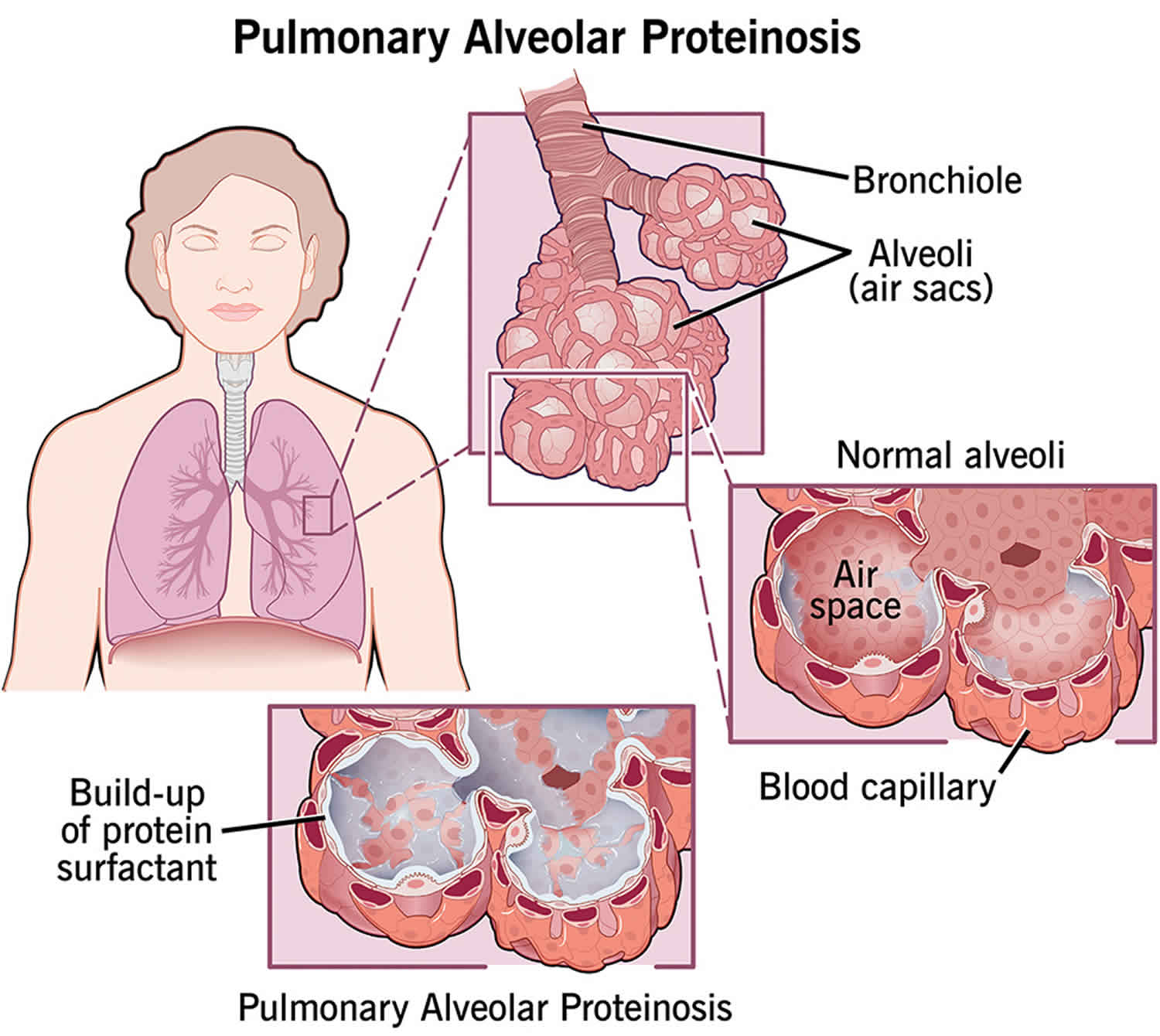
Pulmonary alveolar proteinosis
Pulmonary alveolar proteinosis also known as pulmonary alveolar phospholipoproteinosis or alveolar lipoproteinosis, is a rare disease characterized by an accumulation of a lipoproteinaceous material in the air sacs of the lungs, called the alveoli 1. The alveoli are the part of the lungs that contain air. It is there that gases between the lungs and the blood are exchanged.
Pulmonary alveolar proteinosis is a rare lung disease 2. Prevalence has been reported to be from 3.7 to 40 cases per million depending on the country 3. The incidence has been estimated to be 0.2 cases per million 3.
Pulmonary alveolar proteinosis can be congenital (something you are born with), secondary, and autoimmune 4. All 3 of these pathways result in decreased clearance of surfactant, rather than increased production 3.
- Autoimmune pulmonary alveolar proteinosis is the most common pathophysiologic mechanism accounting for 90% of documented cases 3. Autoimmune pulmonary alveolar proteinosis is initiated by immunoglobulin (Ig)-G anti-granulocyte macrophage colony stimulating factor (anti-GM-CSF) antibodies, which decrease functional alveolar macrophages 4.
- Secondary pulmonary alveolar proteinosis lacks anti-GM-CSF antibodies 5 but has decreased functional macrophages secondary to hematological malignancies (myelodysplastic syndrome, chronic myelogenous leukemia, among others) or primary immunodeficiency diseases (common variable immunodeficiency, DiGeorge syndrome, among others) 3.
- Congenital pulmonary alveolar proteinosis is the least common and results from genetic mutations in GM-CSF receptor proteins or surfactant proteins 6.
Autoimmune pulmonary alveolar proteinosis accounts for approximately 90% of cases, while 4% is secondary pulmonary alveolar proteinosis, 1% is congenital pulmonary alveolar proteinosis, and undetermined pulmonary alveolar proteinosis-like disease represents the remaining 5% 7. Smoking is reported in 53% to 85% of pulmonary alveolar proteinosis patients 8.
In autoimmune pulmonary alveolar proteinosis, males are more commonly affected than females at a 2:1 ratio 7. Median age at time-of-diagnosis is reported from 39 years to 51 years, although ages range from newborn to 72 years old 3.
Secondary pulmonary alveolar proteinosis has been reported with a slightly younger median age at time-of-diagnosis; 37 years to 45 years 5. Gender-ratio is also different in secondary pulmonary alveolar proteinosis, with a 1.2:1 male to female ratio 5.
Pulmonary alveolar proteinosis causes
Autoimmune pulmonary alveolar proteinosis
Autoimmune pulmonary alveolar proteinosis is a direct consequence of antibodies against anti-granulocyte macrophage colony stimulating factor (anti-GM-CSF) 9. There has been speculation that cigarette smoke or infectious diseases stimulate the development of these autoantibodies due to a high prevalence of smoking and infections in patients with pulmonary alveolar proteinosis 10. However, no causal link has been found between cigarette smoke and autoimmune pulmonary alveolar proteinosis 10. Additionally, no causal link has been found between infections and autoimmune pulmonary alveolar proteinosis 10.
Secondary pulmonary alveolar proteinosis
Secondary pulmonary alveolar proteinosis is caused by any disease that reduces the functionally effective alveolar macrophage population 6. In one retrospective analysis, 34.1% of secondary pulmonary alveolar proteinosis was associated with myelodysplastic syndrome, and 15.2% was associated with chronic myelogenous leukemia 5. Acute myeloid leukemia also has a well-established association with pulmonary alveolar proteinosis 3. Less frequent are associations between pulmonary alveolar proteinosis and acute lymphoid leukemia, lymphoma, and myeloma 3. Few case reports have associated pulmonary alveolar proteinosis with non-hematological cancers such as lung cancer, mesothelioma, and glioblastoma 3. Rare reports have associated pulmonary alveolar proteinosis with immunodeficiencies such as severe combined immunodeficiency, agammaglobulinemia, adenosine deaminase deficiency, common variable immunodeficiency, DiGeorge syndrome, dermatomyositis, rheumatoid arthritis, Behcet’s disease, AIDS, GATA2 deficiency, and organ transplantation 11. Secondary pulmonary alveolar proteinosis has also been associated with inhalation of several environmental exposures 3. These environmental exposures include silica, talc, cement, kaolin, aluminum, titanium, indium, and cellulose 3. Several animal studies have induced pulmonary alveolar proteinosis following inhalation of aluminum, fiberglass, indium, nickel, quartz, silica, and titanium 3. French analysis of pulmonary alveolar proteinosis patients found that 39% had an environmental exposure of cement, cereal dust, copper, epoxy, paint, polyvinyl chloride, silica, welding, wood dust, zirconium, or smoke 3. A report from Japan showed that 23% of patients with pulmonary alveolar proteinosis had significant environmental exposures 3. A Korean study reported 53% of pulmonary alveolar proteinosis patients were smokers and 48% had exposure to dust 8. These dust exposures included metal (26.5%), stone or sand (20.6%), chemical or paint (17.7%), farming dust (14.7%), diesel (14.7%), textile (2.9%), and wood (2.9%) 8. One report suggests a link between indium inhalation and anti-GM-CSF antibodies, suggesting these toxic inhalations might induce autoantibodies 3.
Secondary pulmonary alveolar proteinosis cases may also accompany the following disorders 12:
- Infections: infectious causes can include bacteria (Nocardia), fungi, viruses, Mycobacteria, or Pneumocystis carinii
- Neoplasms including lymphomas and leukemias
- Inorganic dust exposure including silicosis and aluminum
- Immunodeficiencies such as HIV infection, lung transplantation, and IgA deficiency
In secondary pulmonary alveolar proteinosis, it is believed that the underlying hematological disease causes either a reduction in the number of alveolar macrophages or a reduction in the functional quality of these macrophages 3. At least 3 patients with acute myeloid leukemia have been found to have a defect in the expression of GM-CSF receptor on alveolar macrophages 3. Following successful treatment of underlying leukemia, the receptor expression returned to normal in these individuals 3.
Congenital pulmonary alveolar proteinosis
Hereditary dysfunction in one of many proteins responsible for surfactant regulation causes congenital pulmonary alveolar proteinosis 6. This includes mutations in the GM-CSF receptor alpha-subunit or beta-subunit, surfactant protein B, surfactant protein C, ATP-binding cassette 3, NK2 homeobox 1, or the lysinuric protein intolerance disease 6.
In congenital pulmonary alveolar proteinosis, surfactant accumulation is a consequence of a genetic mutation resulting in dysfunctional GM-CSF receptor activation 3. In humans, the GM-CSF receptor is composed of an alpha and a beta subunit, each which corresponds to the genes CSF2RA and CSF2RB respectively 3. Mutations in CSF2RA have been reported in young children (newborn to 9 years) with an autosomal recessive inheritance pattern with incomplete penetrance 3. Mutations in CSF2RB are less common and have been reported from newborns up to 36 years old 3. Either mutation leads to an increased GM-CSF level in BALF and serum [4]. Congenital pulmonary alveolar proteinosis has also been described in lysinuric protein intolerance, which is an autosomal recessive disease caused by a mutation in SLC7A7 and leads to defective transport of amino acids at the epithelial membrane 3. This disease presents at an early age with gastrointestinal, renal, and less commonly pulmonary manifestations 3. SLC7A7 mutations lead to dysfunctional arginine transport and dysfunctional alveolar macrophages 13. Although SLC7A7 is a target of GM-CSF, which upregulates it, the GM-CSF pathway does not seem to be altered in this disease or lead to pulmonary alveolar proteinosis 13. Mutations in surfactant protein B, surfactant protein C, ATP-binding cassette 3, or NK2 homeobox can all lead to dysfunctional surfactant release from type II epithelial cells and dysfunctional clearance from the alveoli 14.
Whatever the inciting factor, all of these pathways lead to the same end: accumulation of lipoproteinaceous material in the alveoli due to dysfunctional clearance by alveolar macrophages or type 2 epithelial cells.
Pulmonary alveolar proteinosis symptoms
Pulmonary alveolar proteinosis has the following symptoms:
- Shortness of breath, also called dyspnea
- Chest pain or tightness
- Fever
- Weight loss
- Cough (sometimes, but not always)
- Low levels of oxygen in the blood
- Nail clubbing (abnormal growth of toenails or fingernails)
Clinical presentation of pulmonary alveolar proteinosis varies from indolent to emergent and symptoms are often non-specific 3. Pulmonary proteinosis is asymptomatic in one-third of the patients 12. In symptomatic patients, symptoms are non-specific consisting mainly of cough, dyspnea, fatigue, weight loss, hemoptysis and chest pain. Dyspnea is the most common presenting complaint; present in 39% of patients in one report 3. A cough was reported in 21% of patients 3. Hemoptysis, fever, and chest pain are rare complaints of autoimmune pulmonary alveolar proteinosis and should prompt consideration of another diagnosis 3. Fever may be present in 24% of secondary pulmonary alveolar proteinosis patients due to concomitant hematological malignancies or opportunistic infections 3. Thirty-three percent of patients may be asymptomatic at the time of presentation 4.
A majority of patients are smokers (53% to 85%) 3. Many report occupational exposures (39% to 48%) 8. Even though 90% of pulmonary alveolar proteinosis cases are autoimmune, only 1.7% of patients have other identified autoimmune diseases 3. Physical examination is frequently normal; however, patients can present with cyanosis (25% to 30%), clubbing (30%), or inspiratory crackles (50%) 3.
Pulmonary alveolar proteinosis complications
Patients with pulmonary alveolar proteinosis are at an increased risk of developing an opportunistic infection with approximately 5% of pulmonary alveolar proteinosis patients developing one 15. The lung is the most common site of infection, although extra-pulmonary infections represent 32% of opportunistic infections in pulmonary alveolar proteinosis patients 15. Typical bacterial cases of pneumonia are possible but infrequently reported in pulmonary alveolar proteinosis 15. Nocardia and Mycobacterium tuberculosis are the 2 most commonly reported opportunistic infections in pulmonary alveolar proteinosis 15. Fungal infections reported include Histoplasma, Aspergillus, Cryptococcus, and Blastomyces 15. Other reported infections include Acinetobacter, Coccidiodies, Mucorales, and Streptomyces15.
Pulmonary alveolar proteinosis diagnosis
Given that the clinical presentation of pulmonary alveolar proteinosis is nonspecific, diagnosis of this disease demands appropriate serological, radiological, and bronchoscopic evaluation.
Radiography
Chest radiography may demonstrate bilateral alveolar opacities in a peri-hilar and basilar distribution without air-bronchograms 3. This is sometimes referred to as a “batwing distribution,” or may resemble pulmonary edema without cardiomegaly or pleural effusions 4. Computed tomography reveals intralobular thickening and diffuse ground-glass opacities 3. This pattern is often referred to as “crazy paving.” “Crazy paving” is highly suggestive of pulmonary alveolar proteinosis although not specific or sensitive enough to confirm the diagnosis 4. Lower-lobe predominance has been reported in 22% of cases 3. Pulmonary nodules, mediastinal adenopathy, and focal parenchymal consolidations are absent in pulmonary alveolar proteinosis and should prompt consideration of a different diagnosis 11. Secondary pulmonary alveolar proteinosis is less likely to have interlobular septal thickening (only 33.3% of cases) 5. Congenital pulmonary alveolar proteinosis is also less likely to present with interlobular septal thickening and additionally may present with lung cysts 3.
Biomarkers
Several biomarkers have been studied in pulmonary alveolar proteinosis, including surfactant protein A, B, and D levels, cytokeratin 19, serum carcinoembryonic antigen, serum lactate dehydrogenase, GM-CSF levels, anti-GM-CSF antibodies, and KL-6 6. Serum lactate dehydrogenase is elevated in 50% of pulmonary alveolar proteinosis patients and serum carcinoembryonic antigen is also often elevated 3. Serum levels of the surfactant protein A, B, and D are all increased in pulmonary alveolar proteinosis and seem to be associated with disease severity 3. Overall, these biomarkers have not proven sensitive or specific for diagnosis 3. Testing for IgG anti-GM-CSF antibodies is the only clinically relevant biomarker to date 16. Detection of these antibodies is done via enzyme-linked immunosorbent assay (ELISA) which is the gold-standard and has been validated 17. Serum antibody levels can also be detected via latex-agglutination test with 100% sensitivity and 98% specificity 10. An anti-GM-CSF antibody level of 2.8 micrograms/ml or greater is abnormal and is consistent with pulmonary alveolar proteinosis 18.
Pulmonary Function Testing
Pulmonary function testing is not necessary for diagnosis, nor is it specific for pulmonary alveolar proteinosis; however, the most common pattern on spirometry is a restrictive pattern 3. Spirometry may be normal in 30% of patients with pulmonary alveolar proteinosis and may have a mixed obstruction and restrictive pattern in those who are smokers 3. A significant reduction in diffusion capacity and an increase in the alveolar-arterial gradient are the most common findings on pulmonary function testing in patients with pulmonary alveolar proteinosis 3.
Bronchoscopy
When pulmonary alveolar proteinosis is suspected, bronchoscopy with bronchoalveolar lavage is the gold standard for diagnosis 4. In approximately 75% of suspected cases, BALF examination will confirm a diagnosis 10. The lavage return will often appear milky and opaque 6. Bronchoalveolar lavage fluid cytological examination will reveal large foamy macrophages with amorphous material that stains PAS-positive 10. BALF cellularity is typically lymphocyte-predominant 10. Secondary pulmonary alveolar proteinosis may be associated with opportunistic infections which can lead to mixed cellularity, microbial growth, and potential for a missed diagnosis of pulmonary alveolar proteinosis (only 61.9% of secondary pulmonary alveolar proteinosis cases diagnosed with bronchoscopy) 5.
Lung Biopsy
Biopsies are not necessary for a diagnosis of pulmonary alveolar proteinosis but can be helpful 3. Open-lung biopsy or video-assisted thoracic surgery is rarely done but will demonstrate PAS-positive lipoproteinaceous material 6. Transbronchial biopsies were found to positively diagnose pulmonary alveolar proteinosis in 42% of cases in one study 3. There is at least one report of a bronchoscopic cryobiopsy resulting in a diagnosis of pulmonary alveolar proteinosis in a patient with otherwise normal bronchoalveolar lavage fluid and negative for anti-GM-CSF antibodies 19.
Pulmonary alveolar proteinosis treatment
The current standard of care for autoimmune pulmonary alveolar proteinosis is whole lung lavage 3. The decision to treat is based upon signs of poor gas exchange and symptoms of respiratory distress 4. Patients with mild dyspnea or no symptoms can do well with supportive care and monitoring of pulmonary function tests and chest imaging 3. Whole lung lavage should be considered in patients with dyspnea at rest, resting PaO2 less than 65 mm Hg, resting alveolar-arterial gradient greater than 40 mm Hg or oxygen desaturations on 6-minute walk test 4. whole lung lavage was first performed in 1961 3. Today, the procedure is done under general anesthesia with a double lumen endotracheal tube 4. One lung is ventilated while the contralateral lung is lavaged with warm saline (37 °C) and then allowed to drain to gravity 3. This process is continued until the returning fluid is clear 3. An average of 15 liters of saline is required 3. The procedure is repeated on the contralateral lung after 48 hours or more 3. A global analysis of whole lung lavage therapy showed that centers differ in the time between lavaging the contralateral lung (50% wait 1 to 2 weeks), the choice of lung to begin with, the position of the patient (50% supine, 50% lateral decubitus), chest percussion use during the procedure, and amount of saline used (5 to 40 L) 20. Retrospective studies of whole lung lavage have shown several parameters improve after therapy. Symptomatic improvement occurs in 85% of cases 3. Radiographic findings also improve 3. Forced expiratory volume in 1 second (FEV1) improves by 0.26 L, forced vital capacity (FVC) improves by 0.5 L, diffusion capacity improves by 4.4 mL, and alveolar-arterial gradient improves by 30 mm Hg 3. Retrospective data also suggests a mortality benefit (94% at 5 years with whole lung lavage compared to 85% at 5 years without whole lung lavage) 3. whole lung lavage has not been verified in randomized prospective trials 10. Approximately 50% of patients will require a second whole lung lavage procedure 5. The average duration of benefits following whole lung lavage is 15 months 10. A review found that 70% of patients underwent whole lung lavage within 5 years of diagnosis and that on average patients required two whole lung lavages 13.
GM-CSF Replacement Therapy
Clinical trials for GM-CSF replacement therapy were performed in the late 1990s 10. Subcutaneous injections of GM-CSF showed a positive response in 48% of a small cohort of 25 patients 3. Improvement following GM-CSF injections was much slower than the standard whole lung lavage, and therefore, this therapy has fallen out of favor 3. In a small trial of 12 patients of inhaled GM-CSF therapy, 11 of patients (91%) showed improvement 3. However, a larger subsequent trial of 35 patients resulted in only 68% of patients showing improvement 3. These therapies are safe and show some improvement in autoimmune pulmonary alveolar proteinosis; they were ineffective in congenital pulmonary alveolar proteinosis and inferior to whole lung lavage 6. These are currently considered alternative therapies to whole lung lavage 10.
Immunomodulation Therapy
Systemic corticosteroids were trialed in autoimmune pulmonary alveolar proteinosis but were ineffective, and they were found to increase the risk of pulmonary infections 3. Plasmapheresis has been reported to decrease circulating anti-GM-CSF in 2 cases successfully 3. One of these patients demonstrated significant clinical improvements while the other did not 3. Rituximab, an anti-CD20 therapy, was studied in 2010 in 10 patients 3. Eight of these patients had significant clinical improvement despite no change in serum anti-GM-CSF levels 3. Both Rituximab and plasmapheresis are considered alternative therapies for pulmonary alveolar proteinosis refractory to whole lung lavage 3.
Secondary pulmonary alveolar proteinosis
The only proven therapy for secondary pulmonary alveolar proteinosis is the treatment of the underlying disease 6. Case reports have demonstrated return of functional alveolar macrophages following the appropriate treatment of leukemia 3. whole lung lavage does not seem to benefit this population as only two of 14 patients studied had clinical improvement 3. Allogeneic hematopoietic stem cell transplant is a potential treatment option with two reported cases of complete resolution of secondary pulmonary alveolar proteinosis after transplant 11.
Congenital pulmonary alveolar proteinosis
Whole lung lavage may benefit patients with congenital pulmonary alveolar proteinosis, but is not curative 3. GM-CSF replacement therapies have not shown effectiveness 3. Corticosteroids and steroid-sparing agents such as azathioprine have been used at some centers with mixed results 3. Animal models show promise with gene therapy and direct pulmonary macrophage transplantation resulting in complete resolution of pulmonary alveolar proteinosis 6. At least one patient has had successful hematopoietic stem cell transplant, which cured their pulmonary alveolar proteinosis, but this therapy carries potential risks of graft-versus-host disease and opportunistic infections 9.
Pulmonary alveolar proteinosis prognosis
Pulmonary alveolar proteinosis disease course varies, and the prognosis is unpredictable. When treated with whole lung lavage, the 5-year survival of autoimmune pulmonary alveolar proteinosis is 95% 3. Previously spontaneous remission had been reported as frequently as 50% of the time 21. However, more recent and more extensive analyses have concluded that spontaneous remission occurs in less than 10% of patients 20. In one prospective cohort study of 39 asymptomatic pulmonary alveolar proteinosis patients, 64% of patients remained stable while 7% progressed 3. In a retrospective analysis of mortality in pulmonary alveolar proteinosis patients, 72% of deaths occurred as a direct result of respiratory failure while 20% were due to secondary infections 10. The prognosis for secondary pulmonary alveolar proteinosis is worse with a median time for survival less than 15 months 5. The causes of death in secondary pulmonary alveolar proteinosis patients include the underlying hematological disease (33%), infection (25%), respiratory failure (25%), and bleeding complications (13%) 3.
References- Mlika M, Triki M, Kwas H, Braham E, Ghedira H, Mezni F. When the bronchoalveolar lavage makes the diagnosis of interstitial pneumonia. Clin Respir J. 2018 Jan;12(1):352-353.
- McCarthy C, Lara Gallego B, Trapnell BC, McCormack FX. Epidemiology of Rare Lung Diseases: The Challenges and Opportunities to Improve Research and Knowledge. Adv. Exp. Med. Biol. 2017;1031:419-442.
- Borie R, Danel C, Debray MP, Taille C, Dombret MC, Aubier M, Epaud R, Crestani B. Pulmonary alveolar proteinosis. Eur Respir Rev. 2011 Jun;20(120):98-107.
- Kamboj A, Lause M, Duggirala V. Severe Pulmonary Alveolar Proteinosis in a Young Adult. Am. J. Med. 2018 May;131(5):e199-e200.
- Zhang D, Tian X, Feng R, Guo X, Wang P, Situ Y, Xiao Y, Xu KF. Secondary pulmonary alveolar proteinosis: a single-center retrospective study (a case series and literature review). BMC Pulm Med. 2018 Jan 25;18(1):15.
- McElvaney OJ, Horan D, Franciosi AN, Gunaratnam C, McElvaney NG. Pulmonary alveolar proteinosis. QJM. 2018 Mar 01;111(3):185-186.
- Campo I, Mariani F, Rodi G, Paracchini E, Tsana E, Piloni D, Nobili I, Kadija Z, Corsico A, Cerveri I, Chalk C, Trapnell BC, Braschi A, Tinelli C, Luisetti M. Assessment and management of pulmonary alveolar proteinosis in a reference center. Orphanet J Rare Dis. 2013 Mar 13;8:40.
- Hwang JA, Song JH, Kim JH, Chung MP, Kim DS, Song JW, Kim YW, Choi SM, Cha SI, Uh ST, Park CS, Jeong SH, Park YB, Lee HL, Shin JW, Lee EJ, Jegal Y, Lee HK, Park JS, Park MS. Clinical significance of cigarette smoking and dust exposure in pulmonary alveolar proteinosis: a Korean national survey. BMC Pulm Med. 2017 Nov 21;17(1):147.
- Frémond ML, Hadchouel A, Schweitzer C, Berteloot L, Bruneau J, Bonnet C, Cros G, Briand C, Magnani A, Pochon C, Delacourt C, Cavazzana M, Moshous D, Fischer A, Blanche S, Blic J, Neven B. Successful haematopoietic stem cell transplantation in a case of pulmonary alveolar proteinosis due to GM-CSF receptor deficiency. Thorax. 2018 Jun;73(6):590-592.
- Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N. Engl. J. Med. 2003 Dec 25;349(26):2527-39.
- Tanaka-Kubota M, Shinozaki K, Miyamoto S, Yanagimachi M, Okano T, Mitsuiki N, Ueki M, Yamada M, Imai K, Takagi M, Agematsu K, Kanegane H, Morio T. Hematopoietic stem cell transplantation for pulmonary alveolar proteinosis associated with primary immunodeficiency disease. Int. J. Hematol. 2018 May;107(5):610-614.
- Mlika M, Basit H, Kaul P. Alveolar Proteinosis. [Updated 2019 Oct 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537215
- Barilli A, Rotoli BM, Visigalli R, Bussolati O, Gazzola GC, Kadija Z, Rodi G, Mariani F, Ruzza ML, Luisetti M, Dall’Asta V. In Lysinuric Protein Intolerance system y+L activity is defective in monocytes and in GM-CSF-differentiated macrophages. Orphanet J Rare Dis. 2010 Nov 26;5:32.
- Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 2010;61:105-19.
- Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J. Infect. 2012 Aug;65(2):173-9.
- Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc. Natl. Acad. Sci. U.S.A. 1994 Jun 07;91(12):5592-6.
- Uchida K, Nakata K, Carey B, Chalk C, Suzuki T, Sakagami T, Koch DE, Stevens C, Inoue Y, Yamada Y, Trapnell BC. Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis. J. Immunol. Methods. 2014 Jan 15;402(1-2):57-70.
- Nishimura M, Yamaguchi E, Takahashi A, Asai N, Katsuda E, Ohta T, Ohtsuka Y, Kosaka K, Matsubara A, Tanaka H, Yokoe N, Kubo A, Konno S, Baba K. Clinical significance of serum anti-GM-CSF autoantibody levels in autoimmune pulmonary alveolar proteinosis. Biomark Med. 2018 Feb;12(2):151-159.
- Gando S, Duré R, Violi D, Vazquez B, Labarca G, Fernandez-Bussy S. Bilateral lung disease, extensive and diffuse. Diagnosis of pulmonary alveolar proteinosis by bronchoscopic cryobiopsy. Respir Med Case Rep. 2017;22:260-262.
- Campo I, Luisetti M, Griese M, Trapnell BC, Bonella F, Grutters J, Nakata K, Van Moorsel CH, Costabel U, Cottin V, Ichiwata T, Inoue Y, Braschi A, Bonizzoni G, Iotti GA, Tinelli C, Rodi G., WLL International Study Group. Whole lung lavage therapy for pulmonary alveolar proteinosis: a global survey of current practices and procedures. Orphanet J Rare Dis. 2016 Aug 31;11(1):115.
- Matsuura H, Yamaji Y. Pulmonary Alveolar Proteinosis: Crazing-Paving Appearance. Am. J. Med. 2018 Apr;131(4):e153-e154.





{kind=link}