Toxic encephalopathy
Toxic encephalopathy is a term generally used to indicate brain dysfunction caused by exposure to exogenous substances such as toxic chemicals, solvents, illicit drugs, toxins, poisons, radiation, paints, industrial chemicals, certain metals and medications 1. Toxic encephalopathy includes a spectrum of symptomatology ranging from subclinical deficits to overt clinical disorders. The clinical manifestations of toxic encephalopathy are related to the affected brain regions and cell types 1. Toxic encephalopathy is clinically characterized by changes in cognitive function, level of consciousness, and vigilance. In addition dementia, seizures, headache, hydrocephalus, cerebellar syndromes, tremor, and disturbed visual, auditory, vestibular, or olfactory functions may be encountered.
Metabolic encephalopathy is intended to describe encephalopathy due to such things as fever, dehydration, electrolyte imbalance, acidosis, infection and organ failure. Toxic-metabolic encephalopathy is sometimes used to describe the combination of toxic and metabolic effects 2. Septic encephalopathy is a term that describes brain dysfunction as a manifestation of severe sepsis. Uremic and hepatic encephalopathies are well-known clinical manifestations of renal and hepatic failure.
Toxic encephalopathy is morphologically characterized by cerebral edema (cytotoxic edema: membrane damage (heavy metals), damage of the membrane enzyme system (heavy metals, cyanide), vascular edema [alcohol]), neuronal and axonal injury (impaired axonal transport [aluminum]), neuronal injury (disturbed energy metabolism [carbon monoxide, ethanol]), injury of white matter (myelin damage [therapeutic agents, drugs, environmental toxins]), focal necrosis (carbon monoxide, hyperbaric oxygen, methotrexate), impaired cholinergic transmission (botulinum, tetanus toxins, alcohol, aluminum), impaired noradrenergic transmission (alcohol, amphetamines, cocaine), impaired receptor function (gamma-aminobutyric acid [GABA] and acetylcholine), teratogenic effects of the fetal nervous system (ethanol), carcinogenic effects (cadmium). Pathogenetic mechanisms include impairment of: (1) oxidative metabolism; (2) protein synthesis; (3) cytoskeletal structure, as well as injury of capillaries and astroglial and microglial reactions.
Clinically toxic encephalopathy presents with one of more of the following neurologic or psychiatric symptoms:
- Decreased concentration and consciousness;
- Excitability and convulsions;
- Motor and sensory disturbances;
- Extrapyramidal movement disorders;
- Disturbance of specific senses;
- Disturbance of coordination; and
- Behavioral and psychological changes (Bonhoeffer types).
Most of the intoxications of the central nervous system (CNS) are acute and require immediate treatment.
Chemicals capable of damaging the central nervous system (CNS) are ubiquitous in the environment, particularly in occupational settings. Industrial processes are major sources of some of the most well-known neurotoxins. According to the United States Environmental Protection Agency, more than 65,000 commercial chemicals are currently used in the US, and 2,000-3,000 new chemicals are added to this list each year 3. People may be exposed to these neurotoxins due to their occupations, or occasionally at home or through other inadvertent mechanisms.
The major clinical syndromes of toxic encephalopathy include diffuse acute or chronic toxic encephalopathy, cerebellar syndrome, parkinsonism, and vascular encephalopathy 1. Various neurotoxins, including heavy metals, organic solvents and other chemicals, have been found to be responsible for these relatively specific neurological syndromes 4.
Well known examples of toxic encephalopathy include 5:
- Lathyrus sativus peas, spastic paraparesis; the toxic agent was identified to be beta-N-methylamino-L-alanine (BMAA);
- Guamalian type of Parkinsonism, caused by the seeds of Cycas circinalis; the toxic agent was identified as beta-N–oxalylmethylamino-L-alanine (BOMAA);
- Hexachloraphene encephalopathy;
- Monosodium glutamate in baby food;
- Minamata disease, mercury encephalopathy;
- Housepainters dementia, organic solvents;
- Methylphenyltetrahydropyridine (MPTP), “synthetic heroin,” causing striatal dopamine deficiency and Parkinsonism.
What is encephalopathy?
The National Institute of Neurological Disorders and Stroke has described encephalopathy as a term for “any diffuse disease of the brain that alters brain function or structure” and says the “hallmark of encephalopathy is an altered mental status” 6. Encephalopathy may be caused by infectious agent (bacteria, virus, or prion), metabolic or mitochondrial dysfunction, brain tumor or increased pressure in the skull, prolonged exposure to toxic elements (including solvents, drugs, radiation, paints, industrial chemicals, and certain metals), chronic progressive trauma, poor nutrition, or lack of oxygen or blood flow to the brain. The hallmark of encephalopathy is an altered mental state. Depending on the type and severity of encephalopathy, common neurological symptoms are progressive loss of memory and cognitive ability, subtle personality changes, inability to concentrate, lethargy, and progressive loss of consciousness. Other neurological symptoms may include myoclonus (involuntary twitching of a muscle or group of muscles), nystagmus (rapid, involuntary eye movement), tremor, muscle atrophy and weakness, dementia, seizures, and loss of ability to swallow or speak. Blood tests, spinal fluid examination, imaging studies, electroencephalograms, and similar diagnostic studies may be used to differentiate the various causes of encephalopathy.
There are 2 distinct categories of encephalopathy: acute and chronic encephalopathy. Many sources confuse and confound these categories, lumping them together as one 7.
- Chronic encephalopathies are characterized by chronic mental status alteration that, in most cases, is slowly progressive (anoxic encephalopathy being an exception). They result from permanent, usually irreversible, structural changes within the brain itself. Some may be halted or reversed by early detection and treatment. Examples of the chronic encephalopathies include anoxic brain injury; chronic traumatic encephalopathy; heavy metals (lead, arsenic, mercury, etc); HIV-related; hereditary enzyme deficiencies; Korsakoff; and spongiform 7.
- Acute encephalopathy is characterized by an acute or subacute global, functional alteration of mental status due to systemic factors. It is reversible when these abnormalities are corrected, with a return to baseline mental status. Acute encephalopathy may be further identified as toxic, metabolic, or toxic-metabolic. Toxic encephalopathy describes acute mental status alteration due to medications, illicit drugs, or toxic chemicals. Metabolic encephalopathy is caused by any of a large number of metabolic disturbances. Toxic-metabolic encephalopathy describes a combination of toxic and metabolic factors. Causes of acute toxic and metabolic encephalopathy include acute organ failure such as hepatic and renal; alcohol; dehydration; electrolyte imbalance; fever; hypertension; hypoxemia; illicit drugs; infections including sepsis; medications; toxic chemicals; and Wernicke (thiamine deficiency).
Acute intra-cranial processes (such as stroke or traumatic lesions) alone should not be classified as acute encephalopathy but are more correctly considered an alteration of consciousness (stupor or coma) or concussion 7.
In contrast to the generic term “encephalopathy,” the acute toxic and metabolic encephalopathies as a group are well defined and well described. The 2013 Neurocritical Care Society Practice Update states that “acute encephalopathy is synonymous with acute confusional state, acute organic brain syndrome or delirium…[it] describes the clinical presentation of a global cerebral dysfunction induced by systemic factors.”
Delirium vs. acute encephalopathy
Delirium and acute encephalopathy are essentially 2 different terms describing the same condition 7. Delirium represents the mental manifestation while encephalopathy identifies the underlying pathophysiologic process. This is why the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), classifies acute toxic and metabolic encephalopathic states as delirium and does not use encephalopathy in its definitions.
The coding classifications (ICD-9 and ICD-10) use “encephalopathy” to classify what DSM-5 calls delirium. ICD relegates delirium to a symptom of lesser importance. To permit correct coding for these cases, the term encephalopathy is needed to capture a true picture of the patient’s condition. Clinicians may continue to follow DSM definitions using delirium but should also incorporate the necessary ICD terminology to prevent understating the severity of illness of patients. Examples include:
- Toxic encephalopathy due to phenytoin, causing delirium.
- Delirium due to metabolic encephalopathy.
Acute diffuse toxic encephalopathy
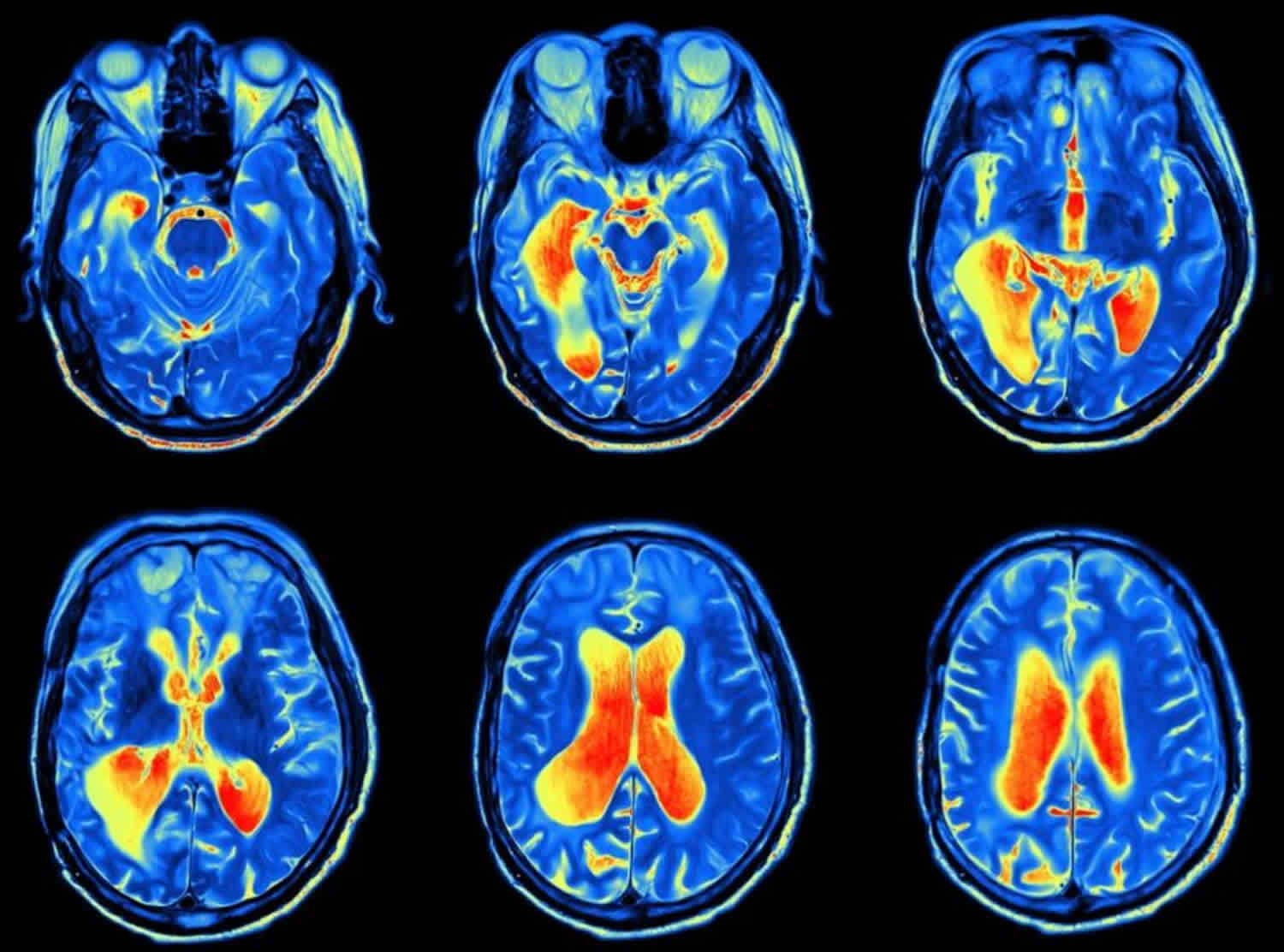
Acute diffuse toxic encephalopathy reflects a global cerebral dysfunction of rapid onset (typically days or weeks), and may be associated with alterations in the level of consciousness. The neurotoxins that produce acute encephalopathy interfere with basic cell functions in the brain 1. Most of these agents gain entry because they are highly lipid soluble and can readily diffuse across membranes. The causative agents include organic solvents, which can alter cellular membrane function, and some gases (e.g., gas anesthetics, carbon monoxide, hydrogen sulfide, and cyanide), which can diffusely affect brain function. Heavy metals can also cause acute encephalopathies; this is more commonly associated with organic metals (e.g., methyl mercury, tetraethyl lead and organic tin) than with inorganic metals (e.g., mercury, lead and tin) 1. Virtually any organic solvent has the potential to produce acute diffuse toxic encephalopathy, the clinical manifestations of which depend on the neurotoxin and the intensity of exposure, and can range from mild euphoria with a normal examination, to stupor, seizure, coma, and even death. In general, the greater the exposure, the more severe the impairment of cerebral function and consciousness. The cerebral cortex is more sensitive to these toxins than is the brainstem: even when consciousness is lost, brainstem function typically remains intact. Diagnosis does not generally present a challenge for acute syndromes, because the exposure and clinical manifestations are likely to be closely linked in time. In patients with severe acute toxic encephalopathy, magnetic resonance imaging (MRI) of the brain may show focal areas, most commonly bilateral basal ganglia, or diffuse areas of edema 8. The treatment of diffuse acute encephalopathy is primarily supportive, starting with removal of the exposure source. For most of the neurotoxins that act diffusely on the brain, recovery from acute exposure is complete 1.
Chronic toxic encephalopathy
Chronic toxic encephalopathy usually represents a chronic persistent diffuse injury to the brain resulting from cumulative or repeated exposures (often over a period of months or years), to solvents or (occasionally) heavy metals. The clinical manifestations of chronic toxic encephalopathy usually involve varying degrees of cognitive impairment 1.
Chronic toxic encephalopathy is an established, internationally recognized condition that results from excessive occupational exposure to solvents via inhalation or skin contact. In 1985, the World Health Organization (WHO) published diagnostic criteria for chronic toxic encephalopathy caused by exposure to solvents 9. The most recent International Classification of Diseases document (ICD 10) defines chronic toxic encephalopathy 10 and the Diagnostic and Statistical Manual for Mental Disorders, Fourth Edition 11 lists the condition as a form of substance-induced persistent dementia.
The severity of chronic toxic encephalopathy is graded as I-III or 1, 2A, 2B, and 3 12:
- Type I chronic toxic encephalopathy and types 1 and 2A chronic toxic encephalopathy include subjective symptoms relating to memory, concentration, and mood. At this stage, clinicians may miss the diagnosis by considering these symptoms as a psychiatric issue due to altered mood 1.
- Type II chronic toxic encephalopathy and type 2B chronic toxic encephalopathy are characterized by objective evidence of attention and memory deficits, decreased psychomotor function 9 and/or learning deficits 12 on neurobehavioral testing. The taking of detailed occupational and medical histories, as well as standardized neurobehavioral testing, are the cornerstones of the standard diagnostic process. Workers with a history of repeated episodes indicative of acute solvent intoxication (e.g., light-headedness, dizziness, headache and nausea) over a period of many years; a history of insidious onset of attention, memory, and mood problems; and objective evidence of impairment on standardized neurobehavioral tests (i.e., deficits in attention, memory, learning and/or psychomotor function) should be considered as meeting the diagnostic criteria for type II chronic toxic encephalopathy or type 2B chronic toxic encephalopathy.
- Type III chronic toxic encephalopathy and type 3 chronic toxic encephalopathy are often accompanied by neurological deficits and neuroradiological findings. This type of chronic toxic encephalopathy often manifests clinical features, whereas types I and II show subclinical deficits. The MRI findings in patients with chronic toxic encephalopathy are nonspecific, although there may be slight brain atrophy; MRI findings mainly support the differential diagnosis of chronic toxic encephalopathy by ruling out other brain diseases. Thus, non-solvent etiologies should be considered if there are major findings on the brain MRI of a patient with suspected chronic toxic encephalopathy 13.
Most cases of chronic toxic encephalopathy are of type II or 2B 14. The Finnish criteria for chronic toxic encephalopathy usually includes the criterion of more than ten years of daily exposure at work 15. Follow-up is also important in diagnosing patients with chronic toxic encephalopathy. Subtle changes in mental functioning due to intoxication often go unrecognized unless the clinician specifically assesses these changes using sophisticated neuropsychological tests 4.
The high index of suspicion gives clues to diagnosis of chronic toxic encephalopathy. The diagnosis of chronic toxic encephalopathy requires a careful clinical assessment that 1) establishes that there is evidence for abnormality, mainly on neuropsychological testing; 2) determines that there is good evidence of a relationship to exposure to a potentially hazardous neurotoxin; and 3) excludes any other underlying causes. Specific therapies for chronic toxic encephalopathy are limited. The patient should be separated from the neurotoxic exposure as soon as possible. Once the toxin has been removed, the reversibility of the brain damage will depend on the grade of chronic toxic encephalopathy 15.
The important question of whether chronic toxic encephalopathy can progress to the development of dementia has not yet been answered. Increasing evidence suggests that most forms of degenerative dementia have a multi-factorial cause involving genetic, biological, and chemical factors 16. Further studies are needed to clarify the issue.
Acute toxic metabolic encephalopathy
Acute toxic-metabolic encephalopathy is an acute condition of global cerebral dysfunction, which includes delirium and confusion, in the absence of primary structural brain disease 17. Toxic metabolic encephalopathy is common among critically ill patients and usually a consequence of systemic illness 18. The electrolyte abnormalities that lead to toxic-metabolic encephalopathy include hypocalcemia or hypercalcemia, hypomagnesemia, and hypophosphatemia 19. Possibly, hypophosphatemia occurs in 10-60% of patients receiving acute renal replacement therapy, especially continuous or prolonged therapy 20. Moderate or severe hypophosphatemia has been recognized as a cause of respiratory muscle weakness but hypophosphatemia-induced toxic metabolic encephalopathy is very rare 21.
Toxic metabolic encephalopathy causes
Acute toxic metabolic encephalopathy is common among critically ill patients and usually a consequence of systemic illness 18. The electrolyte abnormalities that lead to toxic-metabolic encephalopathy include hypocalcemia or hypercalcemia, hypomagnesemia, and hypophosphatemia 19.
Cerebellar syndromes
Gait ataxia, dysarthria, intention tremor, gaze-evoked nystagmus, dysmetria and adiadochokinesia can all result from cerebellar dysfunction 16. Neurotoxin-induced cerebellar syndrome, which is a clinical entity that can be differentiated from solvent-induced chronic toxic encephalopathy or carbon-disulfide-induced vascular encephalopathy 16, is sometimes accompanied by other neurological findings. If a patient presents with cerebellar dysfunction, a detailed history of his or her occupation and neurotoxin exposure should be obtained.
Methyl mercury intoxication (Minamata disease)
Methyl mercury intoxication, known as Minamata disease, causes damage to the granule cell layer in the cerebellum, bilateral diffuse cerebellar atrophy, and microscopically diffuse loss of the granule cell layer in the cerebellar cortex 22. The major clinical features of the disease include progressive cerebellar ataxia and disturbance of the sensory functions of the cerebral cortex. Cerebellar ataxia manifests as gait ataxia, dysarthria, intention tremor, gaze nystagmus, dysmetria and dysdiadochokinesia. In addition, injuries to the somatosensory, visual, auditory or olfactory cortexes of the cerebrum can manifest as visual impairment, hearing impairment, olfactory problems, gustatory disturbance and cerebral cortex-related somatosensory disturbances 22. Concentric constrictions of the visual fields are characteristic findings due to damage to the calcarine cortex 16. In Minamata disease, atrophy of the visual calcarine cortex and the cerebellum has been demonstrated on computed tomography (CT) and MRI 23 and significantly decreased blood flow has been shown in the cerebellum on single-photon emission computed tomography (SPECT) 23. Fetal Minamata disease is a typical congenital toxic encephalopathy. Serious disturbances in mental and motor development are observed in all cases of fetal Minamata disease. Affected individuals show significant bilateral impairments in chewing, swallowing, speech, gait, other coordination and involuntary movement such as dystonia. These symptoms have been associated with the brain damage that is typical of Minamata disease 24.
Methyl bromide intoxication
Methyl bromide is a highly toxic gas that is used widely as an insecticidal fumigant for dry foodstuffs. It can be toxic to both the CNS and the peripheral nervous system 25. Most neurological manifestations of methyl bromide intoxication occur as a result of inhalation. Chronic exposure can cause peripheral polyneuropathy, optic neuropathy and cerebellar dysfunction, sometimes with neuropsychiatric disturbances 25. Typically, occupational history is vital to the diagnosis of bromide intoxication.
Organic tin intoxication
Organic tins, such as the dimethyl and trimethyl compounds, are widely used as polyvinyl-chloride stabilizers, catalysts and biocides 26. Selective cerebellar dysfunction is most prominent upon recovery from coma due to acute severe organic tin intoxication 26. It is easy to diagnose acute organic tin intoxication in patients whose work history and circumstances of exposure are known, and whose signs and symptoms are typical and consistent with those reported in the literature.
A fluid attenuated inversion recovery (FLAIR) MRI taken 15 days after an acute organic tin intoxication showed extensive symmetrical high-signal lesions throughout the white matter of the brain, indicating diffuse brain edema 8. In a follow-up study three years after an acute organic tin poisoning case, brain MRI showed cerebellar atrophy and 18F-fluorodeoxyglucose positron emission tomography (PET)/CT revealed mildly decreased metabolic activity in the pons and in both cerebellar hemispheres 26.
Parkinsonism
Manganese intoxication (manganism)
Manganism is one of the most typical forms of parkinsonism. Chronic excessive exposure to manganese (Mn) can affect the globus pallidus, resulting in parkinsonian signs and symptoms, sometimes along with psychiatric features called locura manganica or Mn madness. Historically, miners developed psychosis due to exposure to Mn at levels of up to several hundred milligrams per cubic meter 27.
The clinical course of manganism can be divided into three stages: at the first stage, patients with manganism usually have prodromal neuropsychiatric symptoms such as asthenia, apathy, somnolence, irritability, emotional lability, or frank psychoses. At the second stage, bradykinetic-rigid parkinsonian syndrome with dystonia, which is reversible, presents as the main clinical feature 1. Patients in the last stage are notable for aggravation of the signs and symptoms described as above. The clinical progression has been found to be irreversible and persistent after the cessation of exposure in some cases 28. Early diagnosis of manganism is therefore important.
The mechanism underlying this response to manganese exposure is not yet clear, but it has been suggested that an initial insult to the globus pallidus during Mn neurotoxicity can result in increased activity in the subthalamic nucleus, which is normally under tonic inhibition by the globus pallidus in the basal ganglia circuitry 29. Diagnosis of classical manganism requires a history of occupational manganese exposure, typical neurological findings such as bradykinesia, rigidity and postural instability, and the exclusion of other neurological diseases related to the basal ganglia, such as Parkinson’s disease, secondary parkinsonism due to traumatic, vascular, or iatrogenic damage, and atypical parkinsonism syndromes 30.
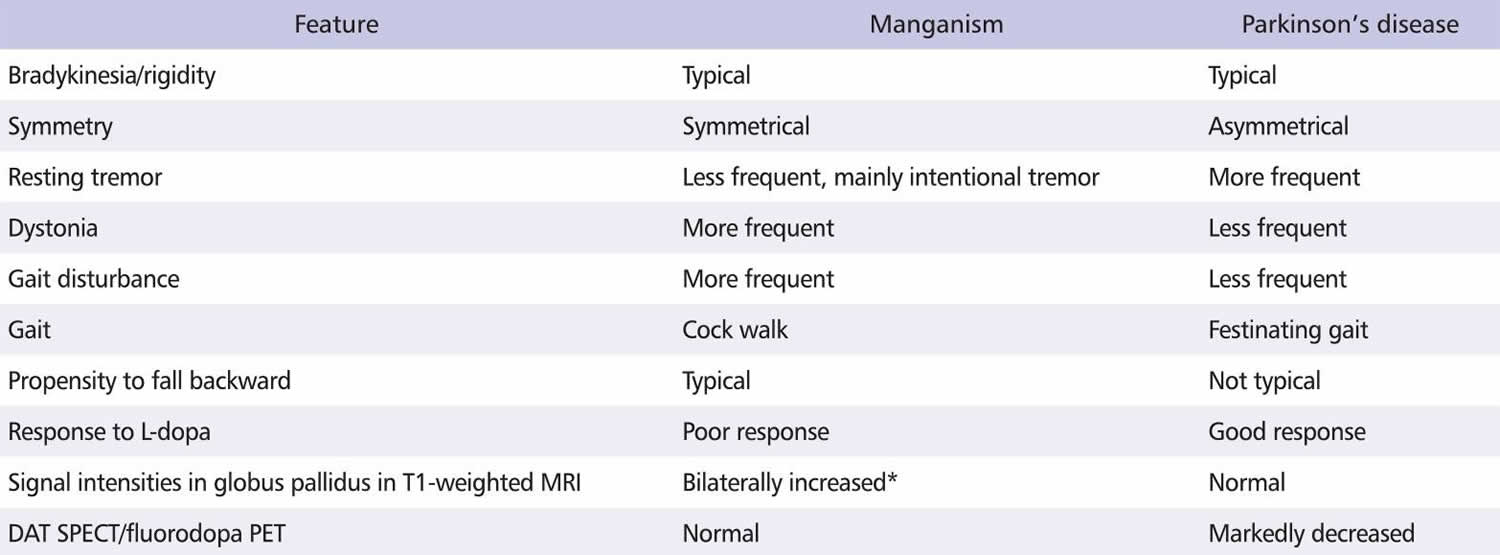
The differential diagnoses of this disorder can be summarized using clinical features and neuroimaging data (Table 1). The pathological lesions caused by manganism are typically degenerative lesions of the globus pallidus, sometimes with less-frequent and less-severe injuries to the substantia nigra. By contrast, in Parkison’s disease the substantia nigra is typically involved while the pallidostriatal complex is spared 31.
A manganese-induced, bilateral and symmetrical increase in signal intensity, confined mainly to the globus pallidus and midbrain, can be observed on T1-weighted MRI in manganese-exposed individuals, but no alterations are typically seen on T2-weighted MRI or CT scans 32. Increased signals on T1-weighted MRI were observed in both asymptomatic manganese-exposed workers and in patients with experimental or occupational manganese poisoning 33. However, these increased signal intensities generally resolved 6-12 months after the cessation of manganese exposure 34. Thus, a high T1 signal on MRI may reflect the target organ dose of recent occupational manganese exposure, but may not necessarily reflect manganism in the spectrum of manganese symptomatology 35.
At lower exposure levels, less severe, subtle, and preclinical neurobehavioral effects have been widely reported in various occupational and environmental settings 36. Concerns have been raised about whether chronic exposure to low levels of manganese can induce Parkison’s disease 37. In fact, Parkison’s disease is not a single disease, but rather a heterogeneous group of clinically similar conditions. It is possible that some individuals diagnosed with Parkison’s disease have neurotoxin-related Parkison’s disease that is likely to have been overlooked because most cases are not attributable to neurotoxin exposure. However, future work will be required to clarify whether manganese exposure induces Parkison’s disease and/or affects the progress of this condition.
Table 1. Comparison of the features of manganism and Parkinson’s disease
Others
Acute carbon monoxide poisoning can result in a delayed extrapyramidal syndrome that begins two to three weeks after recovery from the initial exposure. The parkinsonian features can be progressive and are associated with symmetrical degeneration of the globus pallidus 38. Abnormalities may be seen in brain CT and MRI 39. Carbon monoxide poisoning can also result in cognitive impairment and akinetic mutism associated with subcortical white matter lesions, especially in the bifrontal area 39. Parkinsonian features have occasionally been associated with methanol 40, carbon disulfide 41, paraquat and rotenone 42 and cyanide poisoning 43.
Vascular encephalopathy
Carbon disulfide poisoning is a highly typical and frequently encountered vascular encephalopathy 4. Patients with carbon disulfide poisoning exhibit various clinical characteristics, including multiple brain infarctions 44, peripheral neuropathy 45, coronary heart disease 46, retinopathy including microaneurysm of the fundus 47, hypertension 48, glomerulosclerosis of the kidney 49, and parkinsonian symptoms 41. These findings indicate that the basic mechanisms underlying carbon disulfide poisoning involve atherosclerotic changes in blood vessels [56,64]. The clinical manifestations of vascular encephalopathy (e.g., hemiparesis and speech disturbance) in cases of chronic carbon disulfide poisoning are similar to those observed in patients with atherosclerotic cerebrovascular disorders 50. Many patients presenting with acute cerebrovascular stroke-like symptoms, sometimes with hypertension or diabetes, have been misdiagnosed as having suffered cerebrovascular attacks. Thus, the possibility of carbon disulfide poisoning must not be overlooked when physicians make differential diagnoses in patients with vascular encephalopathy.
Historical records of carbon disulfide poisoning in Japan suggest the presence of a dose-response relationship. Psychosis and peripheral neuropathy due to carbon disulfide poisoning among rayon industry workers were first reported in 1932 and 1934, respectively 51. In the 1930s, when the Japanese first began producing rayon, carbon disulfide poisoning was the most common occupational disease in Japan 52, and psychosis resulting from very high exposure was a predominant health problem. Since 1949, rayon manufacturers have collaborated with university researchers in Japan to control carbon disulfide concentrations in the workplace and to monitor the incidence of carbon disulfide poisoning [52. By the 1960s, atherosclerosis (which is associated with moderate exposure levels) became the main type of carbon disulfide poisoning in Japan 52. Since 1980s, similar clinical features (e.g., atherosclerosis) have been observed in Korean workers with carbon disulfide poisoning, probably due to the transfer of the rayon industries from Japan in the 1960s 53.
Neurodegenerative diseases
Amyotrophic lateral sclerosis (ALS)
ALS is a neurodegenerative disease with an annual worldwide incidence of 2-4 cases per 100,000 individuals 54. A few cases have been reported in Korea 55. The association between ALS and exposure to solvents or lead is unclear, and even the best-designed incidence studies have produced conflicting results 56.
Other neurodegenerative diseases
It is reported that exposure to solvents, aluminum, mercury, or pesticides is implicated in the development of Alzheimer’s disease, which is the most common neurodegenerative disease 57. However, evidence for this causal relationship is limited and further studies are required. Parkison’s disease was dealt with in the manganism section above.
Toxic encephalopathy diagnosis
A diagnosis of toxic encephalopathy can be made after documentation of the following 58:
- A sufficiently intense or prolonged exposure to the neurotoxin;
- A neurological syndrome appropriate for the putative neurotoxins;
- Evolution of symptoms and signs over a compatible temporal course; and
- Exclusion of other neurological disorders that may account for a similar syndrome.
The exposure history, physical examination, neurological examination, and additional laboratory and radiological studies are particularly important for diagnosing a toxic encephalopathy. An overt toxic encephalopathy is not difficult to recognize if a patient develops a well-described clinical syndrome after exposure to a well-known neurotoxin, or if other workers at the same site develop similar clinical pictures. The more difficult (and more common) situation is when a symptomatic individual presents with either an unclear history of exposure or an apparently trivial exposure to a known or suspected neurotoxin. In this situation, careful evaluation of the case is essential 59.
Detailed exposure history
The patient’s exposure history is central to an accurate clinical diagnosis. Many problems can be overlooked or misdiagnosed because the person has not been questioned about his or her job and its related hazards. The occupational history should include information about the person’s current occupation, job task, place of employment, and dates of attendance on that job 60. Exposure data such as workplace airborne concentrations are crucial. A detailed evaluation of the nature, duration, and intensity of the exposure is essential for every evaluation. A description of the availability and use of personal protective equipment will provide further information about the extent of possible exposure. It is also important to ask questions about hobbies, and inadvertent exposure from any source should be considered 60. For the diagnosis of toxic encephalopathy, it is likely to be helpful if information on similar problems observed in others at the worksite is available.
Neurological examination
After a careful history is obtained, clinical examination should be carried out to establish the type and degree of dysfunction. The physical examination should include a general examination followed by a detailed neurological examination. Nonneurological signs may be a clue to toxic exposure; examples of systemic clues include blue gums in lead intoxication, Mees’ lines in arsenic poisoning, and acrodynia in mercury poisoning 61. The neurological examination will generally comprise assessment of mental function (mental status examination), cranial nerve function, muscle strength and tone, reflexes (muscle stretch and cutaneous), sensation, station and gait 59. A complete and rigorous neurological examination is necessary to properly define the clinical neurological syndrome involved. Once defined, a differential diagnosis can be entertained, and occupational versus non-occupation or toxic versus non-toxic causes can be determined.
Lab tests
When a patient is seen close to the time of exposure it may be possible to measure the offending chemical such as lead and mercury or its metabolite in blood or urine. Biomarkers can demonstrate that there has been exposure to the relevant toxin and that the exposure was of sufficiently severity to give rise to a clinical syndrome. It is obligatory in those cases in which there is an acute illness in relation to exposure. However, problems will undoubtedly occur in those cases where there is a delay between exposure and the development of clinical symptoms. Furthermore, for most neurotoxins biomarkers are not readily available 1.
There may be occasionally be paraclincal features in hematological and biochemical tests indicating red blood cell changes as in the case of lead poisoning, or liver function test abnormalities as in the case of some organic solvent poisoning. For most of neurotoxins, however, clinical laboratory tests are not helpful for diagnosis 1.
Neurobehavioral testing
Neurobehavioral (neuropsychological) testing, which is an accepted methodology for assessing the functional integrity of the CNS, has been used extensively to evaluate subclinical neurotoxic effects on cognition, memory, alertness, executive function, mood and psychomotor skills 62. There is a wide spectrum of neuropsychological tests, and the selection must be tailored to each situation. Neurobehavioral testing is generally administered by an examiner, as in the Neurobehavioral Core Test Battery from the WHO 63. Recently, however, many of the tests have been adapted for use on a personal computer. In toxic encephalopathy due to various neurotoxins (e.g., heavy metals or organic solvents), neuropsychological studies have been useful in evaluating subclinical findings 64. Indeed, since the 1990s, subclinical neuropsychological deficits detected by neurobehavioral testing have replaced overt clinical findings as the basis of occupational exposure limits for various neurotoxicants 65. Neurobehavioral tests are also used as diagnostic criteria for chronic toxic encephalopathy.
Electroencephalography (EEG)
EEG, which records the electric activity of the brain, has been used to evaluate occupational neurotoxic exposures 66. The changes that are most obvious on EEG, such as diffuse slowing, are often associated with toxic encephalopathy 1. However, the observed abnormalities are not specific, meaning that EEG has only a limited value in detecting and characterizing toxic encephalopathy Biological principles of chemical neurotoxicity. In: Spencer PS, Schaumburg HH, Ludolph AC, editors. Experimental and clinical neurotoxicology. 2nd ed. Oxford: Oxford University Press; 2000. pp. 3–54.)). With the advent of recent technological advances in neuroimaging, EEG is now used less frequently as a neurodiagnostic method, and more often in evaluating epilepsy.
Evoked potentials (EVPs)
Sensory evoked potentials (EVPs) are widely used in clinical neurology as an index of the integrity of the sensory CNS pathways. Compared to EEG, EVPs can provide more quantitative information and can be used to assess a sensory pathway from the receptor to the cortex. Among the EVPs, the visual evoked potential (VEP), auditory evoked potential (AEP), and somatosensory evoked potential (SEP) are most often used in evaluating neurotoxic disease and other neurological disorders 66. However, many variables can confound interpretation, and the results are not specific to neurotoxic disease 1 and thus should be interpreted with caution 67. Marked auditory evoked potential (AEP) abnormalities have been associated with toluene exposure 68, but these studies were performed in toluene abusers, who are exposed to much higher levels than those usually found in occupational settings.
Neuroimaging studies
Since the invention of CT and MRI scanners, tremendous progress has been made in the medical imaging of the human body. Neuroimaging can be divided into two groups: morphological neuroimaging (anatomy-based imaging) such as CT and MRI, and functional neuroimaging (physiology-based imaging) such as magnetic resonance spectroscopy (MRS), functional MRI (fMRI), diffusion tensor imaging (DTI), SPECT, and PET. At present, with the introduction of new technologies and the solving of technical problems related to the local production of radioisotopes, neuroimaging is shifting from morphological to functional 69.
CAT scan
Modern CT scanners are capable of performing multiple slices with rapid data acquisition and overlapping sections using continuously moving X-ray emitters (spiral CT). These methods enable the rapid production of exquisite images with three-dimensional reconstruction capabilities. In the brain, the use of X-ray contrast agents and angiography has improved intracranial imaging, but CT remains almost exclusively an anatomical imaging tool. In terms of neurotoxin-related damage, CT is valuable for ruling out other naturally occurring disorders of the nervous system, and it can reveal nonspecific changes (e.g., cortical atrophy) in individuals chronically exposed to organic solvents in the workplace 70.
MRI
MRI provides images that enhance either the fatty component (so-called ‘T1-weighted’ images) or the water component (so-called ‘T2-weighted’ images) of tissues. It has emerged as the pre-eminent imaging modality for visualizing neurological diseases in the central nervous system, because it can distinguish gray and white matter, zones of demyelination, and brain edema. The distinction between gray and white matter lesions is crucial because gray matter is more vulnerable to anoxic or ischemic insults due to its higher metabolic demands for oxygen and glucose 71. White matter changes, such as leukoencephalopathy, are usually better seen on MRI, whereas calcifications and hemorrhages are readily detectable by CT. However, the severity and extension of brain lesions on morphological neuroimaging do not necessarily match the severity of clinical status 71. T1-weighted MRI can detect paramagnetic metals, such as Mn, making it uniquely useful in Mn neurotoxicology. On T1-weighted MRI, Mn exposure causes bilateral symmetrical increases in signal intensity that are confined to the globus pallidus and midbrain 37.
Magnetic resonance spectroscopy (MRS)
Numerous whole-body MR scanners now operate at magnetic fields of 1.5 Tesla (T) or above, meaning that they can perform localized proton magnetic resonance spectroscopy without additional hardware 72. The latest very-high-field strength (i.e., 3 T or more) MRI systems generate images that combine anatomical and physiological measurements. In vivo proton magnetic resonance spectroscopy ([1H]-magnetic resonance spectroscopy) is an image-guided, noninvasive method for monitoring neurochemical metabolites in the brain 72. Currently, [1H]-magnetic resonance spectroscopy is most commonly employed to obtain metabolic information that may aid in the diagnosis of many neurological diseases, and also allows the evaluation of disease progression and treatment response 73. Although magnetic resonance spectroscopy permits noninvasive in vivo measurement of brain metabolites, only a few magnetic resonance spectroscopy investigations have assessed the neurological effects of neurotoxins in environmental or occupational health. Aydin et al. 74 demonstrated decreased N-acetylaspartate (NAA) in the cerebellar white matter and centrum semiovale along with increased myoinositol (mI) in toluene abusers. Several recent reports have analyzed the impact of lead exposure on brain metabolism in vivo in adults and children 75, while two other studies employed magnetic resonance spectroscopy to investigate the potential neurotoxic effects of chronic manganese exposure on the brain 76. In parparticular, Guilarte et al. 77 assessed the toxic effects of chronic manganese exposure on the levels of brain metabolites in non-human primates. This [1H]-magnetic resonance spectroscopy study found that the N-acetylaspartate/creatine (NAA/Cr) ratios in the parietal cortex and frontal white matter were decreased after Mn exposure, indicating ongoing neuronal degeneration or dysfunction. N-acetylaspartate (NAA) is known to serve as a neuronal marker 78 and a reduction in brain NAA levels can be interpreted as indicating neuronal dysfunction or loss 79. However, Kim et al. 76 found no significant difference between welders and control subjects in this measure. Similarly, Chang et al. 80 recently showed that the NAA/Cr ratios in both the anterior cingulate cortex and parietal white matter did not differ significantly between welders and controls. However, they found that the mI levels in the anterior cingulate cortex, but not in the parietal white matter, were significantly lower in welders compared with control individuals. Furthermore, in the frontal lobe of the brain, the mI/Cr ratio was significantly correlated with verbal memory scores and blood Mn concentrations. This study therefore suggested that the depletion of mI in welders may reflect a possible glial cell effect (rather than a neuronal effect) associated with long-term exposure to Mn. More recently, Dydak et al. 81 used the MEGA-PRESS sequence to determine γ-aminobutyric acid (GABA) levels in the thalamus, and found that manganese-exposed subjects showed significant decreases in the NAA/Cr ratio of the frontal cortex, and significant increases in the GABA level of the thalamus. Further magnetic resonance spectroscopy-based studies will be required to fully assess the various brain metabolites in manganese-exposed workers.
Functional MRI
Functional MRI (fMRI) uses standard clinical MRI hardware to collect information regarding brain metabolism changes associated with neuronal activity. As neuronal activity and the resulting demand for oxygen increase, the supply of oxygenated hemoglobin correspondingly increases, along with the MR signal measured on T2* images. This generates blood-oxygenation-level-dependent (BOLD) contrasts 82.
The use of fMRI to study neurological diseases has become much more common over the last decade, but employing fMRI to assess neurotoxicity in humans is a rather novel approach. There have not yet been any reports on functional MRI findings in metal neurotoxicity. Chang et al. 83 were the first to use fMRI and sequential finger-tapping to investigate the behavioral significance of additionally recruited brain regions in welders who had experienced chronic Mn exposure. Their findings suggest that fMRI may help us uncover evidence of compromised brain functioning in patients with subclinical manganism. The observation that the cortical motor network was excessively recruited in the chronically Mn-exposed group is in line with the emerging concept that adaptive neural mechanisms are used to compensate for latent dysfunctions in the basal ganglia. Chang et al. 84 also combined fMRI with two-back memory tests to assess the neural correlates of Mninduced memory impairment in response to subclinical dysfunction in the working memory networks of welders exposed to Mn for extended periods of time. These fMRI findings indicated that welders might need to recruit more neural resources to their working memory networks in order to compensate for subtle working memory deficits and alterations in working memory processes.
Diffusion Tensor Imaging (DTI)
Diffusion Tensor Imaging (DTI), which is a unique method for characterizing white matter micro-integrity 85, can reveal the orientation of white matter tracts in vivo and yields indices of microstructural integrity by quantifying the directionality of water diffusion 86. A few previous studies have explored the toxic encephalopathy associated with exposure to environmental neurotoxins, such as mercury 87, manganese 88, methanol 89 and carbon monoxide 90 using diffusion-weighted image (DWI) analysis. However, few studies have reported DTI-detected alterations of microscopic integrity within the white matter of subjects experiencing environmental neurotoxic exposure 91. Kim et al. 92 used DTI to investigate whether welders exposed to Mn exhibited differences in white matter integrity. White matter microstructural abnormalities (decreased fractional anisotropy [FA]), which correlated with deficits in motor and cognitive neurobehavioral performance, were observed in welders, as compared to controls.
Single-photon emission computerized tomography (SPECT) scan
Single-photon emission computerized tomography (SPECT) scan is a widely distributed functional imaging modality that is more easily accessible and less expensive than PET, but has a lower resolution 32. SPECT allows the imaging of regional blood flow, metabolism and neurotransmitter receptors with relatively good spatial resolution. SPECT of patients with Minamata disease showed significantly decreased blood flow in the cerebellum 23. In a case of acute lithium intoxication, the CT and MRI were normal, but a SPECT scan indicated significant focal perfusion defects, predominantly involving the left temporo-parietal area and the right posterior parietooccipital area 93. Huang et al. 94 reported that the striatal 99mTc-TRODAT-1 uptake in dopamine transporter (DAT) SPECT was nearly normal in manganese-induced parkinsonism, but was markedly reduced in Parkinson’s disease. Various ligands that bind to DAT, such as [123I]-β-CIT, [123I]-fluoropropyl-CIT and 99mTc-TRODAT-1, have been used in SPECT studies to elucidate the function of the dopaminergic nigrostriatal pathway, and thus to differentiate manganism from Parkinson’s disease 95.
Positron emission tomography (PET) scan
Positron emission tomography (PET) scan uses short-lived positron-emitting isotopes to mark biologically active compounds. PET isotopes have such short half-lives that they must be produced in on-site cyclotrons to be available in quantities viable for clinical use. PET relies on a visibly labeled ligand to provide image specificity. Specific ligands have been developed to help elucidate the function of the dopaminergic nigrostriatal pathway. For example, the ligand [18F]-dopa provides information about the conversion of L-dopa to dopamine 96. In nonhuman primates and humans with manganism, the [18F]-dopa PET scan (which provides an index of the integrity of the dopaminergic nigrostriatal pathway) is normal 97; by contrast, reduced dopamine uptake occurs in the striatum and particularly the posterior putamen of Parkison’s disease patients 98. Thus, [18F]-dopa PET scans have been used to differentiate manganism from Parkison’s disease 95. In addition, 18F-fluorodeoxyglucose (18F-FDG) PET may be used to show metabolic activity in the brain. In a follow-up study of an acute organic tin poisoning case three years after diagnosis, 18F-FDG PET/CT revealed mildly decreased metabolic activity in the pons and in both cerebellar hemispheres 26. Similarly, decreased metabolism in the thalamus, basal ganglia, temporal lobe and inferior parietal lobe have been observed in hydrogen sulfide poisoning using 18F-FDG PET 99.
Toxic encephalopathy treatment
Toxic encephalopathy treatment is symptomatic and varies, according to the type and severity of the encephalopathy. Your physician can provide specific instructions for proper care and treatment. Anticonvulsants may be prescribed to reduce or halt any seizures. Changes to diet and nutritional supplements may help some patients. In severe cases, dialysis or organ replacement surgery may be needed.
Toxic encephalopathy prognosis
Treating the underlying cause of toxic encephalopathy may improve symptoms. However, the encephalopathy may cause permanent structural changes and irreversible damage to the brain. Some encephalopathies can be fatal.
References- Firestone JA, Longstrength WT., Jr . Neurologic and psychiatric disorders. In: Rosenstock L, Cullen M, Brodkin C, Redlich C, editors. Textbook of clinical occupational and environmental medicine. 4th ed. Philadelphia (PA): Saunders; 2004. pp. 645–660.
- Encephalopathy. https://acphospitalist.org/archives/2010/09/coding.htm
- US Environmental Protection Agency. Toxic Substances Control Act (TSCA). Chemical substance inventory-revised inventory synonym and preferred name file. Washington, DC: Office of Pollution, Prevention, and Toxics; 2000.
- Kim Y, Jeong KS, Yun YH, Oh MS. Occupational neurologic disorders in Korea. J Clin Neurol. 2010;6:64–72.
- Valk J, van der Knaap MS. Toxic encephalopathy. AJNR Am J Neuroradiol. 1992;13(2):747–760. http://www.ajnr.org/content/ajnr/13/2/747.full.pdf
- Encephalopathy Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Encephalopathy-Information-Page
- Encephalopathy. https://acphospitalist.org/archives/2015/01/coding.htm
- Yoo CI, Kim Y, Jeong KS, Sim CS, Choy N, Kim J, Eum JB, Nakajima Y, Endo Y, Kim YJ. A case of acute organotin poisoning. J Occup Health. 2007;49:305–310.
- World Health Organization (WHO); Nordic Council of Ministers. Chronic effects of organic solvents on the central nervous system and diagnostic criteria : report on a joint WHO/Nordic Council of Ministers Working Group; 1985 Jun 10-14; Copenhagen, Denmark. Copenhagen: WHO, Regional Office for Europe, Nordic Council of Ministers; 1985.
- World Health Organization (WHO) The ICD 10 classification of mental and behavioural disorders: Clinical descriptions and diagnostic guidelines. Geneva (Switzerland): WHO; 1992.
- American Psychiatric Association. Diagnostic and statistical manual for mental disorders. 4th ed. Washington, DC: American Psychiatric Press; 1994.
- Baker EL, Seppäläinen AM. Proceedings of the Workshop on neurobehavioral effects of solvents. October 13-16, 1985, Raleigh, North Carolina, U.S.A. Neurotoxicolog. 1986;7:1–95.
- Keski-Säntti P, Mäntylä R, Lamminen A, Hyvärinen HK, Sainio M. Magnetic resonance imaging in occupational chronic solvent encephalopathy. Int Arch Occup Environ Health. 2009;82:595–602.
- van der Hoek JA, Verberk MM, Hageman G. Criteria for solvent-induced chronic toxic encephalopathy: a systematic review. Int Arch Occup Environ Health. 2000;73:362–368.
- Kaukiainen A, Akila R, Martikainen R, Sainio M. Symptom screening in detection of occupational solvent-related encephalopathy. Int Arch Occup Environ Health. 2009;82:343–355.
- Bates D. Diagnosis of neurotoxic syndromes. In: Blain PG, Harris JB, editors. Medical neurotoxicology. London (UK): Arnold; 1999. pp. 3–11.
- Han SA, Park HY, Kim HW, et al. Severe Hypophosphatemia-Induced Acute Toxic-Metabolic Encephalopathy in Continuous Renal Replacement Therapy. Electrolyte Blood Press. 2019;17(2):62–65. doi:10.5049/EBP.2019.17.2.62 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6962441
- Ely EW, Stephens RK, Jackson JC, et al. Current opinions regarding the importance, diagnosis, and management of delirium in the intensive care unit: a survey of 912 healthcare professionals. Crit Care Med. 2004;32:106–112.
- Young GB, DeRubeis DA. Metabolic encephalopathies. In: Young GB, Ropper AH, Bolton CF, editors. Coma and Impaired Consciousness. McGraw-Hill; 1998. p. 307.
- Bellomo R, Cass A, Cole L, et al. The relationship between hypophosphataemia and outcomes during low-intensity and high-intensity continuous renal replacement therapy. Crit Care Resusc. 2014;16:34–41.
- Gravelyn TR, Brophy N, Siegert C, et al. Hypophosphatemia-associated respiratory muscle weakness in a general inpatient population. Am J Med. 1998;84:870–876.
- Ekino S, Susa M, Ninomiya T, Imamura K, Kitamura T. Minamata disease revisited: an update on the acute and chronic manifestations of methyl mercury poisoning. J Neurol Sci. 2007;262:131–144.
- Itoh K, Korogi Y, Tomiguchi S, Takahashi M, Okajima T, Sato H. Cerebellar blood flow in methylmercury poisoning (Minamata disease) Neuroradiology. 2001;43:279–284.
- Takeuchi T. Pathology of Minamata disease. In: Study Group of Minamata Disease, editor. Minamata disease. Kumamoto (Japan): Kumamoto University; 1968. pp. 141–228.
- Geyer HL, Schaumburg HH, Herskovitz S. Methyl bromide intoxication causes reversible symmetric brainstem and cerebellar MRI lesions. Neurology. 2005;64:1279–1281.
- Kim SH, Yoo CI, Kwon JH, Bae JH, Weon YC, Kim Y. A case of cerebellar dysfunction after acute organotin poisoning. Korean J Occup Environ Med. 2009;21:289–292.
- Rodier J. Manganese poisoning in Moroccan miners. Br J Ind Med. 1955;12:21–35.
- Huang CC, Chu NS, Lu CS, Chen RS, Calne DB. Long-term progression in chronic manganism: ten years of follow-up. Neurology. 1998;50:698–700.
- Fitsanakis VA, Au C, Erikson KM, Aschner M. The effects of manganese on glutamate, dopamine and gamma-aminobutyric acid regulation. Neurochem Int. 2006;48:426–433.
- Lucchini R, Kim Y. Health effects of manganese. In: Vojtisek M, Prakash R, editors. Metals and neurotoxicity. Jalgaon (India): Society for science and environment; 2009. pp. 119–147.
- Yamada M, Ohno S, Okayasu I, Okeda R, Hatakeyama S, Watanabe H, Ushio K, Tsukagoshi H. Chronic manganese poisoning: a neuropathological study with determination of manganese distribution in the brain. Acta Neuropathol. 1986;70:273–278.
- Kim Y. Neuroimaging in manganism. Neurotoxicology. 2006;27:369–372.
- Kim Y, Kim KS, Yang JS, Park IJ, Kim E, Jin Y, Kwon KR, Chang KH, Kim JW, Park SH, Lim HS, Cheong HK, Shin YC, Park J, Moon Y. Increase in signal intensities on T1-weighted magnetic resonance images in asymptomatic manganese-exposed workers. Neurotoxicology. 1999;20:901–907.
- Kim Y, Kim JW, Ito K, Lim HS, Cheong HK, Kim JY, Shin YC, Kim KS, Moon Y. Idiopathic parkinsonism with superimposed manganese exposure: utility of positron emission tomography. Neurotoxicology. 1999;20:249–252.
- Park J, Kim Y, Kim JW. High signal intensities on T1-weighted MRI in the spectrum of manganese symptomatology. In: Webster LR, editor. Neurotoxicity syndrome. New York (NY): Nova Biomedical Books; 2007. pp. 249–260.
- Zoni S, Albini E, Lucchini R. Neuropsychological testing for the assessment of manganese neurotoxicity: a review and a proposal. Am J Ind Med. 2007;50:812–830.
- Kim Y, Kim JM, Kim JW, Yoo CI, Lee CR, Lee JH, Kim HK, Yang SO, Chung HK, Lee DS, Jeon B. Dopamine transporter density is decreased in parkinsonian patients with a history of manganese exposure: what does it mean? Mov Disord. 2002;17:568–575.
- Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol. 1983;40:433–435.
- Lee WK, Yu ZH, Lee CC. Delayed neurological sequelae after carbon monoxide poisoning. Aust N Z J Psychiatry. 2008;42:430.
- Ley CO, Gali FG. Parkinsonian syndrome after methanol intoxication. Eur Neurol. 1983;22:405–409.
- Huang CC. Carbon disulfide neurotoxicity: Taiwan experience. Acta Neurol Taiwan. 2004;13:3–9.
- Thiruchelvam M, Richfield EK, Goodman BM, Baggs RB, Cory-Slechta DA. Developmental exposure to the pesticides paraquat and maneb and the Parkinson’s disease phenotype. Neurotoxicology. 2002;23:621–633.
- Rosenow F, Herholz K, Lanfermann H, Weuthen G, Ebner R, Kessler J, Ghaemi M, Heiss WD. Neurological sequelae of cyanide intoxication–the patterns of clinical, magnetic resonance imaging, and positron emission tomography findings. Ann Neurol. 1995;38:825–828.
- Huang CC, Chu CC, Chu NS, Wu TN. Carbon disulfide vasculopathy: a small vessel disease. Cerebrovasc Dis. 2001;11:245–250.
- Chu CC, Huang CC, Chen RS, Shih TS. Polyneuropathy induced by carbon disulphide in viscose rayon workers. Occup Environ Med. 1995;52:404–407.
- Hernberg S, Partanen T, Nordman CH, Sumari P. Coronary heart disease among workers exposed to carbon disulphide. Br J Ind Med. 1970;27:313–325.
- Karai I, Sugimoto K, Goto S. A fluorescein angiographic study on carbon disulfide retinopathy among workers in viscose rayon factories. Int Arch Occup Environ Health. 1983;53:91–99.
- Tolonen M. Vascular effects of carbon disulfide: a review. Scand J Work Environ Health. 1975;1:63–77.
- Yamagata Y, Yuda A, Suzuki K, Nemoto T, Takahashi M, Tuchida H, Saito K, Kusunoki N. Carbon disulphide nephrosclerosis, with special reference to the similarity to diabetic glomerulosclerosis: Renal biopsy findings in 17 patients. J Jpn Diabetical Soc. 1966;9:208–217. Japanese.
- Chuang WL, Huang CC, Chen CJ, Hsieh YC, Kuo HC, Shih TS. Carbon disulfide encephalopathy: cerebral microangiopathy. Neurotoxicology. 2007;28:387–393.
- Miura T. Work and health in rayon and staple industry. In: Miura T, editor. History of work and health. Vol. 4. Kawasaki (Japan): Institute for Science of Labour; 1981. pp. 203–236. Japanese.
- Harada M. Gold and mercury. Tokyo (Japan): Kodansha; 2002. Japanese.
- Park J, Hisanaga N, Kim Y. Transfer of occupational health problems from a developed to a developing country: lessons from the Japan-South Korea experience. Am J Ind Med. 2009;52:625–632.
- Johnson FO, Atchison WD. The role of environmental mercury, lead and pesticide exposure in development of amyotrophic lateral sclerosis. Neurotoxicology. 2009;30:761–765.
- Kim EA, Kang SK. Occupational neurological disorders in Korea. J Korean Med Sci. 2010;25(Suppl):S26–S35.
- Weisskopf MG, Morozova N, O’Reilly EJ, McCullough ML, Calle EE, Thun MJ, Ascherio A. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2009;80:558–561.
- Dobbs MR. Clinical neurotoxicology: Syndromes, substances, environments. 1st ed. Philadelphia (PA): Saunders; 2009. pp. 3–6.
- So YT. Neurotoxicology. In: Ladou J, editor. Current occupational and environmental medicine. 2nd ed. New York (NY): McGraw Hill; 2007. pp. 373–383.
- Rosenberg NL. Recognition and evaluation of work-related neurologic disorders. In: Rosenberg NL, editor. Occupational and environmental neurology. Boston (MA): Butterworth-Heinemann; 1995. pp. 9–45.
- Feldman RG. Occupational neurology. Yale J Biol Med. 1987;60:179–186.
- Dobbs MR. Toxic encephalopathy. Semin Neurol. 2011;31:184–193.
- World Health Organization (WHO) Neurotoxicity risk assessment for human health: Principles and approaches; Environmental health criteria 223. Geneva (Switzerland): WHO; 2001.
- Anger WK, Cassitto MG. Individual-administered human behavioral test batteries to identify neurotoxic chemicals. Environ Res. 1993;61:93–106.
- Meyer-Baron M, Blaszkewicz M, Henke H, Knapp G, Muttray A, Schäper M, van Thriel C. The impact of solvent mixtures on neurobehavioral performance: conclusions from epidemiological data. Neurotoxicology. 2008;29:349–360.
- American Conference of Governmental Industrial Hygienists (ACGIH) Documentation of the threshold limit values and biological exposure indices. 7th ed. Cincinnati (OH): ACGIH; 2011.
- Seppäläinen AM. Neurophysiological approaches to the detection of early neurotoxicity in humans. Crit Rev Toxicol. 1988;18:245–298.
- Arezzo JC, Simson R, Brennan NE. Evoked potentials in the assessment of neurotoxicity in humans. Neurobehav Toxicol Teratol. 1985;7:299–304.
- Rosenberg NL, Spitz MC, Filley CM, Davis KA, Schaumburg HH. Central nervous system effects of chronic toluene abuse–clinical, brainstem evoked response and magnetic resonance imaging studies. Neurotoxicol Teratol. 1988;10:489–495.
- Walker RC, Purnell GL, Jones-Jackson LB, Thomas KL, Brito JA, Ferris EJ. Introduction to PET imaging with emphasis on biomedical research. Neurotoxicology. 2004;25:533–542.
- Jensen PB, Nielsen P, Nielsen NO, Olivarius BD, Hansen JH. Chronic toxic encephalopathy following occupational exposure to organic solvents. The course after cessation of exposure illustrated by a neurophysiological follow-up study. Ugeskr Laeger. 1984;146:1387–1390.
- Hantson P, Duprez T. The value of morphological neuroimaging after acute exposure to toxic substances. Toxicol Rev. 2006;25:87–98.
- Rosen Y, Lenkinski RE. Recent advances in magnetic resonance neurospectroscopy. Neurotherapeutics. 2007;4:330–345.
- Ross AJ, Sachdev PS, Wen W, Brodaty H, Joscelyne A, Lorentz LM. Prediction of cognitive decline after stroke using proton magnetic resonance spectroscopy. J Neurol Sci. 2006;251:62–69.
- Aydin K, Sencer S, Ogel K, Genchellac H, Demir T, Minareci O. Single-voxel proton MR spectroscopy in toluene abuse. Magn Reson Imaging. 2003;21:777–785.
- Weisskopf MG, Hu H, Sparrow D, Lenkinski RE, Wright RO. Proton magnetic resonance spectroscopic evidence of glial effects of cumulative lead exposure in the adult human hippocampus. Environ Health Perspect. 2007;115:519–523.
- Kim EA, Cheong HK, Choi DS, Sakong J, Ryoo JW, Park I, Kang DM. Effect of occupational manganese exposure on the central nervous system of welders: 1H magnetic resonance spectroscopy and MRI findings. Neurotoxicology. 2007;28:276–283.
- Guilarte TR, McGlothan JL, Degaonkar M, Chen MK, Barker PB, Syversen T, Schneider JS. Evidence for cortical dysfunction and widespread manganese accumulation in the nonhuman primate brain following chronic manganese exposure: a 1H-MRS and MRI study. Toxicol Sci. 2006;94:351–358.
- Birken DL, Oldendorf WH. N-acetyl-L-aspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. Neurosci Biobehav Rev. 1989;13:23–31.
- Vion-Dury J, Meyerhoff DJ, Cozzone PJ, Weiner MW. What might be the impact on neurology of the analysis of brain metabolism by in vivo magnetic resonance spectroscopy? J Neurol. 1994;241:354–371.
- Chang Y, Woo ST, Lee JJ, Song HJ, Lee HJ, Yoo DS, Kim SH, Lee H, Kwon YJ, Ahn HJ, Ahn JH, Park SJ, Weon YC, Chung IS, Jeong KS, Kim Y. Neurochemical changes in welders revealed by proton magnetic resonance spectroscopy. Neurotoxicology. 2009;30:950–957.
- Dydak U, Jiang YM, Long LL, Zhu H, Chen J, Li WM, Edden RA, Hu S, Fu X, Long Z, Mo XA, Meier D, Harezlak J, Aschner M, Murdoch JB, Zheng W. In vivo measurement of brain GABA concentrations by magnetic resonance spectroscopy in smelters occupationally exposed to manganese. Environ Health Perspect. 2011;119:219–224.
- Song AW, Huettel SA, McCarthy G. Functional neuroimaging: Basic principles of functional MRI. In: Cabeza R, Kingstone A, editors. Handbook of functional neuroimaging of cognition. 2nd ed. Cambridge (MA): The MIT Press; 2006. pp. 21–52.
- Chang Y, Song HJ, Lee JJ, Seo JH, Kim JH, Lee HJ, Kim HJ, Kim Y, Ahn JH, Park SJ, Kwon JH, Jeong KS, Jung DK. Neuroplastic changes within the brains of manganese-exposed welders: recruiting additional neural resources for successful motor performance. Occup Environ Med. 2010;67:809–815.
- Chang Y, Lee JJ, Seo JH, Song HJ, Kim JH, Bae SJ, Ahn JH, Park SJ, Jeong KS, Kwon YJ, Kim SH, Kim Y. Altered working memory process in the manganese-exposed brain. Neuroimage. 2010;53:1279–1285.
- Beaulieu C. The basis of anisotropic water diffusion in the nervous system – a technical review. NMR Biomed. 2002;15:435–455.
- Le Bihan D, Mangin JF, Poupon C, Clark CA, Pappata S, Molko N, Chabriat H. Diffusion tensor imaging: concepts and applications. J Magn Reson Imaging. 2001;13:534–546.
- Kinoshita Y, Ohnishi A, Kohshi K, Yokota A. Apparent diffusion coefficient on rat brain and nerves intoxicated with methylmercury. Environ Res. 1999;80:348–354.
- McKinney AM, Filice RW, Teksam M, Casey S, Truwit C, Clark HB, Woon C, Liu HY. Diffusion abnormalities of the globi pallidi in manganese neurotoxicity. Neuroradiology. 2004;46:291–295.
- Peters AS, Schwarze B, Tomandl B, Probst-Cousin S, Lang CJ, Hilz MJ. Bilateral striatal hyperintensities on diffusion weighted MRI in acute methanol poisoning. Eur J Neurol. 2007;14:e1–e2.
- Sener RN. Acute carbon monoxide poisoning: diffusion MR imaging findings. AJNR Am J Neuroradiol. 2003;24:1475–1477.
- Lo CP, Chen SY, Chou MC, Wang CY, Lee KW, Hsueh CJ, Chen CY, Huang KL, Huang GS. Diffusion-tensor MR imaging for evaluation of the efficacy of hyperbaric oxygen therapy in patients with delayed neuropsychiatric syndrome caused by carbon monoxide inhalation. Eur J Neurol. 2007;14:777–782.
- Kim Y, Jeong KS, Song HJ, Lee JJ, Seo JH, Kim GC, Lee HJ, Kim HJ, Ahn JH, Park SJ, Kim SH, Kwon YJ, Chang Y. Altered white matter microstructural integrity revealed by voxel-wise analysis of diffusion tensor imaging in welders with manganese exposure. Neurotoxicology. 2011;32:100–109.
- Sheehan W, Thurber S. SPECT and neuropsychological measures of lithium toxicity. Aust N Z J Psychiatry. 2006;40:277.
- Huang CC, Weng YH, Lu CS, Chu NS, Yen TC. Dopamine transporter binding in chronic manganese intoxication. J Neurol. 2003;250:1335–1339.
- Kim J, Kim JM, Kim YK, Shin JW, Choi SH, Kim SE, Kim Y. Dopamine transporter SPECT of a liver cirrhotic with atypical parkinsonism. Ind Health. 2007;45:497–500.
- Pogge A, Slikker W., Jr Neuroimaging: new approaches for neurotoxicology. Neurotoxicology. 2004;25:525–531.
- Shinotoh H, Snow BJ, Chu NS, Huang CC, Lu CS, Lee C, Takahashi H, Calne DB. Presynaptic and postsynaptic striatal dopaminergic function in patients with manganese intoxication: a positron emission tomography study. Neurology. 1997;48:1053–1056.
- Brooks DJ, Salmon EP, Mathias CJ, Quinn N, Leenders KL, Bannister R, Marsden CD, Frackowiak RS. The relationship between locomotor disability, autonomic dysfunction, and the integrity of the striatal dopaminergic system in patients with multiple system atrophy, pure autonomic failure, and Parkinson’s disease, studied with PET. Brain. 1990;113:1539–1552.
- Schneider JS, Tobe EH, Mozley PD, Jr, Barniskis L, Lidsky TI. Persistent cognitive and motor deficits following acute hydrogen sulphide poisoning. Occup Med (Lond) 1998;48:255–260.





{kind=link}