Stargardt disease
Stargardt disease also called Stargardt macular degeneration or juvenile macular dystrophy, is a genetic eye disorder and is the most common childhood inherited macular dystrophy that causes progressive vision loss 1. Stargardt macular degeneration affects the retina, the specialized light-sensitive tissue that lines the back of the eye. Specifically, Stargardt macular degeneration affects a small area near the center of the retina called the macula. The macula is responsible for sharp central vision, which is needed for detailed tasks such as reading, driving, and recognizing faces. In most people with Stargardt macular degeneration, a fatty yellow pigment (lipofuscin) builds up in cells underlying the macula. Over time, the abnormal accumulation of this substance can damage cells that are critical for clear central vision. In addition to central vision loss, people with Stargardt macular degeneration have problems with night vision that can make it difficult to navigate in low light. Some affected individuals also have impaired color vision. The signs and symptoms of Stargardt macular degeneration typically appear in late childhood to early adulthood when kids notice difficulties in reading or adapting to bright light that worsens over time.
Stargardt disease has a genetic basis due to mutations in the ABCA4 gene, and results from the accumulation of visual cycle kinetics-derived byproducts in the retinal pigmented epithelium (RPE) with secondary photoreceptor dysfunction and death. A rarer disease called autosomal dominant Stargardt-like macular dystrophy, similar to Stargardt disease, is caused by the gene ELOVL4.
Stargardt macular degeneration is the most common form of juvenile macular degeneration, the signs and symptoms of which begin in childhood. The estimated prevalence of Stargardt macular degeneration is 1 in 8,000 to 10,000 individuals 2.
Stargardt disease, like other forms of macular degeneration, does not have a cure yet. It is sometimes treated with intraocular injections of anti-VEGF drugs, similar to “wet” Age-related Macular Degeneration treatments, if there is the proliferation or leakage of blood vessels. Nutrition and eye protection (especially sunglasses which block UV A and B and blue light) may delay the progression of this disease. When vision begins to change – whether that is in the teen years or the twenties – vision aids become essential, maximizing the use of peripheral vision.
Note that excessive amounts of Vitamin A – which is usually considered a good nutrient for the eyes – can actually be toxic to the eyes in people with Stargardt disease because the vitamin is not metabolized by cells in the eye. Check with a health practitioner about nutrition.
Find a doctor who sees patients with Inherited Retinal Diseases here: https://www.fightingblindness.org/retinal-specialists
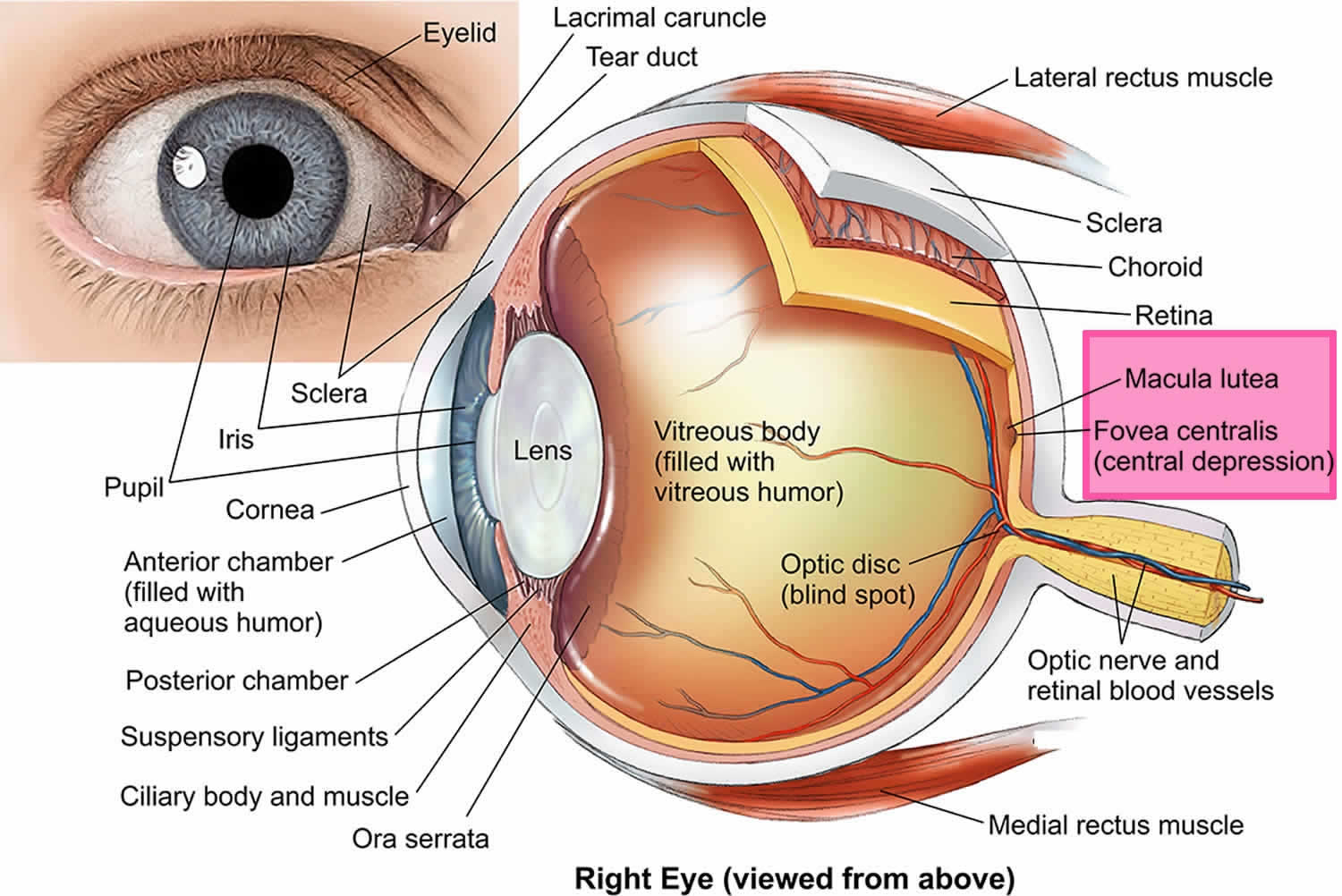
What is the macula?
The macula is the small central area of your retina where the light entering your eye is focused. The retina is made up of cells called photoreceptors which are cells that are sensitive to light.
The macula is a specialized area that contains a high concentration of photoreceptor cells called cone cells. Cone cells work best in bright light and allow you to see fine detail for activities such as reading and watching television, as well as seeing color. Therefore, the macula is very important and is responsible for:
- what you see straight in front of you
- the vision you need for detailed activities such as reading and writing, and
- your ability to appreciate color.
Away from the central macula is the peripheral retina, made up of mostly the other type of photoreceptor called rod cells. Rod cells enable you to see in dim conditions and provide peripheral (side) vision outside of the main line of sight.
Figure 1. Human eye anatomy
How does Stargardt disease affect my sight?
Stargardt disease is sometimes called a juvenile macular dystrophy as it can first appear in childhood. However Startgardt disease can also begin later in adulthood.
At first Stargardt disease will make your vision unclear or blurry. Things may sometimes appear distorted or wavy. You can have problems with your central, detailed vision which can make activities such as reading and recognizing faces difficult. Your colour perception may also be affected. If you’ve had Stargardt disease for a number of years then you may have a blank patch in the centre of your vision. This blank patch will not move and will always be in the very center of your field of vision.
Stargardt disease doesn’t usually affect other parts of your retina so your peripheral or side vision is not normally affected. Since you use your peripheral vision when you’re moving around, most people with Stargardt disease can manage to continue getting out and about on their own.
Stargardt disease can also cause problems with light, such as glare and difficulties adapting to changing light conditions.
How does Stargardt disease affect your eye?
Stargardt disease causes changes to the appearance of the macula area of your retina. When the ophthalmologist (hospital eye doctor) looks into your eye to examine your retina they may notice differences which can help them to diagnose the condition:
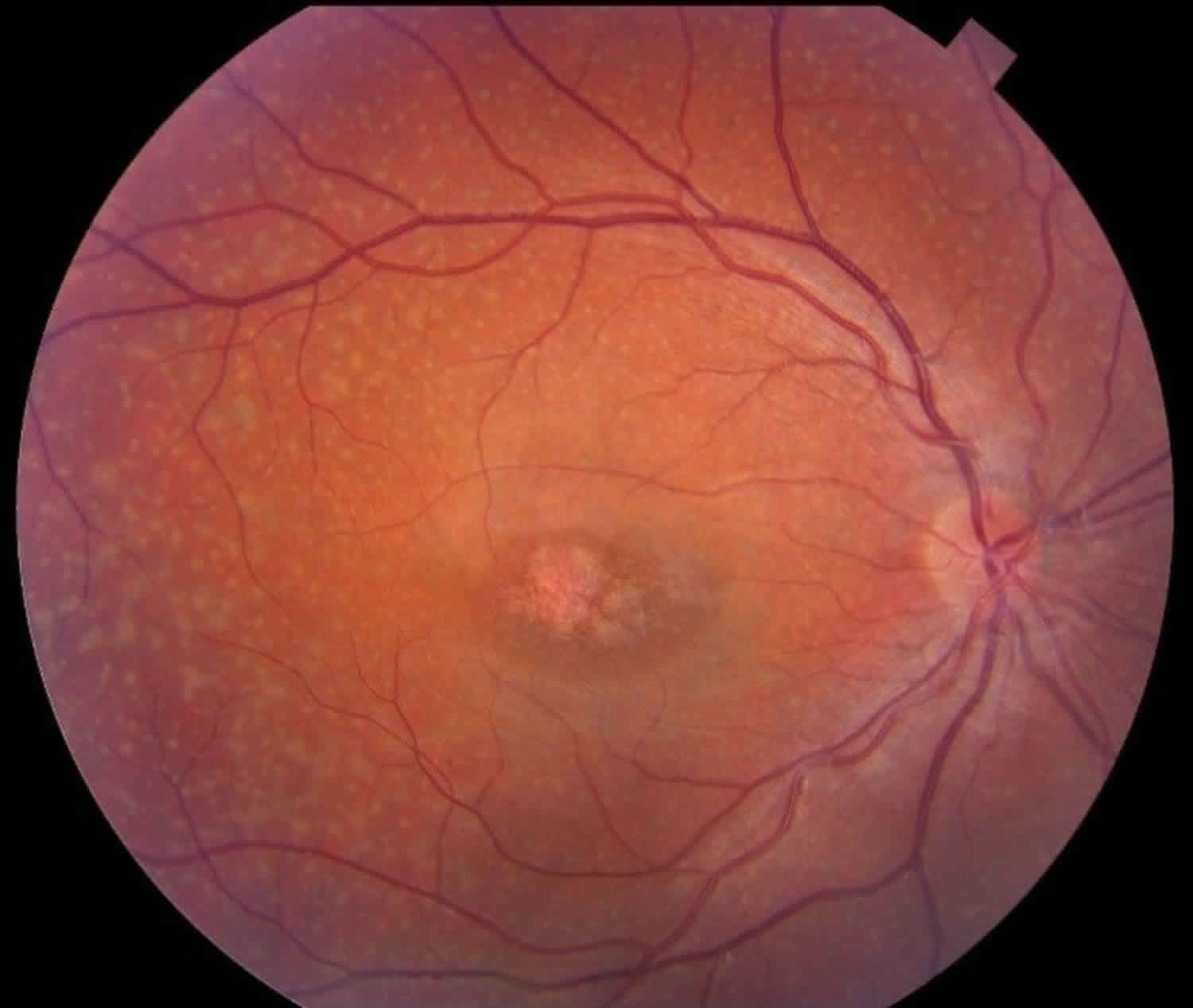
- Yellowish flecks which surround your macula are very characteristic of Stargardt disease. These yellow flecks are lipofuscin which is a by-product of cell activity.
- As the disease progresses, an oval lesion can be seen which is often referred to as “beaten bronze” in appearance within your macular area.
Sometimes people have just the flecks without the macular lesion, and in the past, these people may have been diagnosed with an eye condition called fundus flavimaculatus. However, researchers now believe that these two problems, the macular lesion and the yellow flecks, are part of the same genetic problem, but are just expressed in different ways.
It would seem that some people with fundus flavimaculatus can develop more severe sight problems than people with more classic forms of Stargardt disease.
Should the child of a person with Stargardt disease be evaluated?
While vision care and eye exams should be a part of every child’s routine medical care, meeting with a specialist (such as an ophthalmologist or optometrist) is recommended for children with a family history of a genetic eye disease 3. A specialist can advise parents regarding if and when a child should be evaluated (and how often); and can recommend specific types of eye exams that may be beneficial or appropriate for children with a family history of Stargardt disease.
Stargardt disease is most commonly inherited in an autosomal recessive manner. Therefore in most cases, the child of a person with autosomal recessive Stargardt disease will be an unaffected carrier, and generally only at risk to develop the condition if their other parent is a carrier (or is affected). If an affected parent has autosomal dominant Stargardt disease (which is rare), each of his/her children has a 50% chance to inherit the mutated gene responsible for the condition.
Genetic testing for unsymptomatic minors (children who currently don’t have symptoms) is usually not recommended for Stargardt disease and other genetic conditions that are not treatable 4.
Is genetic testing available for Stargardt disease?
Yes. Genetic testing may help distinguish the type of Stargardt disease a person has and provide information about the mode of inheritance and risks to other family members. The Genetic Testing Registry provides information about the genetic tests available for Stargardt disease. The intended audience for the Genetic Testing Registry is health care providers and researchers. Patients and consumers with specific questions about genetic testing for this condition should speak with their ophthalmologist or a genetics professional.
Find a doctor who sees patients with Inherited Retinal Diseases here: https://www.fightingblindness.org/retinal-specialists
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Is Stargardt disease cureable?
Stargardt disease remains an incurable condition. Current therapeutic options include photoprotection and low-vision aids. Pharmacological slow-down of the visual cycle and gene therapy represent prospects of long-term visual rescue.
Are there any dietary supplements or natural substances that may slow Stargardt disease progressive vision loss?
At present there is no cure for Stargardt disease, and there is very little that can be done to slow its progression. Wearing sunglasses to protect the eyes from UVa, UVb and bright light may be of some benefit. Animal studies have shown that taking excessive amounts of vitamin A and beta carotene could promote the additional accumulation of lipofuscin, as well as a toxic vitamin A derivative called A2E; it is typically recommended that these be avoided by individuals with Stargardt disease. There are possible treatments for Stargardt disease that are being tested, including a gene therapy treatment, which has been given orphan drug status by the European Medicines Agency (EMEA, similar to the FDA). You can read more about this treatment by clicking here (https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/08/609-public-summary-positive-opinion-orphan-designation-adeno-associated-viral-vector-serotype-5_en.pdf). There are also clinical trials involving embryonic stem cell treatments 5.
What is the latest research on Stargardt disease?
Over the past several decades, researchers have identified hundreds of genes that contribute to inherited eye diseases, including Stargardt disease. This information has led to better diagnostic tests, and is providing insight into possible treatments.
Scientists are also working to find new mutations in the ABCA4 gene, and in other genes, that might contribute to Stargardt disease. National Eye Institute’s National Ophthalmic Disease Genotyping and Phenotyping Network (https://eyegene.nih.gov/) is an important resource for these efforts. The eyeGENE network gives patients access to genetic studies and clinical trials, while giving researchers access to patient DNA samples and clinical information.
Many studies continue to explore the biology and genetics of Stargardt disease, and of macular degeneration more generally. For example, National Eye Institute is conducting a natural history study of Stargardt and other ABCA4-related diseases. The study is following 45 individuals with ABCA4 mutations for five years. The main goals are to better understand the natural course of the disease, to make contact with people who may be interested in future clinical trials, and to collect blood, skin, and DNA samples from those people. These samples can be studied in the lab to explore the mechanisms of Stargardt disease.
Such mechanistic studies can lead to new treatment strategies. One strategy currently under study is to reduce the build-up of lipofuscin and other toxic byproducts in the retina. An NEI-funded group based at Columbia University is working with a synthetic form of vitamin A, called ALK-001, that isn’t readily converted into lipofuscin. In mice and larger animal models, an oral form of ALK-001 slows the formation of lipofuscin deposits. ALK-001 is being tested for safety in healthy volunteers before testing begins on people with Stargardt disease.
A group based at Case Western Reserve University and supported by the NEI Translational Research Program on Therapy for Visual Disorders is taking another approach. The group has tested a panel of drugs already deemed safe by the Food and Drug Administration (FDA) in a mouse model of Stargardt. They found that some FDA-approved drugs can reduce retinal damage in the mice. They are now evaluating chemical compounds with a similar structure to FDA-approved drugs, and exploring different modes of drug delivery.
Gene therapy — that is, repairing or replacing the defective ABCA4 gene — also holds promise for treating Stargardt disease. Gene replacement therapy requires a method for delivering the gene of interest into cells, and for some diseases, the solution has been to package the gene inside a small, harmless virus called the adeno-associated virus (AAV). The ABCA4 gene is too large to fit within this virus. Researchers funded by NEI therefore modified a larger virus from the lentivirus family, and engineered it to carry ABCA4. Tests in the Stargardt mouse model showed that this approach can reduce lipofuscin accumulation. Oxford Biomedica has refined this technology, and licensed it under the name StarGen. Human safety trials of StarGen began in 2011.
Finally, stem cell-based therapies are showing promise for Stargardt disease in clinical trials. Stem cells are immature cells that can generate many mature cells types in the body, including the photoreceptors that die off in Stargardt disease. Human stem cells can be derived from embryonic or adult tissues. Both kinds of cells are being tested in patients with Stargardt disease and age-related macular degeneration (AMD), which is a leading cause of vision loss in the United States.
A U.S. company called Advanced Cell Technology (ACT) is conducting a trial of retinal pigment epithelium (RPE) cells for AMD and Stargardt disease. These cells provide support and nourishment to the retina. The RPE cells under study in the ACT trial are derived from human embryonic stem cells.
Other clinical and laboratory studies are making use of adult cells that have been reprogrammed into stem cells, called induced pluripotent — or iPS — cells. In 2014, Japanese scientists launched the first clinical trial of iPS cells to treat AMD, and indeed the first trial ever of iPS cells. The goal is to coax the iPS cells into making RPE cells. NEI scientists are planning a similar trial and going a step further — by seeding the RPE cells onto a scaffold so that they form a sheet, similar to how they arrange themselves in the eye. NEI scientists also plan to make iPS cells from the skin cells collected in the Stargardt natural history study. These iPS cells could be used to make photoreceptors and RPE cells — for use in potential cell therapies and as a research tool to study how potential drugs will affect these cell types.
Stargardt disease cause
In most cases, Stargardt macular degeneration is caused by mutations in the ABCA4 gene. Less often, mutations in the ELOVL4 gene cause this condition. The ABCA4 and ELOVL4 genes provide instructions for making proteins that are found in light-sensing (photoreceptor) cells in the retina.
The ABCA4 protein transports potentially toxic substances out of photoreceptor cells. These substances form after phototransduction, the process by which light entering the eye is converted into electrical signals that are transmitted to the brain. Mutations in the ABCA4 gene prevent the ABCA4 protein from removing toxic byproducts from photoreceptor cells. These toxic substances build up and form lipofuscin in the photoreceptor cells and the surrounding cells of the retina, eventually causing cell death. Loss of cells in the retina causes the progressive vision loss characteristic of Stargardt macular degeneration.
The ELOVL4 protein plays a role in making a group of fats called very long-chain fatty acids. The ELOVL4 protein is primarily active (expressed) in the retina, but is also expressed in the brain and skin. The function of very long-chain fatty acids within the retina is unknown. Mutations in the ELOVL4 gene lead to the formation of ELOVL4 protein clumps (aggregates) that build up and may interfere with retinal cell functions, ultimately leading to cell death.
The retina contains light-sensing cells called photoreceptors. There are two types of photoreceptors: rods and cones. Together, rod and cones detect light and convert it into electrical signals, which are then “seen” by the brain. Rods are found in the outer retina and help us see in dim and dark lighting. Cones are found in the macula and help us see fine visual detail and color. Both cones and rods die away in Stargardt disease, but for unclear reasons, cones are more strongly affected in most cases.
Stargardt disease inheritance pattern
Stargardt macular degeneration can have different inheritance patterns.
When mutations in the ABCA4 gene cause Stargardt disease, it is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations (see Figure 2). The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Autosomal recessive mutations in the ABCA4 gene account for about 95 percent of Stargardt disease. The other five percent of cases are caused by rarer mutations in different genes that play a role in lipofuscin function. Some of these mutations are autosomal dominant.
In autosomal recessive inheritance, it takes two copies of the mutant gene to give rise to the disease. An individual who has one copy of a recessive gene mutation is known as a carrier. When two carriers have a child, there is a:
- 1 in 4 chance of having a child with the disease
- 1 in 2 chance of having a child who is a carrier
- 1 in 4 chance of having a child who neither has the disease nor is a carrier
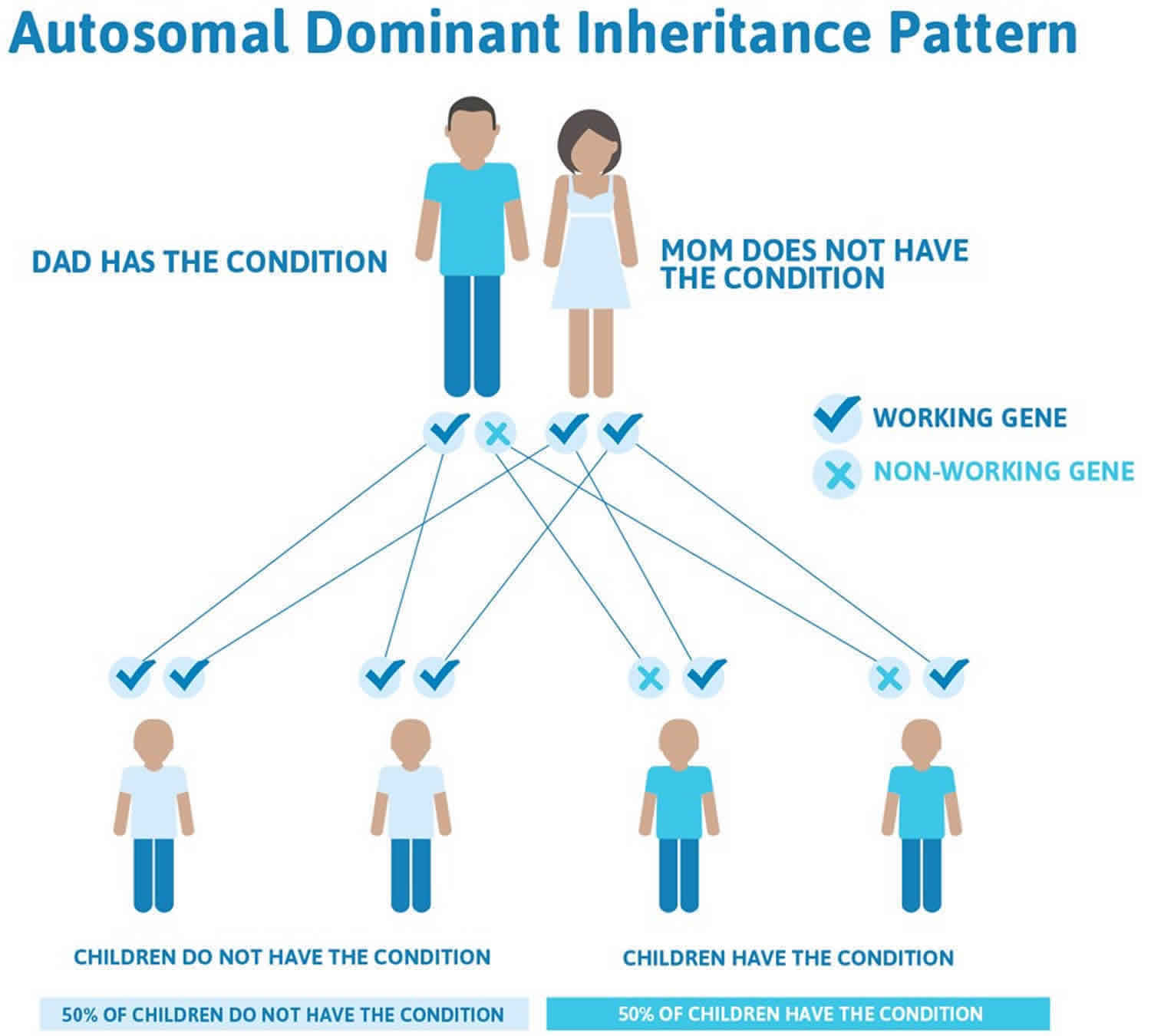
When Stargardt disease is caused by mutations in the ELOVL4 gene, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder (see Figure 3).
Figure 2. Stargardt disease autosomal recessive inheritance pattern
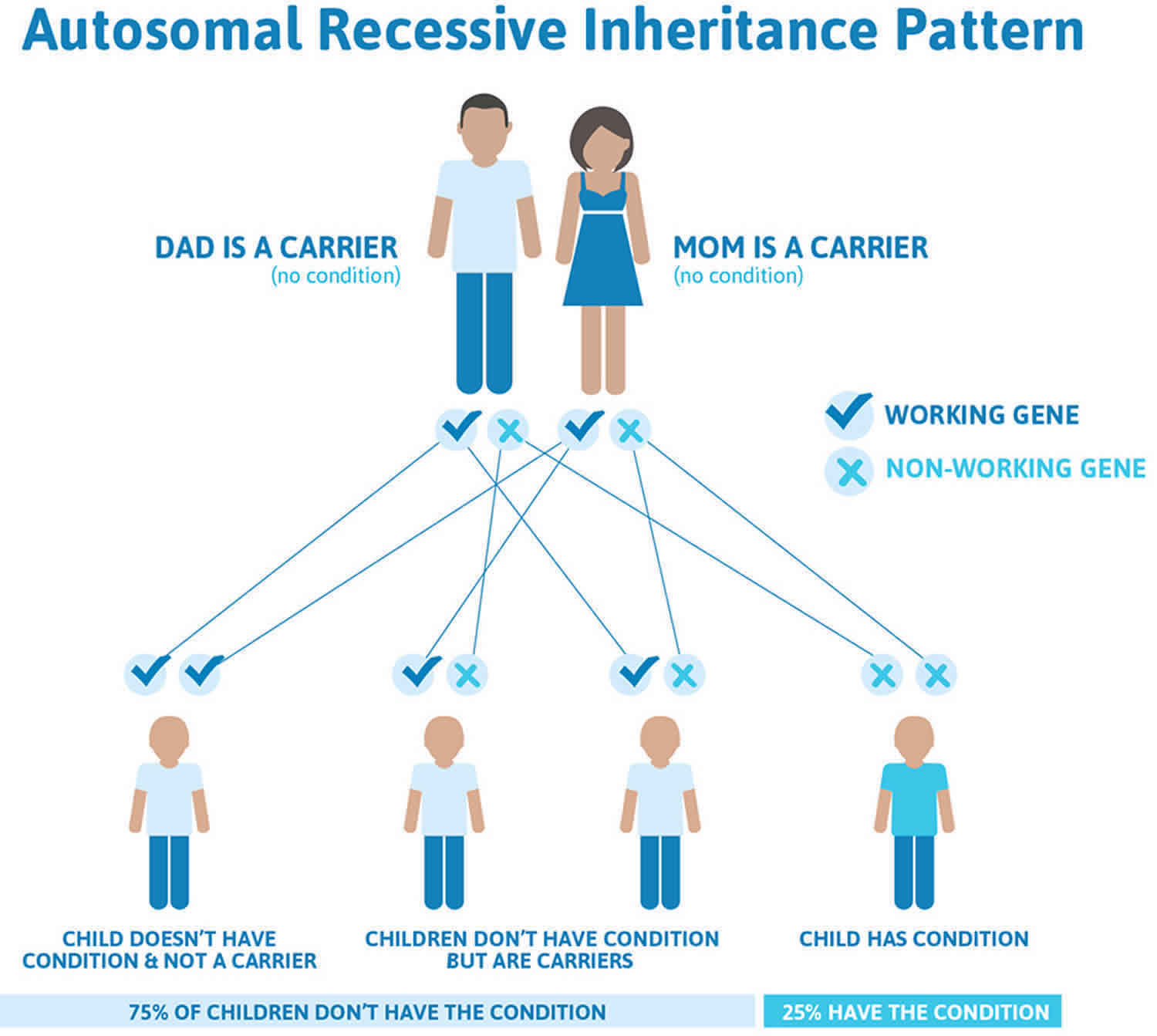
Figure 3. Stargardt disease autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Stargardt disease symptoms
The most common symptom of Stargardt disease is variable, often slow loss of central vision in both eyes. Visual acuity (the ability to distinguish details and shape) may decrease slowly at first, accelerate, and then level off. Some peripheral vision is usually maintained. People with Stargardt disease might notice gray, black, or hazy spots in the center of their vision, or that it takes longer than usual for their eyes to adjust when moving from light to dark environments. Their eyes may be more sensitive to bright light. Some people also develop color blindness later in the disease.
The progression of symptoms in Stargardt disease is different for each person. People with an earlier onset of disease tend to have more rapid vision loss. Vision loss may decrease slowly at first, then worsen rapidly until it levels off. Most people with Stargardt disease will end up with 20/200 vision or worse. People with Stargardt disease may also begin to lose some of their peripheral (side) vision as they get older.
A retinal doctor examining the retinas of a person with Stargardt disease will see characteristic yellowish flecks in the retinal pigment epithelium (RPE). The flecks are deposits of lipofuscin, a byproduct of normal retinal cell activity. However, in Stargardt disease, lipofuscin accumulates abnormally.
Stargardt disease diagnosis
An eye care professional can make a positive diagnosis of Stargardt disease by examining the retina. Lipofuscin deposits can be seen as yellowish flecks in the macula. The flecks are irregular in shape and usually extend outward from the macula in a ring-like pattern. The number, size, color, and appearance of these flecks are widely variable.
A standard eye chart and other tests may be used to assess symptoms of vision loss in Stargardt disease, including:
- Visual field testing. Visual fields testing attempts to measure distribution and sensitivity of field of vision. Multiple methods are available for testing; none is painful and most share a requirement for the patient to indicate ability to see a stimulus / target. This process results in a map of the person’s visual field, and can point to a loss of central vision or peripheral vision.
- Color Testing: There are several tests that can be used to detect loss of color vision, which can occur late in Stargardt disease. Three tests are often used to get additional information: fundus photography combined with autofluorescence, electroretinography, and optical coherence tomography.
- A fundus photo is a picture of the retina. These photos may reveal the presence of lipofuscin deposits. In fundus autofluorescence (FAF), a special filter is used to detect lipofuscin. Lipofuscin is naturally fluorescent (it glows in the dark) when a specific wavelength of light is shined into the eye. This test can detect lipofuscin that might not be visible with standard fundus photography, making it possible to diagnose Stargardt disease earlier.
- Electroretinography (ERG) measures the electrical response of rods and cones to light. During the test, an electrode is placed on the cornea and light is flashed into the eye. The electrical responses are viewed and recorded on a monitor. Abnormal patterns of light response suggest the presence of Stargardt disease or other diseases that involve retinal degeneration.
- Optical coherence tomography (OCT) is a scanning device that works a little like ultrasound. While ultrasound captures images by bouncing sound waves off of living tissues, OCT does it with light waves. The patient places his or her head on a chin rest while invisible, near-infrared light is focused on the retina. Because the eye is designed to allow light in, it’s possible to get detailed pictures deep within the retina. These pictures are then analyzed for any abnormalities in the thickness of the retinal layers, which could indicate retinal degeneration. OCT is sometimes combined with infrared scanning laser ophthalmoscope (ISLO) to provide additional surface images of the retina.
Stargardt disease treatment
Currently, there is no treatment for Stargardt disease. Research and developments into gene therapy and stem cells is very active and it’s hoped that this may lead to treatments becoming available at some stage in the future.
- Gene therapy aims to replace the faulty gene such as ABCA4 within the affected retinal cells with a new gene that works properly. The “normal” gene is injected into the eye of the person with Stargardt disease. The hope is that the affected cells then begin to work correctly thereby stopping the progression of the disease.
- Stem cells are cells that can divide many times and can replace damaged or missing cells in different organs and tissues of the body. If stem cells can be turned into the specialised retinal cells, it may be possible to replace the cells that have been damaged in Stargardt disease.
Another area of research is looking at medications that can reduce the amount of vitamin A by-product that builds up in the eyes of those with Stargardt disease. If you have Stargardt disease you aren’t able to clear vitamin A by-product in your eye due to the faulty gene. These by-products collect in the macula and this affects how well the cells work, leading to your sight being affected.
Researchers have reported that exposure to ultraviolet (UV) light may theoretically cause further retinal damage. It has been demonstrated that di-retinoid-pyridinium-ethanolamine (A2E) does not accumulate in the retinal pigmented epithelium of ABCA4 knockout mice kept in total darkness. Thus, Stargardt patients should be advised to avoid direct sunlight exposure. Ultraviolet-blocking sunglasses are a useful option. Therefore protecting your eyes from UV and blue light with sunglasses that have 100 per cent UV filtering is important. Wraparound styles provide protection from light coming in from the sides and tops.
Some ophthalmologists encourage people with Stargardt disease to wear dark glasses and hats when out in bright light to reduce the buildup of lipofuscin. Cigarette smoking and second hand smoke should be avoided. Animal studies suggest that high-dose vitamin A may increase lipofuscin accumulation and potentially accelerate vision loss. Therefore, supplements containing more than the recommended daily allowance of vitamin A should be avoided, or taken only under a doctor’s supervision. There is no need to worry about getting too much vitamin A through food.
A number of services and devices can help people with Stargardt disease carry out daily activities and maintain their independence. Low-vision aids can be helpful for many daily tasks and range from simple hand-held lenses to electronic devices such as electronic reading machines or closed circuit video magnification systems. Because many people with Stargardt disease will become visually disabled by their 20s, the disease can have a significant emotional impact. Work, socializing, driving and other activities that may have come easily in the past are likely to become challenging. So counseling and occupational therapies often need to be part of the treatment plan.
Isotretinoin has reportedly been capable of dampening A2E deposition in the retinal pigmented epithelium of ABCA4 knockout mice. Considerable side effects associated with chronic intake of isotretinoin prevent its chronic use in humans.
Stargardt disease is a preferred target for gene replacement therapy. Like in Leber Congenital Amaurosis (LCA), from which the most astonishing results of human ocular gene therapy have been obtained, the replacement of the mutant ABCA4 gene by its wild-type counterpart may produce positive results, as those observed in RPE65 gene trials.
Animal testing using gene therapy for Stargardt disease remains limited. Kong and co-workers were able to successfully treat the Stargardt phenotype in abca4 knockout mice using lentiviral gene therapy. Each mouse received a single unilateral subretinal injection of ABCA4-carrying equine infectious anemia viral vectors, which resulted in significant rescue of the retinal phenotype. Treated eyes showed marked reduction in retinal A2E accumulation. Moreover, 1 year after gene transfer, A2E accumulation in treated eyes matched the A2E levels of normal wild-type controls.
The relatively large ABCA4 gene (6.8 kb) presents a unique packaging challenge with the available viral vectors. Lentiviral vectors are the most suitable for ABCA4 gene transfer. Other packaging options include AAV2/5 chimeras and new vector systems, such as the Hd-Ad vectors or other non-viral vectors.
Given Stargardt is a retinal degeneration, identification of viable photoreceptors plays a central role in the selection of patients amenable to gene therapy. Thus, retinal imaging is essential to assess photoreceptor viability, when selecting patients for gene therapy. High-definition OCT and Adaptive Optics technology enhances lateral resolution in retinal images up to 3-4μm, allowing the visualization of individual photoreceptors.
Years of gene therapy research for Leber Congenital Amaurosis and underlying RPE65 mutations, have produced incomparable breakthroughs, which will invariably serve as a foundation for further research involving other retinal dystrophies. In December 2009, Oxford BiomedicaTM announced that StarGenTM, a gene-based therapy that uses the company’s LentiVector® technology for the treatment of Stargardt disease, has received orphan designation from the Committee for Orphan Medicinal Products of the European Medicines Agency (EMEA). In collaboration with Sanofi-Aventis, both companies advanced StarGenTM into PhaseI/II development in 2010. The US charity, Foundation Fighting Blindness, is also supporting the programme and previously funded preclinical development. With this initiative, Oxford BiomedicaTM is deemed to bring considerable hope for the 600 new cases of Stargardt disease diagnosed every year and for many other Stargardt patients that currently await treatment for their visually-debilitating condition.
References- Stargardt disease/Fundus flavimaculatus. https://eyewiki.org/Stargardt_disease/Fundus_flavimaculatus
- Stargardt macular degeneration. https://ghr.nlm.nih.gov/condition/stargardt-macular-degeneration
- Your Child’s Vision. https://kidshealth.org/en/parents/vision.html
- Recommendations for Genetic Testing of Inherited Eye Diseases – 2014. https://www.aao.org/clinical-statement/recommendations-genetic-testing-of-inherited-eye-d
- Stargardt Disease Defined. https://www.macular.org/stargardt-disease





{kind=link}