
Wolman disease
Wolman disease is a congenital disease characterized by an impaired metabolism of the fats (lipids). Wolman disease is the most severe type of lysosomal acid lipase deficiency 1. The lysomal acid lipase deficiency causes a buildup of lipids (fats) in body organs and calcium deposits in the adrenal glands. Common symptoms of Wolman disease in infants include enlarged liver and spleen, poor weight gain, low muscle tone, jaundice, vomiting, diarrhea, developmental delay, anemia, and poor absorption of nutrients from food. Wolman disease is caused by mutations in the LIPA gene which provides instructions to make the lysosomal acid lipase. Inheritance is autosomal recessive 2. Wolman disease is severe and life-threatening, however enzyme replacement therapy, available for the treatment of lysosomal acid lipase deficiencies, in the United States, the European Union, and Japan, have shown improvement of symptoms, including liver problems, as well as an increased life expectancy 3. Liver transplantation can be considered in some cases when the liver disease is severe. Reports of treatment with bone marrow transplantation have shown mixed results, correcting the metabolic disease in a few cases, but not in others 4.
Wolman disease is an extremely rare disorder that affects males and females in equal numbers. More than 50 cases have been reported in the medical literature. However, cases may go undiagnosed or misdiagnosed making it difficult to determine the disorder’s true frequency in the general population. Wolman disease is named after one of the physicians who first identified the disorder in the medical literature in 1956.
Is Wolman disease curable?
While there is not a universal cure for all individuals with Wolman disease, some therapeutic options have resulted in correction of the underlying disease process. Studies have shown that children with Wolman disease have had successful early treatment with transplantation of hematopoietic stem cells and umbilical cord stem cells 5. The use of hematopoietic stem cells transplantation is controversial as it can be associated with complications. More studies are needed to determine the overall efficacy of these treatment options 6.
Wolman disease causes
Wolman disease is caused by mutations in the lysosomal acid lipase (LIPA) gene. The LIPA gene contains instructions for producing the enzyme lysosomal acid lipase. This enzyme is essential for breaking down (metabolizing) certain fats in the body, especially cholesterol (specifically cholesteryl esters) and to a lesser degree triglycerides. Without proper levels of this enzyme, these fats abnormally accumulate in and damage various tissues and organs of the body. Mutations in the LIPA gene that cause Wolman disease result in the lack of production of the LIPA enzyme or production of a defective, inactive form of the LIPA enzyme.
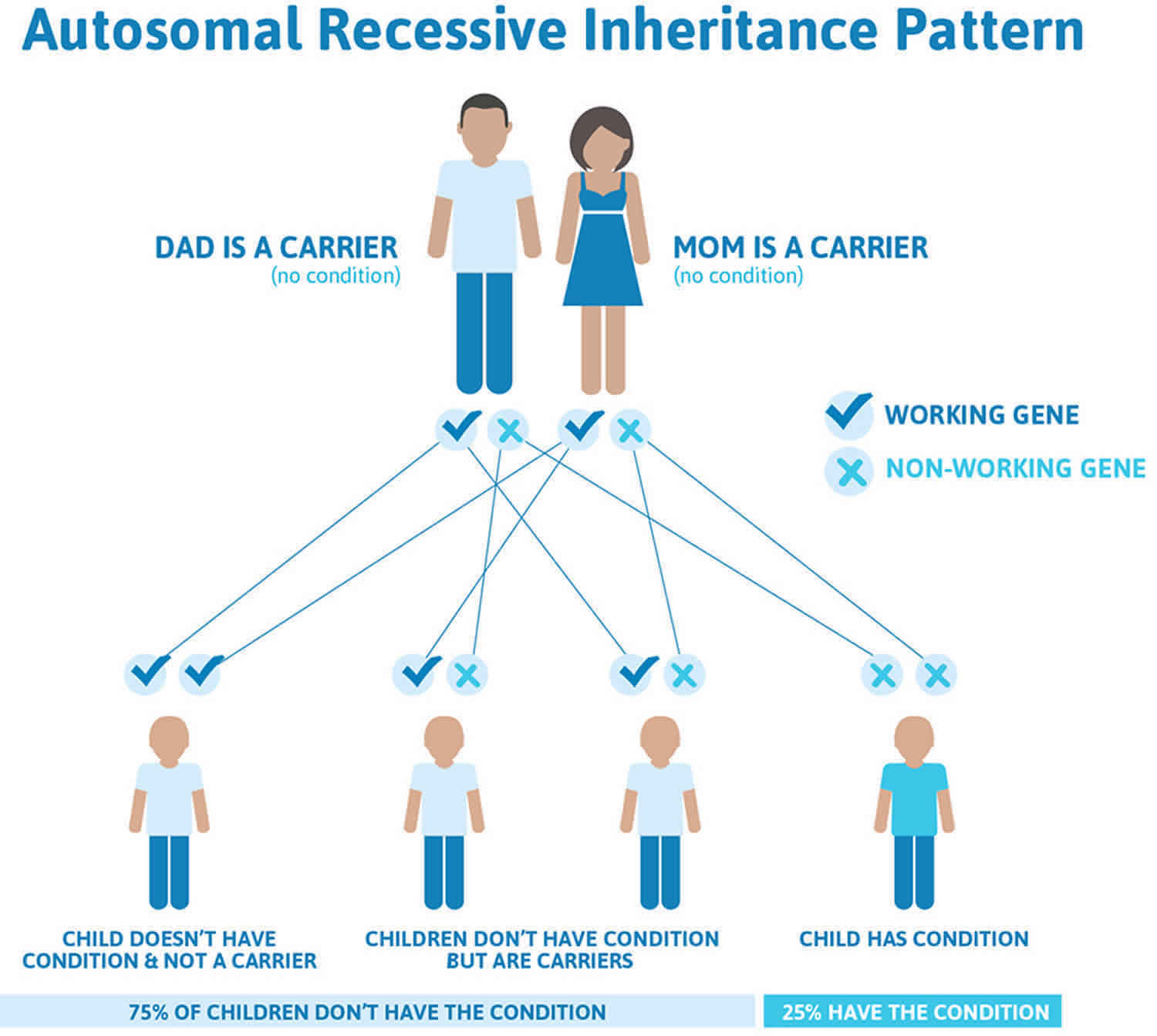
Wolman disease inheritance pattern
Wolman disease is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Figure 1. Wolman disease autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Wolman disease symptoms
The symptoms of Wolman disease usually become apparent shortly after birth, usually during the first few weeks of life with vomiting, steatorrhea, and abdominal distention 7. Other infants may come to medical attention within weeks to months due to failure to thrive 8. Affected infants may develop bloating or swelling of the stomach (abdominal distention) and may have significant enlargement of the liver and spleen (hepatosplenomegaly). Hepatomegaly, the result of build-up of cholesterol esters and triglycerides in macrophages of the liver, is common and can be dramatic. Splenomegaly, the result of the same mechanism, may also be present. Scarring (fibrosis) of the liver may also occur. In some cases, fluid may accumulate in the abdominal cavity (ascites).
Increased lipid deposition along the gastrointestinal tract leads to thickened bowel walls with resultant malnutrition and wasting 9.
Steatosis may progress to liver failure.
Enlarged adrenal glands with calcification, a classic finding in Wolman disease, can lead to adrenal cortical insufficiency.
Infants with Wolman disease have serious digestive abnormalities including malabsorption, a condition in which the intestines fail to absorb nutrients and calories form food. Malabsorption associated with Wolman disease causes persistent and often forceful vomiting, frequent diarrhea, foul-smelling, fatty stools (steatorrhea) and malnutrition. Because of these digestive complications, affected infants usually fail to grow and gain weight at the expected rate for their age and sex (failure to thrive).
Enlargement of the liver and spleen and protrusion of the abdomen can cause umbilical hernia, a condition in which the contents of the stomach may push through an abnormal opening or tear in the abdominal wall near the bellybutton. Additional symptoms may also occur in Wolman disease including yellowing of the skin, mucous membranes and whites of the eyes (jaundice), a persistent low-grade fever, and poor muscle tone (hypotonia). Infants may exhibit delays in the development of motor skills.
A distinct finding associated with Wolman disease is the hardening of adrenal gland tissue due to the accumulation of calcium (calcification). The adrenal glands are located on top of the kidneys and produce two hormones called epinephrine and norepinephrine. Other hormones produced by the adrenal glands help to regulate the fluid and electrolyte balance in the body. Calcification of the adrenal glands is not detectable by physical examination, but can be seen with x-ray study. Calcification may prevent the adrenal glands from producing enough essential hormones and can affect metabolism, blood pressure, the immune system and other vital processes of the body.
Infants with Wolman disease may experience the loss of previously acquired skills required the coordination of muscle and motor skills (psychomotor regression). The symptoms of Wolman disease often get progressively worse eventually leading to life-threatening complications during infancy including extremely low levels of circulating red blood cells (severe anemia), liver (hepatic) dysfunction or failure, and physical wasting away and severe weakness often associated with chronic disease and marked by weight loss and loss of muscle mass (cachexia or inanition).
Wolman disease diagnosis
A diagnosis of Wolman disease may be suspected in newborn infants based upon identification of characteristic symptoms such as abnormally enlarged liver and gastrointestinal problems. A diagnosis may be confirmed by a thorough clinical evaluation, a detail patient history (including family history) and specialized tests that reveal absence or deficient activity of the enzyme lysosomal lipase acid (LIPA) in certain cells and tissues of the body. Molecular genetic testing for mutations in the LIPA gene is also available.
Wolman disease treatment
In December 2015, the U.S. Food and Drug Administration (FDA) approved Kanuma (sebelipase alfa) as the first treatment for people with lysosomal acid lipase (LAL) deficiency.
Other treatment is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Proper nutrition can be maintained intravenously. If the adrenal glands are not functioning properly, medications may be used to supplement the hormones normally produced by these glands.
A team approach for individuals with Wolman disease may be necessary and may include special social support and other medical services. Genetic counseling is recommended for affected individuals and their families.
Investigational therapies
In the medical literature, a few children with Wolman disease were treated with hematopoietic stem cell transplantation (HSCT). Hematopoietic stem cells are specialized cells found in the bone marrow (the soft spongy material found in long bones). These blood stem cells grow and eventually develop into one of the three main types of blood cells– red blood cells, white blood cells or platelets. A transplant is done to replace the bone marrow (and consequently the whole blood system) of an affected individual with marrow from a person who does not have a particular disorder. The healthy cells produced by the new marrow contain sufficient levels of lysosomal acid lipase required to breakdown cholesterol and triglycerides. Individuals with Wolman disease treated with hematopoietic stem cell transplantation have shown dramatic improvement of existing symptoms and avoidance of additional complications such as liver failure. Researchers speculate that early diagnosis and prompt treatment with a hematopoietic stem cell transplant increases the chances of preserving liver function and preventing cognitive decline. More research is necessary to determine the long-term safety and effectiveness of this potential therapy for infants with Wolman disease. Hematopoietic stem cell transplants are not without drawbacks. The procedure is expensive and carries the risk of serious complications including graft-versus-host disease and other long-term and late effects.
Researchers have been studying enzyme replacement therapy for lysosomal storage diseases such as Wolman disease. Enzyme replacement therapy involves replacing a missing enzyme in individuals who are deficient or lack the particular enzyme in question. Synthetic versions of missing enzymes have been developed and used to treat individuals with certain lysosomal diseases including Hurler syndrome, Fabry syndrome and Gaucher disease.
Gene therapy is also being studied as another possible approach to therapy for some lysosomal storage disorders. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the production of active enzyme and prevent the development and progression of the disease in question. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a “cure.” However, at this time, there are many technical difficulties to resolve before gene therapy can succeed.
Wolman disease prognosis
Wolman disease is usually fatal by age 1 10.
References- Wolman Disease. https://rarediseases.org/rare-diseases/wolman-disease
- Lysosomal acid lipase deficiency. https://ghr.nlm.nih.gov/condition/lysosomal-acid-lipase-deficiency
- Erwin AL. The role of sebelipase alfa in the treatment of lysosomal acid lipase deficiency. Therapeutic Advances in Gastroenterology. 2017; 10(7):553-562. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5484437
- Gramatges MM, Dvorak C, Regula D, Enns G, Weinberg K, Agarwal R. Pathological evidence of Wolman’s disease following hematopoietic stem cell transplantation despite correction of lysosomal acid lipase activity. Bone Marrow Transplant. 2009;44:449–50.
- Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant. 2009;43(1):21–27. doi:10.1038/bmt.2008.273
- Hoffman EP, Barr ML, Giovanni MA, et al. Lysosomal Acid Lipase Deficiency. 2015 Jul 30 [Updated 2016 Sep 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK305870
- Shome DK, Al-Jishi E, Greally JF, Malik N, Zainaldeen HA, Das NS. The Middle-East connection of Wolman Disease. Saudi Med J. 2002;23:597–601.
- Browne M, Somers G, Savoia H, Kukuruzovic R. Wolman’s disease in an infant. Br J Haematol. 2003;122:522.
- Nchimi A, Rausin L, Khamis J. Ultrasound appearance of bowel wall in Wolman’s disease. Pediatr Radiol. 2003;33:284–5.
- Acid Lipase Disease Information Page. ttps://www.ninds.nih.gov/Disorders/All-Disorders/Acid-Lipase-Disease-Information-Page





{kind=link}