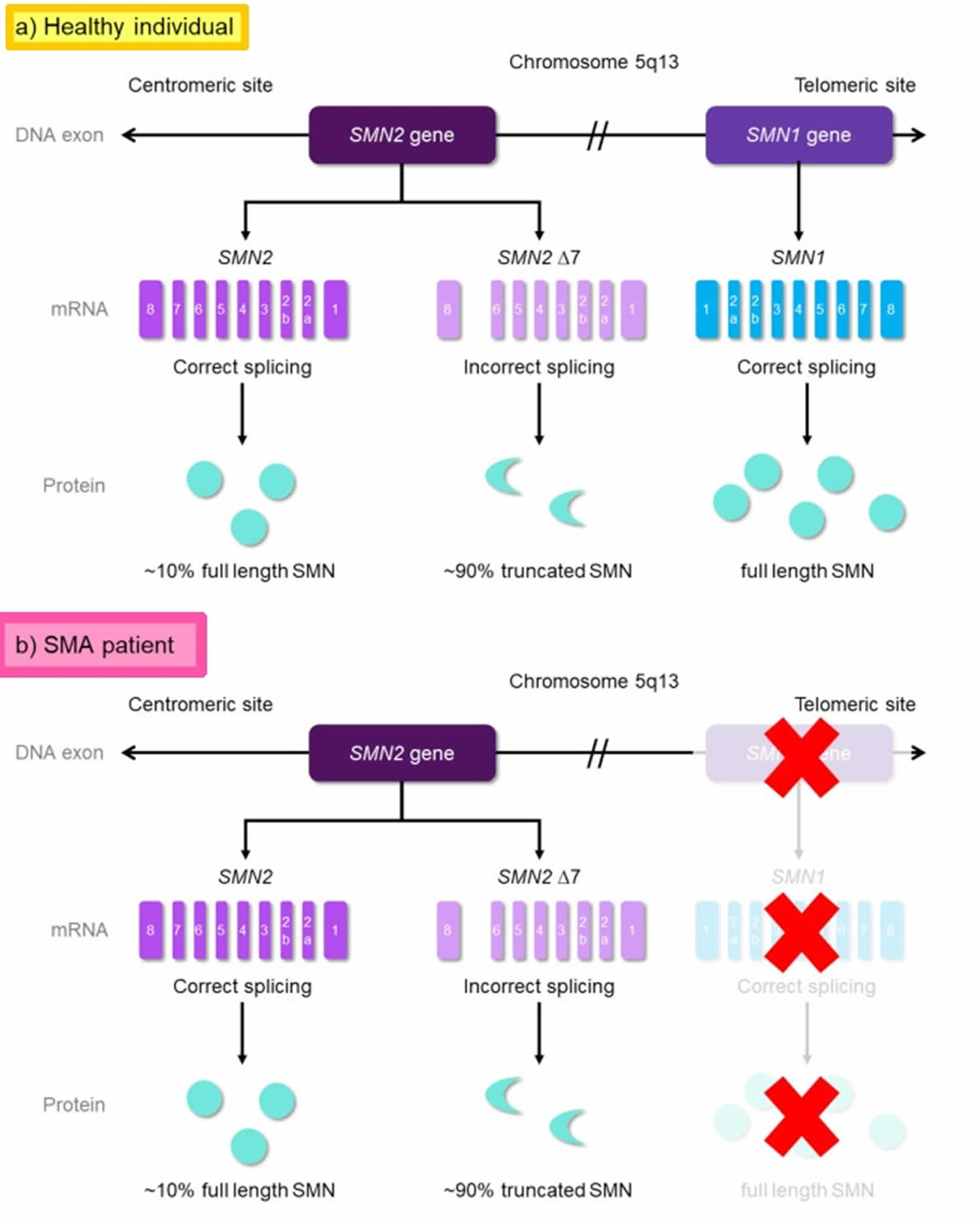
Footnotes: Genetic cause of spinal muscular atrophy (SMA). The SMN1 and SMN2 genes are located in an inverse duplicated region on chromosome 5q13.2. On DNA level, the two genes only differ by one functionally relevant nucleotide exchange within exon 7. This transition from C to T results in the generation of an exonic splicing silencer (ESS) site leading to exon 7 skipping on mRNA level. SMN1 gene codes for the functional, full-length SMN (FL-SMN) protein; while the SMN2 gene produces ~10% FL-SMN protein and ~90% truncated SMN protein (SMNΔ7) [non-functional version of the SMN protein] due to incorrect splicing. SMA is caused by homozygous deletions of SMN1 resulting in highly reduced SMN protein levels. However, the number of SMN2 copies that determines the amount of functional SMN protein can modify disease severity.
(a) In healthy individuals, both SMN genes are present. (b) In SMA patients, the absence of the SMN1 gene, due to mutations, causes no FL-SMN protein production from SMN1 (This condition is indicated as red ‘X’). The production solely depends on the SMN2 gene, resulting insufficient production.
[Source 5 ]Figure 2. SMA subtypes

Footnotes: Classification of spinal muscular atrophy (SMA) sub-types. * All SMA patients, regardless of the subtypes, have no functional copies of survival motor neuron 1 (SMN1). SMA can be classified into five types (0 to 4) ranging from the most severe form to a milder form. (a) SMA Type 0 (type 1A) is the most severe form and in-utero onset. They normally have limited life expectancy. (b) SMA Type 1 infants display clinical symptoms at birth or by the age of six months. They never develop the ability to sit and if no intervention is provided results in death by two years. (c) SMA Type 2 patients are diagnosed within six to 18 months of age and they do develop the ability to sit but they never walk unaided. However, they are able to survive well into adulthood. (d) SMA Type 3 can be further classified into 3a (onset between 18 months to three years old) and 3b (onset between ages of three to 30 years old). They have a normal life expectancy. (e) SMA Type 4 is the mildest form and adult-onset. Patients with SMA type 4 have a normal life expectancy.
[Source 5 ]Figure 3. SMA patients global distribution

Footnote: Global distribution of SMA patients who have registered under TREAT-NMD, an international network for the neuromuscular field. The number of registries is rounded to two decimal places in percentage and Russia is included in the ‘Rest of the World’ herein.
[Source 57 ]Spinal muscular atrophy types
There is a wide range of impairment seen in spinal muscular atrophy caused by defects in the SMN1 gene, from onset before birth with breathing difficulties at birth to mild weakness in adults. Accordingly, this most common form of spinal muscular atrophy can be classified into four types, based on highest motor milestone achieved.
Figure 4. SMA types

Footnotes: Figure 2 depicts a classification of five types of SMA that are characterized by the SMN2 copy number. Such a gene is theoretically correlated with the SMN protein level, therefore relating to the onset and severity of different subtypes of SMA. Despite genetic confirmation of SMN1 absence or mutations in all patients, spinal muscular atrophy (SMA) presentation ranges from very compromised neonates (SMA type 0 also known as SMA type 1A) to adults with minimal manifestations (MM) depending on the number of SMN2 copies and full-length protein produced by each patient and modulated by negative or positive modifiers that influence the final phenotype.
Abbreviations: SMN1 = survival motor neuron 1 gene; SMN2 = survival motor neuron 2 gene; SMA = spinal muscular atrophy; MM = minimal manifestations; SMN = survival motor neuron.
[Source 37 ]Table 1. Spinal muscular atrophy types
| SMA type | Age of Onset | Milestones achieved | Evolution / natural history | Typical SMN2 copy number |
|---|---|---|---|---|
| 1A (also known as type 0) | Prenatal | None | Death in weeks to less than 6 months, contractures, cardiopathy Other findings:
| 1 |
| 1B | Less than 3 months | Poor or absent head control | Feeding and respiratory problems, linear decline. Death by second or third year of life | 2 |
| 1C | Older than 3 months | Head control, sit with support only | Feeding and respiratory problems. Plateau in first two years | 3 |
| 2 | Older than 6 months | Able to sit unaided | Scoliosis. Survival to adolescence or adulthood (70% alive at age 25 yrs) Other findings:
Weaker cases may lose sitting capability (2a) and stronger cases may stand with support (2b) | 3 |
| 3a | Between 18 and 36 months | Walking unaided | Scoliosis Early loss of ambulation Normal lifespan | 3 |
| 3b | Older than 3 years | Walking unaided | Later loss of ambulation Normal lifespan | 3–4 |
| 4 | Second or third decade of life | Walking unaided | Ambulant until late in life Normal lifespan | 4 |
Spinal muscular atrophy type 0
Spinal muscular atrophy type 0 also known as SMA type 1A is evident before birth and is the rarest and most severe form of spinal muscular atrophy, occurs with minimal presence of the SMN2 gene 58, 59. Affected infants move less in the womb, and as a result they are often born with arthrogryposis (joint deformities or contractures). They have extremely weak muscle tone (hypotonia) in particular respiratory and heart muscles at birth. Their respiratory muscles are very weak and they have respiratory distress at birth and they often do not survive past infancy due to respiratory failure (rarely survive past age six months) 60, 61. Some infants with spinal muscular atrophy type 0 (SMA type 1A) have respiratory failure, facial diplegia (facial paralysis) and/or heart defects such as atrial septal defects that are present from birth (congenital), leading to death during the infancy stage 62, 63, 64.
There have not been any published reports of infants with SMA type 0 (SMA type 1A) who have been treated with nusinersen or gene therapy 6.
Spinal muscular atrophy type 1
Spinal muscular atrophy type 1 also called Werdnig-Hoffmann disease or infantile-onset spinal muscular atrophy, is the most common form of spinal muscular atrophy (~45% of cases) 65. Spinal muscular atrophy type 1 is a severe form of the disorder with muscle weakness evident at birth or within the first few months of life, is evident usually before 6 months of age at most 58. Most affected children, typically present with two or three copies of the SMN2 gene 66 and cannot control their head movements or sit unassisted. Proximal, symmetric muscle weakness, lack of motor development with regression of motor function, reduced or absent deep tendon reflexes, and poor muscle tone are the major clinical manifestations. Mild contractures are often noted at the knees and, rarely, at the elbows. Children with spinal muscular atrophy type 1 may have swallowing problems that can lead to difficulty feeding and poor growth. They can also have breathing problems due to weakness of intercostal respiratory muscles with relative preservation of diaphragm musculature leads to an abnormally “bell-shaped” chest that prevents the lungs from fully expanding and paradoxic respiration (abdominal breathing) 67, 68, 69, 70. The mean age of symptom onset is 2.5 months 71. Cognitive function is normal. Without any treatment, affected children never sit or stand and the vast majority usually die of respiratory failure before the age of 2 years. Most children with spinal muscular atrophy type 1 do not survive past early childhood due to respiratory failure.
Symptoms of spinal muscular atrophy type 1 include hypotonia (reduced muscle tone), diminished limb movements, lack of tendon reflexes, fasciculation of the tongue, swallowing and feeding difficulties, leading to growth failure and recurrent aspiration 58. These children also develop scoliosis (curvature of the spine) or other skeletal abnormalities as they get older. The diaphragm is not involved until late in the course of disease. Children with spinal muscular atrophy type 1 now live longer and can reach higher motor milestones like sitting and even walking with more proactive clinical care and newly available disease modifying treatment.
With supportive care only, prospective studies of children with SMA type 1 have shown median survival of 24 months 72; however, more recent studies have shown a median time to either death or >16 hours/day of ventilation of 8 to 13.5 months 36, 73. With proactive respiratory and nutritional supportive care, survival is improving 74. Severe symptomatic bradycardia has been noted in a study of the long-term survival of ventilator-dependent individuals with SMA type 1 75. Promising new treatments are changing the natural history of SMA type 1, particularly when treatment is initiated before onset of symptoms.
Spinal muscular atrophy type 2
Spinal muscular atrophy type 2 also called Dubowitz disease or the intermediate spinal muscular atrophy, is characterized by muscle weakness that develops in children between ages 6 and 18 months; the mean age of symptom onset is 8.3 months 71, 58. Children with SMA type 2 can sit without support, although they may need help getting to a seated position, but are unable to stand or walk unaided, and some may lose the ability to stay seated independently over time without treatment. Although poor muscle tone may be evident at birth or within the first few months of life, individuals with SMA type 2 may gain motor milestones slowly until about age five years. However, as the muscle weakness worsens later in childhood, affected individuals may need support to sit. With supportive care only, the maximum motor milestone attained is the ability to sit independently when placed. Affected individuals then have a slow decline in motor function and on average lose the ability to sit independently by the mid-teens 76. Cognition is normal. Cardiac abnormalities are unlikely to develop 77.
Individuals with spinal muscular atrophy type 2 cannot stand or walk unaided. Deep tendon reflexes are decreased to absent. They often have involuntary trembling (tremors) in their fingers, a spine that curves side-to-side (scoliosis), and progressive respiratory muscle weakness that can be life-threatening 78, 79, 80. They may have respiratory difficulties including hypoventilation in sleep 68. The progression of disease is variable without treatment. Life expectancy of individuals with spinal muscular atrophy type 2 varies, but many people with this condition live into their twenties or thirties. With disease modifying treatment and proactive clinical care, children with spinal muscular atrophy type 2 have improved motor outcomes. A review of life expectancy of 240 individuals with SMA type 2 from Germany and Poland found that 68% of individuals with SMA type 2 were alive at age 25 years 81. The ability to stand is directly correlated with better pulmonary function and long-term survival. This natural history, however, will likely be improved by newer treatments.
Spinal muscular atrophy type 3
Approximately 30% of SMA patients are from spinal muscular atrophy type 3 also called Kugelberg-Welander disease or Wohlfart-Kugelberg-Welander disease, which has an onset of muscle weakness after age 18 months with a mean age of onset of 39 months ± 32.6 months 78, 67, 82, 71. SMA Type 3 is further classified into Type 3a with onset between 18 months to three years old and 3b with onset between ages of 3 to 30 years old. Typically, patients develop a variable degree of muscle weakness, resulting in heterogeneous physical symptoms 83. Although most of them are able to walk independently, some present with progressive proximal weakness and lose ambulation after early childhood, and the disease is usually associated with foot deformity, difficulty of climbing stairs and muscle cramps 58, 78, 84.
The legs are more severely affected than the arms. Individuals with spinal muscular atrophy type 3 can stand and walk unaided, but over time, walking and climbing stairs may become increasingly difficult. They first show difficulty walking and running, climbing steps, or rising from a chair. The proximal leg muscles are most often affected first, with a tremor seen in the hands. With supportive care only, individuals walk independently but proximal muscle weakness may lead to more frequent falls or trouble walking up and down stairs. Fatigue can adversely affect quality of life and function significantly. Most children with SMA type 3 treated only with supportive care make gains in their motor function until about age six years and then experience a slow decline in function until about puberty. Puberty (until age ~20) may be associated with a more rapid decline in function for adolescents with SMA type 3. With supportive care only, adulthood is then associated with another, much slower decline in function 85.
Although individuals with spinal muscular atrophy type 3 develop the ability to walk, the vast majority will lose that ability with time. Many affected individuals require wheelchair assistance later in life. If symptom onset is before age three years, loss of ambulation typically occurs in the second decade. However, if symptom onset is between ages 3 and 12 years, loss of ambulation may occur in the fourth decade 86. Individuals with SMA type 3 have little to no respiratory muscle weakness. Cardiac and cognitive functions are normal.
Complications include scoliosis and joint contractures—chronic shortening of muscles or tendons around joints–caused by abnormal muscle tone and weakness, which prevents the joints from moving freely. Individuals with SMA type 3 may be prone to respiratory infections, but with care most have a normal lifespan. Disease modifying treatment can improve outcomes. People with spinal muscular atrophy type 3 typically have a normal life expectancy. In a retrospective study of individuals with SMA, the life expectancy of 329 individuals with SMA type 3 from Germany and Poland treated only with supportive care was not different from that of the general population 81. This natural history, however, will likely be improved by newer treatments.
Spinal muscular atrophy type 4
Spinal muscular atrophy type 4 is the least common form of SMA and affects fewer than 5% of individuals with SMA and often begins in early adulthood (after 21 years of age) 73, 78. Affected individuals usually experience mild to moderate muscle weakness, tremors, and mild breathing problems. There is a specific pattern of muscle involvement, with weakness disproportionately affecting the deltoids, triceps, and quadriceps. There may be a loss of patellar reflexes, with sparing of the deep tendon reflexes in the upper extremities and Achilles. Individuals may have a hand tremor. Cardiac and cognitive functioning is normal. With supportive care only, findings are similar to but less severe than those described for SMA type 3, and if loss of ambulation occurs, it may be after the fifth decade 87, 88, 81, 86. People with spinal muscular atrophy type 4 have a normal life expectancy 58, 83.
Spinal muscular atrophy variants
Juvenile bulbar palsy, or bulbar hereditary motor neuronopathy (HMN) types I and II
Bulbar hereditary motor neuronopathy I (Vialletto-van Laere syndrome) is an autosomal recessive syndrome that begins in the second decade of life. It is characterized by facial weakness, dysphagia and dysarthria followed by facial weakness and compromised respiratory function. The distinguishing feature of this syndrome is the development of bilateral sensorineural hearing loss.
Bulbar HMN II (Fazio-Londe disease)
Bulbar HMN II (Fazio-Londe disease) is characterized by progressive bulbar paralysis in the first decade of life. Patients present with stridor, dysarthria, and dysphagia. Cranial-nerve involvement leads to facial diplegia, ptosis, and ophthalmoplegia. Generalized weakness of the lower motor neurons and rare corticospinal-tract signs are sometimes observed. Median survival for patients with bulbar hereditary motor neuronopathy II is 18 months 89.
Distal spinal muscular atrophy (spinal Charcot-Marie-Tooth or hereditary motor neuronopathy type II)
Distal spinal muscular atrophy (spinal Charcot-Marie-Tooth or hereditary motor neuronopathy type II) may clinically mimic Charcot-Marie-Tooth (CMT) disease, otherwise known as hereditary motor and sensory neuropathy (HMSN) types 1 and 2: CMT is characterized by peroneal muscular atrophy, weakness, and wasting in the legs. High foot arches (pes cavus) are often present. Deep tendon reflexes are reduced or absent. Distal large fiber sensory loss is found on examination, although patients do not usually present with complaints of subjective sensory loss. Compared with Charcot-Marie-Tooth, patients with distal spinal muscular atrophy do not have sensory loss and the electrodiagnostic examination shows sparing of sensory nerves 90.
X-lined recessive bulbospinal muscular atrophy (Kennedy disease)
X-lined recessive bulbospinal muscular atrophy (Kennedy disease) patients present with bulbar weakness, gynecomastia, and lower motor neuron weakness beginning at age 20-40 years 91. Muscles cramps often precede weakness, and facial and perioral fasciculations are seen in more than 90% of patients. Increased rates of type 2 diabetes, infertility, and hand tremor are associated with Kennedy disease. This condition results from a triple repeat mutation (cytosine-adenine-guanine [CAG]) in exon 1 of the androgen receptor gene on the X chromosome. Because of the X-linked nature of Kennedy disease, daughters of affected patients are obligated carriers; therefore, genetic counseling is indicated.
Scapuloperoneal spinal muscular atrophy type 1
Scapuloperoneal spinal muscular atrophy type 1 (autosomal dominant form) appears at age 14-26, with weakness, distal leg atrophy, and absent tendon reflexes and sparing of intrinsic foot muscles. Facial, bulbar, and pectoral muscles are rarely affected. Progression is slow, with survival into the seventh or eight decade of life.
Scapuloperoneal spinal muscular atrophy type 2
Scapuloperoneal spinal muscular atrophy type 2 (autosomal recessive form): Patients present between birth and age 5 years, with weakness and atrophy of the lower extremities and pectoral girdle. The course is variable, and patients can survive to the fourth decade 92.
X-linked form scapuloperoneal spinal muscular atrophy
X-linked form scapuloperoneal spinal muscular atrophy has been described with an onset before age 10 years. Patients present with weakness of the pectoral girdle and arms with contractures. Cardiac conduction defects and cardiomyopathy are noted. The syndrome is slowly progressive but stabilizes by age 20 years, and patients survive to the sixth decade.
Davidenkow syndrome
Davidenkow syndrome is a form of scapuloperoneal spinal muscular atrophy characterized by weakness of the pectoral girdle and distal leg muscles, pes equinovarus, and distal sensory loss and fasciculations. Autosomal dominant (age of onset, 15-30 y) and autosomal recessive (age of onset, < 15 y) forms have been described. The clinical course is slow in the autosomal dominant form, whereas the course of the autosomal recessive form is unknown.
Fascioscapulohumeral spinal muscular atrophy
Most reports of fascioscapulohumeral spinal muscular atrophy are from Japan. It is an autosomal dominant or sporadic disorder characterized by limb-girdle and facial weakness occurring before age 20 years. The phenotype of fascioscapulohumeral spinal muscular atrophy is similar to that of fascioscapulohumeral dystrophy (FSHD), another unrelated muscular dystrophy. However, fascioscapulohumeral spinal muscular atrophy does not have the chromosome 4 gene deletion seen in fascioscapulohumeral dystrophy. Progression is slow, and the overall prognosis is good.
Scapulohumeral spinal muscular atrophy
Described initially in a Dutch family, scapulohumeral spinal muscular atrophy is a autosomal dominant disorder is characterized by the onset of scapulohumeral weakness and atrophy between the fourth and sixth decades of life. Progression is rapid, with death from respiratory failure occurring within 3 years.
Oculopharyngeal spinal muscular atrophy
Oculopharyngeal spinal muscular atrophy is seen mainly in people of French-Canadian descent and is characterized by bulbar and cranial-nerve weakness followed by myopathic weakness of the limbs. The pattern of inheritance is autosomal dominant with variable penetrance. The onset is usually in the fourth to fifth decades of life, and the disease is slowly progressive.
Ryukyuan spinal muscular atrophy
Ryukyuan spinal muscular atrophy is an autosomal recessive disorder described in men who live in the Japanese community on Ryukyu Islands. The onset is before age 5 years, and the disease is characterized by weakness and atrophy of the lower extremities, skeletal abnormalities (eg, scoliosis), and foot deformities (eg, pes cavus). Deep tendon reflexes are diminished or absent. The course of Ryukyuan spinal muscular atrophy disease is unknown 93.
Spinal muscular atrophy with pontocerebellar hypoplasia (PCH1)
This heterogeneous autosomal recessive disorder is characterized by generalized muscle weakness, global developmental delay, and early death. One study found that 30-40% of patients with milder disease course had protein EXOSC3 mutations 94.
Other variants
Other variants have been described, including multiple long-bone fractures at birth, diaphragmatic paralysis with early respiratory failure, congenital heart defects, arthrogryposis, segmental amyotrophy, vocal-cord paralysis (distal hereditary motor neuronopathy type VII), and disease of the anterior horn cell with agenesis of the corpus callosum (spinal muscular atrophy with respiratory distress) 95. Spinal muscular atrophy with respiratory distress presents with rapid decline over 2 years, followed by a plateau, and is linked with mutation in the IGHMBP2 gene 96. An autosomal dominant late-onset lower motor neuronopathy was discovered in 2 Finnish families with linkage to a mutation on band 22q11.2-q13.2 97. A rare form of autosomal dominant proximal spinal muscular atrophy has been identified with a possible linkage to an SETX gene mutation 98.
Spinal muscular atrophy causes
About 95% of SMA cases are caused by a homologous deletion or mutation of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 region (long arm of chromosome 5), which is the blueprint for the survival motor neuron (SMN) protein (see Figure 1) 78, 99, 100. A normal individual has two forms of the SMN gene, which are telomeric SMN1 and its paralog, centromeric SMN2 (Figure 1) 2, 67. Both SMN1 and SMN2 genes are nearly identical, with only a difference in five base pairs. However, the base pair differences do not alter the amino acid sequence, and they encode the same SMN protein. The number of copies of the SMN2 gene modifies the severity of the condition with a high number of SMN2 copies related to milder phenotypes and helps determine which type develops 25, 26. For example, SMA type 1 patients generally have one or two SMN2 copies, while SMA type 3 or 4 patients have more than four copies 28, 29. However, this inverse relationship is not always true, as a few patients with two SMN2 copies showed milder SMA phenotypes, while there have also been patients with three SMN2 copies that have been defined as type 1 17, 30, 31, 32. Lacking either one SMN gene leads to low levels of SMN protein, though this still allows embryonic development and usually occurs in SMA carriers 13, 101, 102.
The SMN1 and SMN2 genes both provide instructions for making a protein called the survival motor neuron (SMN) protein. Normally, most functional SMN protein is produced from the SMN1 gene, with a small amount produced from the SMN2 gene. Several different versions of the SMN protein are produced from the SMN2 gene, but only one version is functional; the other versions are smaller and quickly broken down. The SMN protein is one of a group of proteins called the SMN complex, which is important for the maintenance of motor neurons. Motor neurons transmit signals from the brain and spinal cord that tell skeletal muscles to tense (contract), which allows the body to move.
Most people with spinal muscular atrophy are missing a piece of the SMN1 gene, which impairs SMN protein production. A shortage of SMN protein leads to motor neuron death, and as a result, signals are not transmitted between the brain and muscles. Muscles cannot contract without receiving signals from the brain, so many skeletal muscles become weak and waste away, leading to the signs and symptoms of spinal muscular atrophy.
Researchers suggest that a shortage of SMN protein leads to the inefficient assembly of the machinery needed to process pre-mRNA. A lack of mature mRNA and subsequently, the proteins needed for normal cell functioning, has damaging effects on motor neuron development and survival. The loss of motor neurons leads to the signs and symptoms of spinal muscular atrophy. However, it is unclear why these cells are particularly sensitive to a reduction in the amount of SMN protein. Some research findings indicate that a shortage of SMN protein impairs the formation and function of axons and dendrites, leading to the death of motor neurons. While the mechanism is not clear, it is apparent that increased SMN2 gene copy number leads to an increase in SMN protein production, which improves the function and survival of motor neurons and results in less severe disease.
Typically, people have two copies of the SMN1 gene and one to two copies of the SMN2 gene in each cell. However, the number of copies of the SMN2 gene varies, with some people having up to eight copies. In people with spinal muscular atrophy, having multiple copies of the SMN2 gene is usually associated with less severe features of the condition that develop later in life. The SMN protein produced by the SMN2 genes can help make up for the protein deficiency caused by SMN1 gene mutations. People with spinal muscular atrophy type 0 typically have zero copies or one copy of the SMN2 gene in each cell, while those with type 1 generally have one or two copies, those with type 2 usually have three copies, those with type 3 have three or four copies, and those with type 4 have four or more copies. Other factors, many unknown, also contribute to the variable severity of spinal muscular atrophy.
Spinal muscular atrophy inheritance pattern
Spinal muscular atrophy types 0, 1, 2, 3, and 4 is inherited in an autosomal recessive pattern, which means both copies of the SMN1 gene in each cell have mutations. In most cases, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. In rare cases, a person with spinal muscular atrophy inherits an SMN1 gene mutation from one parent and acquires a new mutation in the other copy of the gene that occurs during the formation of reproductive cells (eggs or sperm) or in early embryonic development. In these cases, only one parent is a carrier of the SMN1 gene mutation.
Individuals who have more than the usual two copies of the SMN2 gene usually do not inherit the extra copies from a parent. They typically arise during a random error when making new copies of DNA (replication) in an egg or sperm cell or just after fertilization.
Finkel type spinal muscular atrophy is inherited in an autosomal dominant pattern. This means that the person has one copy of the altered gene in each cell that causes the disorder.
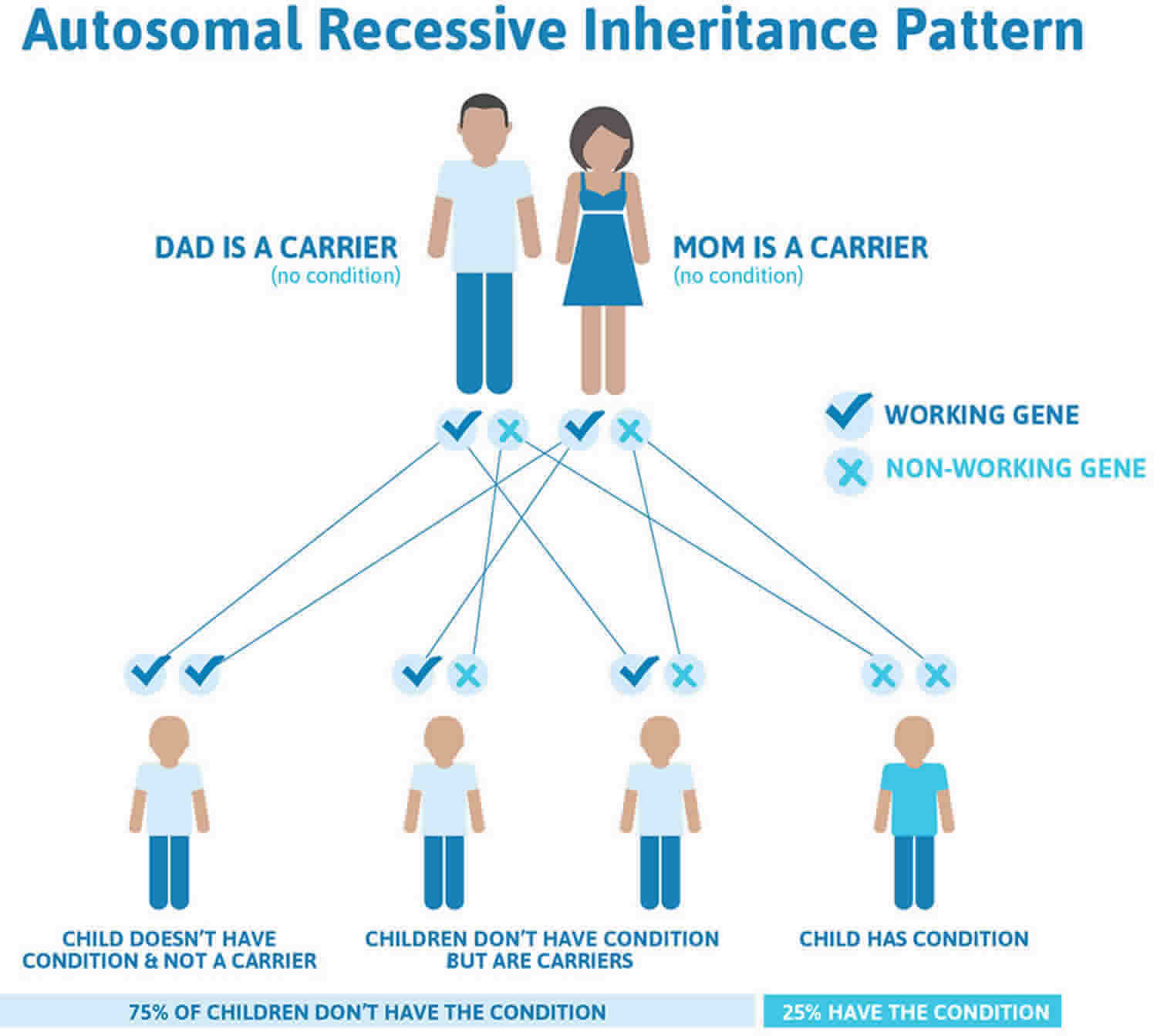
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 5 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 5. Spinal muscular atrophy autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
How can I prevent spinal muscular atrophy?
SMA is an inherited disease. If you or your partner carries the mutated gene that causes SMA, a genetic counselor can explain the chances of your child having SMA or being a carrier.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
You may be able to take steps before pregnancy to lower the risk of passing on SMA. A process called preimplantation genetic diagnosis (PGD) identifies embryos that don’t have the mutated gene. Your doctor implants healthy embryos during in vitro fertilization (IVF). Preimplantation genetic diagnosis (PGD) ensures your child will have two healthy SMN1 genes and not get SMA.
SMA symptoms vary depending on the type. SMA is characterized by a progressive loss of muscle control, muscle movement, and increasing muscle weakness. SMA patients generally start to show symptoms early in life, and the disease becomes more severe over time. Proximal muscles (muscles closest to the center of the body, such as those in the torso and neck) are more severely affected than muscles furthest from the center of the body (distal muscles, such as those in the hands and feet). People with SMA either never acquire or may progressively lose the ability to walk, stand, sit and/or eventually move. In addition, normal growth and development can place additional stress on already weakened muscles. Therefore, SMA patients often develop respiratory illnesses, and bone and/or spinal deformities which may require surgical treatment.
There are four primary types of spinal muscular atrophy:
- SMA type 0 also known as SMA type 1A is the most severe form of spinal muscular atrophy and begins before birth. Usually, the first symptom of type 0 is reduced movement of the fetus that is first seen between 30 and 36 weeks of the pregnancy and as a result they are often born with joint deformities (contractures) and have extremely weak muscle tone (hypotonia). After birth, these newborns have little movement and have difficulties with swallowing and breathing. Their respiratory muscles are very weak and they often do not survive past infancy due to respiratory failure. Some infants with spinal muscular atrophy type 0 (SMA type 1A) also have heart defects that are present from birth (congenital).
- SMA type 1 (severe): About 60% of people with SMA have type 1, also called Werdnig-Hoffman disease. Symptoms appear at birth or within an infant’s first six months of life. Infants with type 1 SMA have difficulty breathing, swallowing and sucking and they are unable to sit without support. They don’t meet typical milestones like holding up their heads or sitting. As muscles continue to weaken, children become more prone to respiratory infections and collapsed lungs (pneumothorax). Most children with type 1 SMA die before their second birthday.
- SMA type 2 (intermediate): Symptoms of type 2 SMA also called Dubowitz disease appear when a child is between six months and 18 months old. This type tends to affect the lower limbs. Children with type 2 SMA may be able to sit up but cannot stand or walk without help. Most children with type 2 SMA live into adulthood.
- SMA type 3 (mild): Symptoms of type 3 SMA also called Kugelbert-Welander disease or juvenile-onset SMA appear after a child’s first 18 months of life. Symptoms appear between early childhood (older than age 1 year) and early adulthood. Some people with SMA type 3 don’t have signs of disease until early adulthood. SMA type 3 symptoms include mild muscle weakness, difficulty walking and frequent respiratory infections. Over time, symptoms can affect their ability to stand and walk. Type 3 SMA doesn’t significantly shorten life expectancy.
- SMA type 4 (adult): The rare adult form of SMA doesn’t typically appear until the mid-30s. Muscle weakness symptoms progress slowly, so most people with type 4 remain mobile and live full lives.
- Finkel type occur in adulthood, usually after age 30. Symptoms of adult-onset spinal muscular atrophy are usually mild to moderate and include muscle weakness, tremor and twitching.
Spinal muscular atrophy complications
Over time, people with SMA experience progressive muscle weakness and loss of muscle control. Potential complications include:
- Bone fractures, hip dislocation and scoliosis (curvature of the spine).
- Malnutrition and dehydration due to problems eating and swallowing that may require a feeding tube.
- Pneumonia and respiratory infections.
- Weak lungs and breathing problems that may require breathing support (ventilation).
Individuals with SMA suffer from respiratory, gastrointestinal, poor weight gain with growth failure, restrictive lung disease, scoliosis, joint contractures and sleep difficulties that affect the quality of life and can potentially be life-threatening, for example, chest infections secondary to aspiration due to inadequate swallowing and muscle weakness 103. Patients with SMA are prone to suffer from metabolic acidosis, especially during periods of illness or fasting, the underlying cause of this predisposition is unknown, and there have been suggestions that dysfunctional glucose metabolism secondary to pancreatic abnormalities may play a role 104, 105. At this time, it is unknown what long-term complications may arise in individuals who receive early and/or presymptomatic targeted treatment.
Spinal muscular atrophy differential diagnosis
Disorders to consider in the spinal muscular atrophy (SMA) differential diagnosis 6:
- Congenital (less than 6months): Pompe disease, Prader-Willi syndrome, Myotonic dystrophy type 1, Sellweger spectrum disorder, Congenital myasthenic syndromes, X-linked infantile spinal muscular atrophy. It is essential to consider congenital myopathies, disorders of metabolism, and disorders of mitochondria.
- Childhood: Botulism, hexosaminidase A deficiency, Guillain-Barré, Duchenne muscular dystrophy, Fazio-Londe syndrome, Hirayama disease
- Adulthood: Amyotrophic lateral sclerosis, spinal, and bulbar muscular atrophy 106
Spinal muscular atrophy diagnosis
Some SMA symptoms resemble those resulting from neuromuscular disorders like muscular dystrophy. A thorough history, examination as well as creatine kinase (CK), electromyogram (EMG), nerve conduction studies (NCV), muscle biopsy, MRI, as well as referral to a geneticist can all be key to determining the correct diagnosis 6, 107 . To find the cause of symptoms, your healthcare provider will perform a physical exam and get a medical history. Your physician may also order one or more of these tests to diagnose SMA 108:
- Blood test: An enzyme and protein blood test can check for high levels of creatine kinase. Deteriorating muscles release this enzyme into the bloodstream.
- Genetic test: This blood test identifies problems with the SMN1 gene. As a diagnostic tool, a genetic test is 95% effective at finding the altered SMN1 gene. Some states test for SMA as part of routine newborn screenings.
- Nerve conduction test: An electromyogram (EMG) measures the electrical activity of nerves muscles and nerves.
- Muscle biopsy: Rarely, a physician may perform a muscle biopsy. This procedure involves removing a small amount of muscle tissue and sending it to a lab for examination. A biopsy can show atrophy, or loss of muscle.
To make a diagnosis of spinal muscular atrophy, symptoms need to be present. When symptoms are present, diagnosis can be made by genetic testing. Gene alterations (mutations) in the SMN1 and VAPB genes cause spinal muscular atrophy. Having extra copies of the SMN2 gene can modify the course of spinal muscular atrophy.
A blood test is available to look for deletions or mutations of the SMN1 gene. This test identifies at least 95 percent of spinal muscular atrophy Types 1, 2, and 3 and may also reveal if a person is a carrier of a defective gene that could be passed on to children. If the SMN1 gene is not found to be abnormal or the individual’s history and examination are not typical of spinal muscular atrophy, other diagnostic tests may include electromyography (which records the electrical activity of the muscles during contraction and at rest), nerve conduction velocity studies (which measure the nerve’s ability to send an electrical signal), muscle biopsy (used to diagnose many neuromuscular disorders), and other blood tests.
Genetic testing for a mutation in the VAPB gene is done to diagnose the Finkel type spinal muscular atrophy.
In some situations other tests such as an electromyography (EMG) or muscle biopsy may be needed because it is not possible to conduct the SMN gene tests or no abnormality is identified.
Newborn screening for SMA is being implemented throughout the United States. As of January 2021, 39 states screen for SMA representing 86% of all infants born in the U.S. Newborn screening facilitates early identification of infants with SMA and thus early implementation of treatment. Infants identified by SMA newborn screening are urgently referred for confirmatory testing, discussion of treatments and care. Early treatment prior to the onset of symptoms provides the best outcomes. Newborn screening will not identify 3-5% of infants with SMA due to having a point mutation in the SMN1 gene. These infants will progress to develop symptoms and require rapid diagnosis and treatment.
Carrier testing for SMA is also available using a molecular genetic test in which the number of copies of the SMN1 gene is determined. The American College of Obstetricians and Gynecologists recommends offering carrier screening for SMA to all women who are considering pregnancy or are currently pregnant.
Figure 6. Spinal muscular atrophy diagnostic algorithm
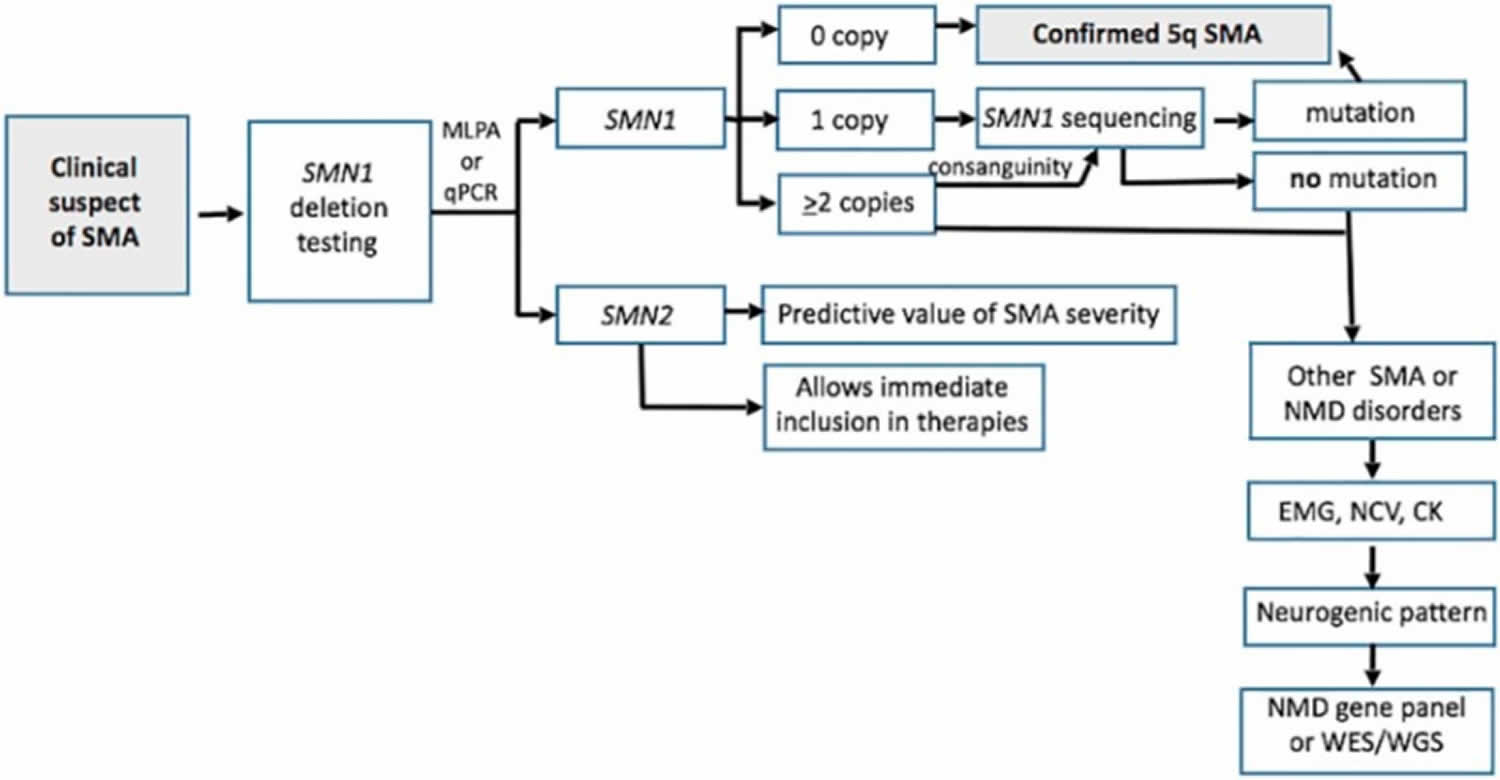
Abbreviations: EMG = electromyography; NCV = nerve conduction studies; CK = creatine kinase
[Source 6 ]Can spinal muscular atrophy be diagnosed during pregnancy?
If you’re pregnant and have a family history of SMA, prenatal tests can determine if the developing fetus has the disease. These tests slightly increase the risk of miscarriage or pregnancy loss. Prenatal tests for SMA include:
- Amniocentesis: During amniocentesis, your obstetrician inserts a thin needle into your belly to draw out a small amount of fluid from the amniotic sac. A lab specialist (pathologist) checks the fluid for SMA. This test takes place after the 14th week of pregnancy.
- Chorionic villus sampling (CVS): Your obstetrician removes a small tissue sample from the placenta through your cervix or stomach. A pathologist checks the sample for SMA. CVS can take place as early as the 10th week of pregnancy.
Spinal muscular atrophy treatment
There is no complete cure for spinal muscular atrophy (SMA). The treatment of SMA requires a multidisciplinary team approach and should notably include neurologists, medical geneticists, physical therapists, speech pathologists, pulmonologists, respiratory therapists, medical social workers, nutritionists, psychologists and specialized nurses.
There are two main components to SMA management: treatment that slows the progression of the disease (disease-modifying therapy) and therapy that helps manage symptoms, prevents complications and improves quality of life (supportive therapy). Many people with SMA benefit from physical and occupational therapy and assistive devices, such as orthopaedic braces, crutches, walkers and wheelchairs.
Genetic counseling is recommended for affected individuals and their families.
Disease-modifying therapy
In December 2016 the U.S. Food and Drug Administration (FDA) approved nusinersen (Spinraza®) as the first drug approved to treat children and adults with spinal muscular atrophy. The drug is administered by injection intrathecally into the cerebrospinal fluid (CSF) surrounding the spinal cord. Nusinersen (Spinraza®) is an antisense oligonucleotide that promotes functional SMN2 production by inhibiting ISS-N1 (an SMN2 exon 7 splicer) and thereby increasing the amount of functional SMN protein produced, which is critical for the maintenance of motor neurons 109. The benefit is best documented in infants and children, particularly when started early 110, 109. Nusinersen (Spinraza®) costs $118,000 for one vial and costs $708,000 for the first year of treatment 111, 112.
In May 2019, the FDA approved onasemnogene abeparovec-xioi (Zolgensma®) gene therapy for children less than 2 years old who have infantile-onset spinal muscular atrophy. This single dose intravenous injection gene therapy utilizes the properties of adeno-associated virus serotype 9 (AAV9) and uses it to deliver the SMN1 gene into cells and thereby allowing the body to produce functioning SMN protein 113. A phase 1/2 study testing safety and efficacy of onasemnogene abeparvovec in 15 patients with SMA type 1 showed all patients were alive at 20 months compared to an expected 8% in historical cohorts. Furthermore, significant motor improvements were seen, with 11/15 able to sit unassisted in a cohort of patients classically defined by their inability to sit 113. Onasemnogene abeparvovec is priced at $2.125 million for a single injection and has been reported to be the most expensive drug in the world 114. Onasemnogene abeparvovec is currently licensed for use in the USA 111, 115.
In 7 August 2020, the FDA approved risdaplam (Evrysdi®) to treat patients two months of age and older with SMA. Risdiplam is the first orally administered drug approved for the treatment of SMA. People take risdaplam daily by mouth (orally). Risdiplam is a SMN2 splicing modifier designed to treat patients with SMA that is caused by mutations in chromosome 5q that lead to SMN protein deficiency 49. In FIREFISH, 41% (7/17) of infants treated with the Risdiplam therapeutic dosage achieved the ability to sit without support for at least 5 seconds as measured by the Bayley Scales of Infant and Toddler Development Third Edition (BSID-III) 50. In addition, 90% (19/21) of infants were alive without permanent ventilation at 12 months of treatment and reached 15 months of age or older 51. In SUNFISH 52, children and adults treated with risdiplam experienced a clinically meaningful and statistically significant improvement in motor function at 12 months (1.55 point mean difference) compared with placebo (1.36 points; –0.19 points, respectively), as measured by a change from baseline in the Motor Function Measure-32 total score.
Based upon these trial results, nusinersen (Spinraza®) has been approved for use in the United States, Europe, Canada, Japan, and several other countries in Asia and the Middle East. Onasemnogene abeparvovec (Zolgensma®) gene theray has been approved for use in the United States, Europe, Japan, and many other countries in South America and Asia. Risdiplam (Evrysdi®) has been approved in the United States and European Union 49, 53. Nusinersen and onasemnogene abeparvovec are also recommended by the National Institute for Health and Care Excellence (NICE) in the United Kingdom 54.
Symptomatic therapy
The symptomatic management of SMA includes physical therapy, occupational therapy, monitoring respiratory function and intervening as clinically indicated, nutritional status monitoring and intervention, spine curvature monitoring and intervention and use of orthotics and adaptive equipment as needed.
Physical therapy, occupational therapy, and rehabilitation may help to improve posture, prevent joint immobility, and slow muscle weakness and atrophy. Stretching and strengthening exercises may help reduce contractures, increase range of motion, and keeps circulation flowing. Some individuals require additional therapy for speech and swallowing difficulties. Assistive devices such as supports or braces, orthotics, speech synthesizers, and wheelchairs may be helpful to improve functional independence.
Proper nutrition and calories are essential to maintaining weight and strength, while avoiding prolonged fasting. People who cannot chew or swallow may require insertion of a feeding tube. Non-invasive ventilation at night can improve breathing during sleep, and some individuals also may require assisted ventilation during the day due to muscle weakness in the neck, throat, and chest.
Restrictive lung disease seen in SMA type 0, 1, and 2 causes respiratory failure and is ultimately the cause of death. Non-invasive ventilation, usually in the form of BiPAP (bilevel positive airway pressure), has been used as a way of increasing quality of life and life expectancy 116, 55. Patients requiring this form of support will also have a weak cough and have an increased risk of respiratory compromise in the form of mucus plugging, aspiration, recurrent infections, and subsequent hypoxemia 116. The involvement of chest physiotherapists is key for cough assessments, mucous clearing, and tracking of forced vital capacity in children >5 years of age 55. Where non-invasive ventilation is no longer sufficient difficult discussions regarding tracheostomy and permanent invasive ventilation need to be made, this should take place in a multi-disciplinary setting with early involvement of palliative care specialists 116.
Due to associated muscle weakness, patients are prone to tire quickly and have swallowing difficulties that can lead to a failure to thrive and have a negative compounding effect on muscle weakness. Other gastrointestinal symptoms include constipation, delayed gastric emptying, and reflux 103. For patients with type 1 SMA early consideration of laparoscopic gastrostomy and Nissen fundoplication is important and can improve nutritional status and decrease the frequency of aspiration 117. Patients with SMA type 2 require close attention to nutritional status as although they may plot as being in the normal range on a growth chart for their age, they are more likely to have increased adiposity, the involvement of nutritionists is therefore vital in ensuring optimal nutritional management 118, 119.
Patients suffer from orthopedic complications such as scoliosis, hip subluxation, and susceptibility to fractures. SMA type 1 and 2 are particularly affected by these complications, with type 3 being variably affected 120. Physiotherapy involvement is important in optimizing and preserving function, and mobility with the use of stretching exercises and passive movement of joints helps to avoid joint contractures 120. Similarly, orthotic specialist involvement is important to utilize frames, orthotics, and wheelchairs to improve quality of life and mobility. Orthopedic surgical monitoring is required for scoliosis with periodic consideration for spinal fusion and bracing 120, 103.
Spinal muscular atrophy prognosis
The quality of life and life expectancy for people with SMA varies depending on the type. SMA type 0 being the worst and individuals dying within the first months of life. Infants with type 1 SMA usually die before their second birthday. Children with type 2 or type 3 SMA may live full lives depending on the severity of symptoms. People who develop SMA during adulthood (type 4) often remain active and enjoy a normal life expectancy 20. However, with the recent introduction of disease-modifying agents such as onasemnogene abeparvovec, there have already been reported cases of SMA type 1 patients living longer than the historical cohort data would suggest, prognosis, therefore could potentially be much improved and is the source of ongoing study 121.
Spinal muscular atrophy life expectancy
Spinal muscular atrophy life expectancy varies depending on the type of spinal muscular atrophy. Some forms of spinal muscular atrophy are fatal without treatment. The mortality and/or morbidity rates of spinal muscular atrophy are inversely correlated with the age at onset. High death rates are associated with early onset disease. People with spinal muscular atrophy may appear to be stable for long periods, but improvement should not be expected without treatment.
Many patients with the severe form of spinal muscular atrophy type 1 die before the age of two 122. However, the life expectancy of SMA type 1 patients really depends on the severity of the disease at diagnosis, as well as treatment choices, which can include respiratory therapy, nutritional support, and physical therapy. Therefore, depending on the care provided and the severity of the disease, some SMA Type 1 patients may live into adulthood.
In contrast, outcomes of juvenile and adult spinal muscular atrophies are difficult to define because the progression of these diseases varies widely. Children with moderate to mild forms of SMA Types 2 and 3 live into adulthood and may have a normal life expectancy depending on the severity of respiratory, nutritional, and orthopedic symptoms 122.
Survival probabilities for types 1 and 2 and probabilities of being ambulatory for type 3 were derived for 445 SMA patients. These patients were subdivided on the basis of ISMAC criteria (ie, developmental milestones and age of onset) 123.
- Spinal muscular atrophy 1: Survival probabilities at ages 2, 4, 10, and 20 years were 32%, 18%, 8%, and 0%, respectively.
- Spinal muscular atrophy 2: Survival probabilities at ages 2, 4, 10, and 20 years were 100%, 100%, 98%, and 77%, respectively.
- Spinal muscular atrophy 3: Results differed, but life expectancy of patients with spinal muscular atrophy type 3 is close to that of the healthy population. Onset before age 3 years: Probabilities of being ambulatory at ages 2, 4, 10, 20, and 40 years were 98%, 94.5%, 73%, 44%, and 34%, respectively. Onset after age 3 years: Probabilities of being ambulatory at ages 2, 4, 10, 20, and 40 years were 100%, 100%, 97%, 89%, and 67%, respectively.
A recent series of 237 patients showed similar survival probabilities 124.
Disease onset after age 2-3 months has been correlated to longer survival time in spinal muscular atrophy type 1 125. Antibiotic treatment has not prolonged survival in spinal muscular atrophy type 1. Birnkrant et al 126 examined the role of noninvasive positive-pressure ventilation and gastrostomy in patients with spinal muscular atrophy type 1. Although these supportive measures can be effective in slowly progressive neuromuscular diseases, they did not alter survival in patients with spinal muscular atrophy type 1. A later study by Lemoine et al 127 concluded that early noninvasive respiratory intervention prolonged survival time compared with supportive care alone.
References- Otsuki N., Arakawa R., Kaneko K., Aoki R., Arakawa M., Saito K. A New Biomarker Candidate for Spinal Muscular Atrophy: Identification of a Peripheral Blood Cell Population Capable of Monitoring the Level of Survival Motor Neuron Protein. PLoS ONE. 2018;13:e0201764. doi: 10.1371/journal.pone.0201764
- Kolb S.J. Spinal Muscular Atrophy. Arch. Neurol. 2011;68:979. doi: 10.1001/archneurol.2011.74
- Jablonka S, Hennlein L, Sendtner M. Therapy development for spinal muscular atrophy: perspectives for muscular dystrophies and neurodegenerative disorders. Neurol Res Pract. 2022 Jan 4;4(1):2. doi: 10.1186/s42466-021-00162-9
- Yang M, Awano H, Tanaka S, Toro W, Zhang S, Dabbous O, Igarashi A. Systematic Literature Review of Clinical and Economic Evidence for Spinal Muscular Atrophy. Adv Ther. 2022 May;39(5):1915-1958. doi: 10.1007/s12325-022-02089-2
- Chong LC, Gandhi G, Lee JM, Yeo WWY, Choi SB. Drug Discovery of Spinal Muscular Atrophy (SMA) from the Computational Perspective: A Comprehensive Review. Int J Mol Sci. 2021 Aug 20;22(16):8962. doi: 10.3390/ijms22168962
- Prior TW, Leach ME, Finanger E. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2020 Dec 3]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352
- Crawford T.O., Pardo C.A. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol. Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010
- Markowitz J.A., Tinkle M.B., Fischbeck K.H. Spinal Muscular Atrophy in the Neonate. J. Obstet. Gynecol. Neonatal Nurs. 2004;33:12–20. doi: 10.1177/0884217503261125
- DiDonato C.J., Parks R.J., Kothary R. Development of a Gene Therapy Strategy for the Restoration of Survival Motor Neuron Protein Expression: Implications for Spinal Muscular Atrophy Therapy. Hum. Gene Ther. 2003;14:179–188. doi: 10.1089/104303403321070874
- Mahajan R. Onasemnogene Abeparvovec for Spinal Muscular Atrophy: The Costlier Drug Ever. Int. J. Appl. Basic Med. Res. 2019;9:127. doi: 10.4103/ijabmr.IJABMR_190_19
- Tisdale S., Pellizzoni L. Disease Mechanisms and Therapeutic Approaches in Spinal Muscular Atrophy. J. Neurosci. 2015;35:8691–8700. doi: 10.1523/JNEUROSCI.0417-15.2015
- Sumner C.J., Crawford T.O. Two Breakthrough Gene-Targeted Treatments for Spinal Muscular Atrophy: Challenges Remain. J. Clin. Investig. 2018;128:3219–3227. doi: 10.1172/JCI121658
- Lunn M.R., Wang C.H. Spinal Muscular Atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6
- Roberts D.F., Chavez J., Court S.D.M. The Genetic Component in Child Mortality. Arch. Dis. Child. 1970;45:33–38. doi: 10.1136/adc.45.239.33
- Wirth B. An Update of the Mutation Spectrum of the Survival Motor Neuron Gene (SMN1) in Autosomal Recessive Spinal Muscular Atrophy (SMA) Hum. Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9
- D’Amico A., Mercuri E., Tiziano F.D., Bertini E. Spinal Muscular Atrophy. Orphanet J. Rare Dis. 2011 doi: 10.1186/1750-1172-6-71
- Ogino S., Leonard D.G.B., Rennert H., Ewens W.J., Wilson R.B. Genetic Risk Assessment in Carrier Testing for Spinal Muscular Atrophy. Am. J. Med Genet. 2002;110:301–307. doi: 10.1002/ajmg.10425
- Prior T.W., Snyder P.J., Rink B.D., Pearl D.K., Pyatt R.E., Mihal D.C., Conlan T., Schmalz B., Montgomery L., Ziegler K., et al. Newborn and Carrier Screening for Spinal Muscular Atrophy. Am. J. Med Genet. Part A. 2010;152A:1608–1616. doi: 10.1002/ajmg.a.33474
- Schorling D.C., Pechmann A., Kirschner J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020;7:1–13. doi: 10.3233/JND-190424
- D’Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011 Nov 2;6:71. doi: 10.1186/1750-1172-6-71
- Spinal muscular atrophy. https://medlineplus.gov/genetics/condition/spinal-muscular-atrophy
- About SMA. https://smafoundation.org/about-sma
- Sugarman EA, Nagan N, Zhu H, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20:27–32. doi: 10.1038/ejhg.2011.134
- Kolb S, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33:831–846. doi: 10.1016/j.ncl.2015.07.004
- Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A Single Nucleotide Difference That Alters Splicing Patterns Distinguishes the SMA Gene SMN1 from the Copy Gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177
- Rad I.A. Mutation Spectrum of Survival Motor Neuron Gene in Spinal Muscular Atrophy. J. Down Syndr. Chromosome Abnorm. 2017;3:1–2. doi: 10.4172/2472-1115.1000118
- SMN2 gene. https://medlineplus.gov/genetics/gene/smn2
- Calucho M., Bernal S., Alías L., March F., Venceslá A., Rodríguez-Álvarez F.J., Aller E., Fernández R.M., Borrego S., Millán J.M., et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 2018;28:208–215. doi: 10.1016/j.nmd.2018.01.003
- Al Dakhoul S. Very Severe Spinal Muscular Atrophy (Type 0) Avicenna J. Med. 2017;7:32. doi: 10.4103/2231-0770.197512
- Feldkötter M., Schwarzer V., Wirth R., Wienker T.F., Wirth B. Quantitative Analyses of SMN1 and SMN2 Based on Real-Time LightCycler PCR: Fast and Highly Reliable Carrier Testing and Prediction of Severity of Spinal Muscular Atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi: 10.1086/338627
- Wirth B., Brichta L., Schrank B., Lochmüller H., Blick S., Baasner A., Heller R. Mildly Affected Patients with Spinal Muscular Atrophy Are Partially Protected by an Increased SMN2 Copy Number. Hum. Genet. 2006;119:422–428. doi: 10.1007/s00439-006-0156-7
- Cuscó I., Barceló M.J., Rojas–García R., Illa I., Gámez J., Cervera C., Pou A., Izquierdo G., Baiget M., Tizzano E.F. SMN2 Copy Number Predicts Acute or Chronic Spinal Muscular Atrophy but Does Not Account for Intrafamilial Variability in Siblings. J. Neurol. 2006;253:21–25. doi: 10.1007/s00415-005-0912-y
- Bertini E, Burghes A, Bushby K, Estournet-Mathiaud B, Finkel RS, Hughes RA, Iannaccone ST, Melki J, Mercuri E, Muntoni F, Voit T, Reitter B, Swoboda KJ, Tiziano D, Tizzano E, Topaloglu H, Wirth B, Zerres K. 134th ENMC International Workshop: Outcome Measures and Treatment of Spinal Muscular Atrophy, 11-13 February 2005, Naarden, The Netherlands. Neuromuscul Disord. 2005 Nov;15(11):802-16. doi: 10.1016/j.nmd.2005.07.005
- Rudnik-Schöneborn S, Forkert R, Hahnen E, Wirth B, Zerres K. Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN gene deletion findings. Neuropediatrics. 1996 Feb;27(1):8-15. doi: 10.1055/s-2007-973741
- Rudnik-Schöneborn S, Hausmanowa-Petrusewicz I, Borkowska J, Zerres K. The predictive value of achieved motor milestones assessed in 441 patients with infantile spinal muscular atrophy types II and III. Eur Neurol. 2001;45(3):174-81. doi: 10.1159/000052118
- Finkel RS, McDermott MP, Kaufmann P, Darras BT, Chung WK, Sproule DM, Kang PB, Foley AR, Yang ML, Martens WB, Oskoui M, Glanzman AM, Flickinger J, Montes J, Dunaway S, O’Hagen J, Quigley J, Riley S, Benton M, Ryan PA, Montgomery M, Marra J, Gooch C, De Vivo DC. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-7. doi: 10.1212/WNL.0000000000000741
- Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017 Sep;24(9):529-533. doi: 10.1038/gt.2017.52
- Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci. 2015;35:8691–8700. doi: 10.1523/JNEUROSCI.0417-15.2015
- Mercuri E, Finkel RS, Muntoni F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis RH, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Qian Y, Sejersen T; SMA Care Group. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-115. doi: 10.1016/j.nmd.2017.11.005
- Schorling DC, Pechmann A, Kirschner J. Advances in treatment of spinal muscular atrophy: new phenotypes, new challenges, new implications for care. J Neuromuscul Dis. 2020;7:1–13. doi: 10.3233/JND-190424
- Wurster C.D., Ludolph A.C. Nusinersen for Spinal Muscular Atrophy. Ther. Adv. Neurol. Disord. 2018;11:175628561875445. doi: 10.1177/1756285618754459
- Tabet R., El Bitar S., Zaidan J., Dabaghian G. Spinal Muscular Atrophy: The Treatment Approved. Cureus. 2017 doi: 10.7759/cureus.1644
- Wurster CD, Ludolph AC. Nusinersen for spinal muscular atrophy. Ther Adv Neurol Disord. 2018;11:1756285618754459
- US Food & Drug Administration. SPINRAZA prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209531s010lbl.pdf
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377:1723–1732. doi: 10.1056/NEJMoa1702752
- ZOLGENSMA prescribing information. https://www.novartis.com/us-en/sites/novartis_us/files/zolgensma.pdf
- Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198
- Day JW, Finkel RS, Chiriboga CA, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:284–293. doi: 10.1016/S1474-4422(21)00001-6
- Risdiplam prescribing information. https://www.gene.com/download/pdf/evrysdi_prescribing.pdf
- Bayley N. Bayley scales of infant and toddler development. 3. New York: Psychological Corporation; 2006.
- Baranello G, Darras BT, Day JW, et al. Risdiplam in type 1 spinal muscular atrophy. N Engl J Med. 2021;384:915–923. doi: 10.1056/NEJMoa2009965
- A Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of Risdiplam (RO7034067) in Type 2 and 3 Spinal Muscular Atrophy (SMA) Participants (SUNFISH). https://classic.clinicaltrials.gov/ct2/show/NCT02908685
- European Medicines Agency. Evrysdi (risdiplam). https://www.ema.europa.eu/en/medicines/human/EPAR/evrysdi
- National Institute for Health and Care Excellence (NICE). Single technology appraisal—nusinersen for treating spinal muscular atrophy [ID1069]—committee papers. London, England: NICE; 2019. https://www.nice.org.uk/guidance/ta588/documents/committee-papers-4
- Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, Trela A; Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007 Aug;22(8):1027-49. doi: 10.1177/0883073807305788
- Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22:1027–1049. doi: 10.1177/0883073807305788
- https://treat-nmd.org/
- Kolb S.J., Kissel J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015;33:831–846. doi: 10.1016/j.ncl.2015.07.004
- Dubowitz V. Very Severe Spinal Muscular Atrophy (SMA Type 0): An Expanding Clinical Phenotype. Eur. J. Paediatr. Neurol. 1999;3:49–51. doi: 10.1016/S1090-3798(99)80012-9
- Dubowitz V. Very severe spinal muscular atrophy (SMA type 0): an expanding clinical phenotype. Eur J Paediatr Neurol. 1999;3(2):49-51. doi: 10.1053/ejpn.1999.0181
- MacLeod MJ, Taylor JE, Lunt PW, Mathew CG, Robb SA. Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. 1999;3(2):65-72. doi: 10.1053/ejpn.1999.0184
- Rudnik-Schoneborn S., Heller R., Berg C., Betzler C., Grimm T., Eggermann T., Eggermann K., Wirth R., Wirth B., Zerres K. Congenital Heart Disease Is a Feature of Severe Infantile Spinal Muscular Atrophy. J. Med Genet. 2008;45:635–638. doi: 10.1136/jmg.2008.057950
- Grotto S., Cuisset J.-M., Marret S., Drunat S., Faure P., Audebert-Bellanger S., Desguerre I., Flurin V., Grebille A.-G., Guerrot A.-M., et al. Type 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients. J. Neuromuscul. Dis. 2016;3:487–495. doi: 10.3233/JND-160177
- Menke L.A., Poll-The B.T., Clur S.-A., Bilardo C.M., van der Wal A.C., Lemmink H.H., Cobben J.M. Congenital Heart Defects in Spinal Muscular Atrophy Type I: A Clinical Report of Two Siblings and a Review of the Literature. Am. J. Med Genet. Part A. 2008;146A:740–744. doi: 10.1002/ajmg.a.32233
- Zerres K. Natural History in Proximal Spinal Muscular Atrophy. Arch. Neurol. 1995;52:518. doi: 10.1001/archneur.1995.00540290108025
- Butchbach M.E.R. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front. Mol. Biosci. 2016;3 doi: 10.3389/fmolb.2016.00007
- Tariq F., Holcik M., MacKenzie A. Neurodegenerative Diseases. InTech; London, UK: 2013. Spinal Muscular Atrophy: Classification, Diagnosis, Background, Molecular Mechanism and Development of Therapeutics.
- Zerres K., Davies K.E. 59th ENMC International Workshop: Spinal Muscular Atrophies: Recent Progress and Revised Diagnostic Criteria 17–19 April 1998, Soestduinen, The Netherlands. Neuromuscul. Disord. 1999;9:272–278. doi: 10.1016/S0960-8966(99)00016-4
- O’Hagen J.M., Glanzman A.M., McDermott M.P., Ryan P.A., Flickinger J., Quigley J., Riley S., Sanborn E., Irvine C., Martens W.B., et al. An Expanded Version of the Hammersmith Functional Motor Scale for SMA II and III Patients. Neuromuscul. Disord. 2007;17:693–697. doi: 10.1016/j.nmd.2007.05.009
- Thomas N.H., Dubowitz V. The Natural History of Type I (Severe) Spinal Muscular Atrophy. Neuromuscul. Disord. 1994;4:497–502. doi: 10.1016/0960-8966(94)90090-6
- Lin CW, Kalb SJ, Yeh WS. Delay in Diagnosis of Spinal Muscular Atrophy: A Systematic Literature Review. Pediatr Neurol. 2015 Oct;53(4):293-300. doi: 10.1016/j.pediatrneurol.2015.06.002
- Oskoui M, Levy G, Garland CJ, Gray JM, O’Hagen J, De Vivo DC, Kaufmann P. The changing natural history of spinal muscular atrophy type 1. Neurology. 2007 Nov 13;69(20):1931-6. doi: 10.1212/01.wnl.0000290830.40544.b9
- Kolb SJ, Coffey CS, Yankey JW, et al. NeuroNEXT Clinical Trial Network on behalf of the NN101 SMA Biomarker Investigators. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. 2017 Dec;82(6):883-891. doi: 10.1002/ana.25101
- Grychtol R, Abel F, Fitzgerald DA. The role of sleep diagnostics and non-invasive ventilation in children with spinal muscular atrophy. Paediatr Respir Rev. 2018 Sep;28:18-25. doi: 10.1016/j.prrv.2018.07.006
- Bach JR. Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am J Phys Med Rehabil. 2007 May;86(5):349-55. doi: 10.1097/PHM.0b013e31804b1d66
- Mercuri E, Finkel R, Montes J, Mazzone ES, Sormani MP, Main M, Ramsey D, Mayhew A, Glanzman AM, Dunaway S, Salazar R, Pasternak A, Quigley J, Pane M, Pera MC, Scoto M, Messina S, Sframeli M, Vita GL, D’Amico A, van den Hauwe M, Sivo S, Goemans N, Kaufmann P, Darras BT, Bertini E, Muntoni F, De Vivo DC. Patterns of disease progression in type 2 and 3 SMA: Implications for clinical trials. Neuromuscul Disord. 2016 Feb;26(2):126-31. doi: 10.1016/j.nmd.2015.10.006
- Finkel RS, Mercuri E, Meyer OH, Simonds AK, Schroth MK, Graham RJ, Kirschner J, Iannaccone ST, Crawford TO, Woods S, Muntoni F, Wirth B, Montes J, Main M, Mazzone ES, Vitale M, Snyder B, Quijano-Roy S, Bertini E, Davis RH, Qian Y, Sejersen T; SMA Care group. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018 Mar;28(3):197-207. doi: 10.1016/j.nmd.2017.11.004
- Arnold W.D., Kassar D., Kissel J.T. Spinal Muscular Atrophy: Diagnosis and Management in a New Therapeutic Era. Muscle Nerve. 2015;51:157–167. doi: 10.1002/mus.24497
- Dawood A.A., Moosa A. Hand and ECG Tremor in Spinal Muscular Atrophy. Arch. Dis. Child. 1983;58:376–378. doi: 10.1136/adc.58.5.376
- Sharawat I.K., Kohli T.S., Saini L. Trembling Hands and Trembling ECG. BMJ Case Rep. 2019;12:230618. doi: 10.1136/bcr-2019-230618
- Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997 Feb 27;146(1):67-72. doi: 10.1016/s0022-510x(96)00284-5
- Piepers S., Berg L.H., Brugman F., Scheffer H., Ruiterkamp-Versteeg M., Engelen B.G., Faber C.G., Visser M., Pol W.-L., Wokke J.H.J. A Natural History Study of Late Onset Spinal Muscular Atrophy Types 3b and 4. J. Neurol. 2008;255:1400–1404. doi: 10.1007/s00415-008-0929-0
- Shorrock H.K., Gillingwater T.H., Groen E.J.N. Overview of Current Drugs and Molecules in Development for Spinal Muscular Atrophy Therapy. Drugs. 2018;78:293–305. doi: 10.1007/s40265-018-0868-8
- Moosa A., Dubowitz V. Spinal Muscular Atrophy in Childhood: Two Clues to Clinical Diagnosis. Arch. Dis. Child. 1973;48:386–388. doi: 10.1136/adc.48.5.386
- Montes J, McDermott MP, Mirek E, et al. Ambulatory function in spinal muscular atrophy: Age-related patterns of progression. PLoS One. 2018 Jun 26;13(6):e0199657. doi: 10.1371/journal.pone.0199657
- Wadman RI, Stam M, Gijzen M, Lemmink HH, Snoeck IN, Wijngaarde CA, Braun KP, Schoenmakers MA, van den Berg LH, Dooijes D, van der Pol WL. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0-4. J Neurol Neurosurg Psychiatry. 2017 Apr;88(4):365-367. doi: 10.1136/jnnp-2016-314292
- Brahe C, Servidei S, Zappata S, Ricci E, Tonali P, Neri G. Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet. 1995 Sep 16;346(8977):741-2. doi: 10.1016/s0140-6736(95)91507-9
- Clermont O, Burlet P, Lefebvre S, Bürglen L, Munnich A, Melki J. SMN gene deletions in adult-onset spinal muscular atrophy. Lancet. 1995 Dec 23-30;346(8991-8992):1712-3. doi: 10.1016/s0140-6736(95)92881-2
- McShane MA, Boyd S, Harding B, et al. Progressive bulbar paralysis of childhood. A reappraisal of Fazio-Londe disease. Brain. 1992 Dec. 115 ( Pt 6):1889-900.
- Harding AE, Thomas PK. Hereditary distal spinal muscular atrophy. A report on 34 cases and a review of the literature. J Neurol Sci. 1980 Mar. 45(2-3):337-48.
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968 Jul. 18(7):671-80
- Kaeser HE. Scapuloperoneal muscular atrophy. Brain. 1965 Jun. 88(2):407-18.
- Kondo K, Tsubaki T, Sakamoto F. The Ryukyuan muscular atrophy. An obscure heritable neuromuscular disease found in the islands of southern Japan. J Neurol Sci. 1970 Oct. 11(4):359-82.
- Rudnik-Schöneborn S, Senderek J, Jen JC, Houge G, Seeman P, Puchmajerová A, et al. Pontocerebellar hypoplasia type 1: clinical spectrum and relevance of EXOSC3 mutations. Neurology. 2013 Jan 29. 80(5):438-46
- Kamoshita S, Takei Y, Miyao M, Yanagisawa M, Kobayashi S, Saito K. Pontocerebellar hypoplasia associated with infantile motor neuron disease (Norman’s disease). Pediatr Pathol. 1990. 10(1-2):133-42.
- Eckart M, Guenther UP, Idkowiak J, Varon R, Grolle B, Boffi P, et al. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics. 2012 Jan. 129(1):e148-56.
- Penttilä S, Jokela M, Hackman P, Maija Saukkonen A, Toivanen J, Udd B. Autosomal dominant late-onset spinal motor neuronopathy is linked to a new locus on chromosome 22q11.2-q13.2. Eur J Hum Genet. 2012 Nov. 20(11):1193-6.
- Rudnik-Schöneborn S, Arning L, Epplen JT, Zerres K. SETX gene mutation in a family diagnosed autosomal dominant proximal spinal muscular atrophy. Neuromuscul Disord. 2012 Mar. 22(3):258-62.
- Campbell L., Potter A., Ignatius J., Dubowitz V., Davies K. Genomic Variation and Gene Conversion in Spinal Muscular Atrophy: Implications for Disease Process and Clinical Phenotype. Am. J. Hum. Genet. 1997;61:40–50. doi: 10.1086/513886
- Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3
- Burghes A.H.M., Beattie C.E. Spinal Muscular Atrophy: Why Do Low Levels of Survival Motor Neuron Protein Make Motor Neurons Sick? Nat. Rev. Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670
- Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossol W., Prior T.W., et al. The Human Centromeric Survival Motor Neuron Gene (SMN2) Rescues Embryonic Lethality in Smn-/-Mice and Results in a Mouse with Spinal Muscular Atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333
- Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin. 2015 Nov;33(4):831-46. doi: 10.1016/j.ncl.2015.07.004
- Kelley RI, Sladky JT. Dicarboxylic aciduria in an infant with spinal muscular atrophy. Ann Neurol. 1986 Dec;20(6):734-6. doi: 10.1002/ana.410200615
- Bowerman M, Swoboda KJ, Michalski JP, Wang GS, Reeks C, Beauvais A, Murphy K, Woulfe J, Screaton RA, Scott FW, Kothary R. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol. 2012 Aug;72(2):256-68. doi: 10.1002/ana.23582
- Darras BT. Spinal muscular atrophies. Pediatr Clin North Am. 2015 Jun;62(3):743-66. doi: 10.1016/j.pcl.2015.03.010
- Han JJ, McDonald CM. Diagnosis and clinical management of spinal muscular atrophy. Phys Med Rehabil Clin N Am. 2008 Aug;19(3):661-80, xii. doi: 10.1016/j.pmr.2008.02.004
- Chien YH, Chiang SC, Weng WC, Lee NC, Lin CJ, Hsieh WS, Lee WT, Jong YJ, Ko TM, Hwu WL. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. J Pediatr. 2017 Nov;190:124-129.e1. doi: 10.1016/j.jpeds.2017.06.042
- Claborn MK, Stevens DL, Walker CK, Gildon BL. Nusinersen: A Treatment for Spinal Muscular Atrophy. Ann Pharmacother. 2019 Jan;53(1):61-69. doi: 10.1177/1060028018789956
- Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, Topaloglu H, Tulinius M, Montes J, Glanzman AM, Bishop K, Zhong ZJ, Gheuens S, Bennett CF, Schneider E, Farwell W, De Vivo DC; ENDEAR Study Group. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017 Nov 2;377(18):1723-1732. doi: 10.1056/NEJMoa1702752
- Malone DC, Dean R, Arjunji R, Jensen I, Cyr P, Miller B, Maru B, Sproule DM, Feltner DE, Dabbous O. Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J Mark Access Health Policy. 2019 May 8;7(1):1601484. doi: 10.1080/20016689.2019.1601484
- Ludolph AC, Wurster CD. Therapeutic advances in SMA. Curr Opin Neurol. 2019 Oct;32(5):777-781. doi: 10.1097/WCO.0000000000000738
- Mendell JR, Al-Zaidy S, Shell R, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med. 2017 Nov 2;377(18):1713-1722. doi: 10.1056/NEJMoa1706198
- Mahajan R. Onasemnogene Abeparvovec for Spinal Muscular Atrophy: The Costlier Drug Ever. Int J Appl Basic Med Res. 2019 Jul-Sep;9(3):127-128. doi: 10.4103/ijabmr.IJABMR_190_19
- Hoy SM. Onasemnogene Abeparvovec: First Global Approval. Drugs. 2019 Jul;79(11):1255-1262. doi: 10.1007/s40265-019-01162-5
- Schroth MK. Special considerations in the respiratory management of spinal muscular atrophy. Pediatrics. 2009 May;123 Suppl 4:S245-9. doi: 10.1542/peds.2008-2952K
- Durkin ET, Schroth MK, Helin M, Shaaban AF. Early laparoscopic fundoplication and gastrostomy in infants with spinal muscular atrophy type I. J Pediatr Surg. 2008 Nov;43(11):2031-7. doi: 10.1016/j.jpedsurg.2008.05.035
- Sproule DM, Montes J, Montgomery M, Battista V, Koenigsberger D, Shen W, Punyanitya M, De Vivo DC, Kaufmann P. Increased fat mass and high incidence of overweight despite low body mass index in patients with spinal muscular atrophy. Neuromuscul Disord. 2009 Jun;19(6):391-6. doi: 10.1016/j.nmd.2009.03.009
- Davis RH, Godshall BJ, Seffrood E, Marcus M, LaSalle BA, Wong B, Schroth MK, Swoboda KJ. Nutritional practices at a glance: spinal muscular atrophy type I nutrition survey findings. J Child Neurol. 2014 Nov;29(11):1467-72. doi: 10.1177/0883073813503988
- Haaker G, Fujak A. Proximal spinal muscular atrophy: current orthopedic perspective. Appl Clin Genet. 2013 Nov 14;6(11):113-20. doi: 10.2147/TACG.S53615
- Chen TH. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int J Mol Sci. 2020 May 7;21(9):3297. doi: 10.3390/ijms21093297
- About SMA:Frequently Asked Questions. https://smafoundation.org/about-sma/faq
- Zerres K, Rudnik-Schöneborn S. Natural History in Proximal Spinal Muscular Atrophy: Clinical Analysis of 445 Patients and Suggestions for a Modification of Existing Classifications. Arch Neurol. 1995;52(5):518–523. doi:10.1001/archneur.1995.00540290108025
- Ge X, Bai J, Lu Y, Qu Y, Song F. The natural history of infant spinal muscular atrophy in China: a study of 237 patients. J Child Neurol. 2012 Apr. 27(4):471-7.
- Farrar MA, Vucic S, Johnston HM, du Sart D, Kiernan MC. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr. 2013 Jan. 162(1):155-9.
- Birnkrant DJ, Pope JF, Martin JE, et al. Treatment of type I spinal muscular atrophy with noninvasive ventilation and gastrostomy feeding. Pediatr Neurol. 1998 May. 18(5):407-10.
- Lemoine TJ, Swoboda KJ, Bratton SL, Holubkov R, Mundorff M, Srivastava R. Spinal muscular atrophy type 1: are proactive respiratory interventions associated with longer survival?. Pediatr Crit Care Med. 2012 May. 13(3):e161-5.





{kind=link}