Arbovirus
Arboviruses also known as arthropod-borne viruses, are viruses that maintain transmission cycles between vertebrate animal reservoirs as main amplifying hosts and insects as primary vectors 1. Arboviruses must replicate in the arthropod vectors, such as mosquitoes, ticks, midges or sandflies, prior to transmission. Female mosquitoes acquire arbovirus during blood feeding of an infected animal and the arbovirus replicates in the mesenteronal epithelial cells. The arbovirus released from the mesenteronal epithelial cells infects salivary glands after secondary amplification in other cells and tissues. Some arboviruses can infect the salivary glands without secondary amplification in other cells and tissues. Subsequently, the arbovirus released from salivary gland epithelium is transmitted during blood feeding of the vertebrate host. Arbovirus or arthropod-borne virus infections are increasingly important causes of neurologic disease in the United States through both endemic transmission and travel-associated infections 2.
Arboviruses are included in different taxonomic families, including Flaviviridae (genus Flavivirus), Bunyaviridae (genus Nairovirus, Orthobunyavirus, Phlebovirus, and Tospovirus), Togaviridae (genus Alphavirus), Rhabdoviridae (genus Vesiculovirus), Orthomyxoviridae (genus Thogotovirus), and Reoviridae (genus Orbivirus and Coltivirus) (see Figure 1). Many of the important zoonotic arboviruses belong to the families Togaviridae and Flaviviridae 3. However, there are many other clinically important human and animal arboviruses belonging to the Bunyaviridae family, such as Crimean-Congo hemorrhagic fever virus (tick-borne) in the genus Nairovirus 4 and Toscana virus (sandfly-borne) and Rift Valley fever virus (mosquito-borne) in the genus Phlebovirus 5. Colorado tick fever virus in the family Reoviridae (genus Coltivirus) is also an important human arbovirus 6. A zoonosis or zoonotic disease is defined as a disease or infection that is naturally transmitted from vertebrate animals to humans 7.
Figure 1. Classification of arboviruses
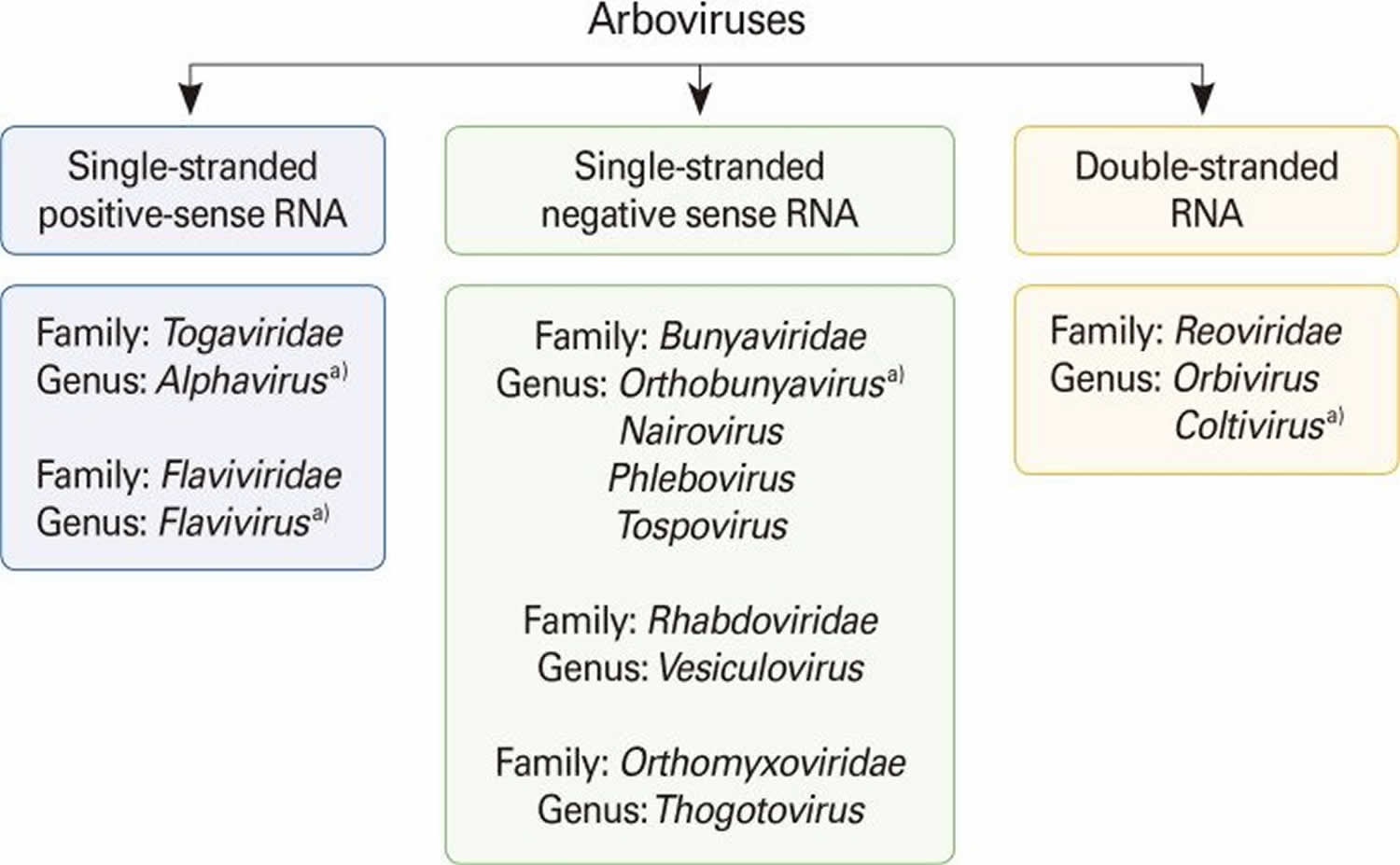
Footnote: Arboviruses are included in six different taxonomic virus families. a)Arboviruses that cause human encephalitides belong to four genera in four virus families.
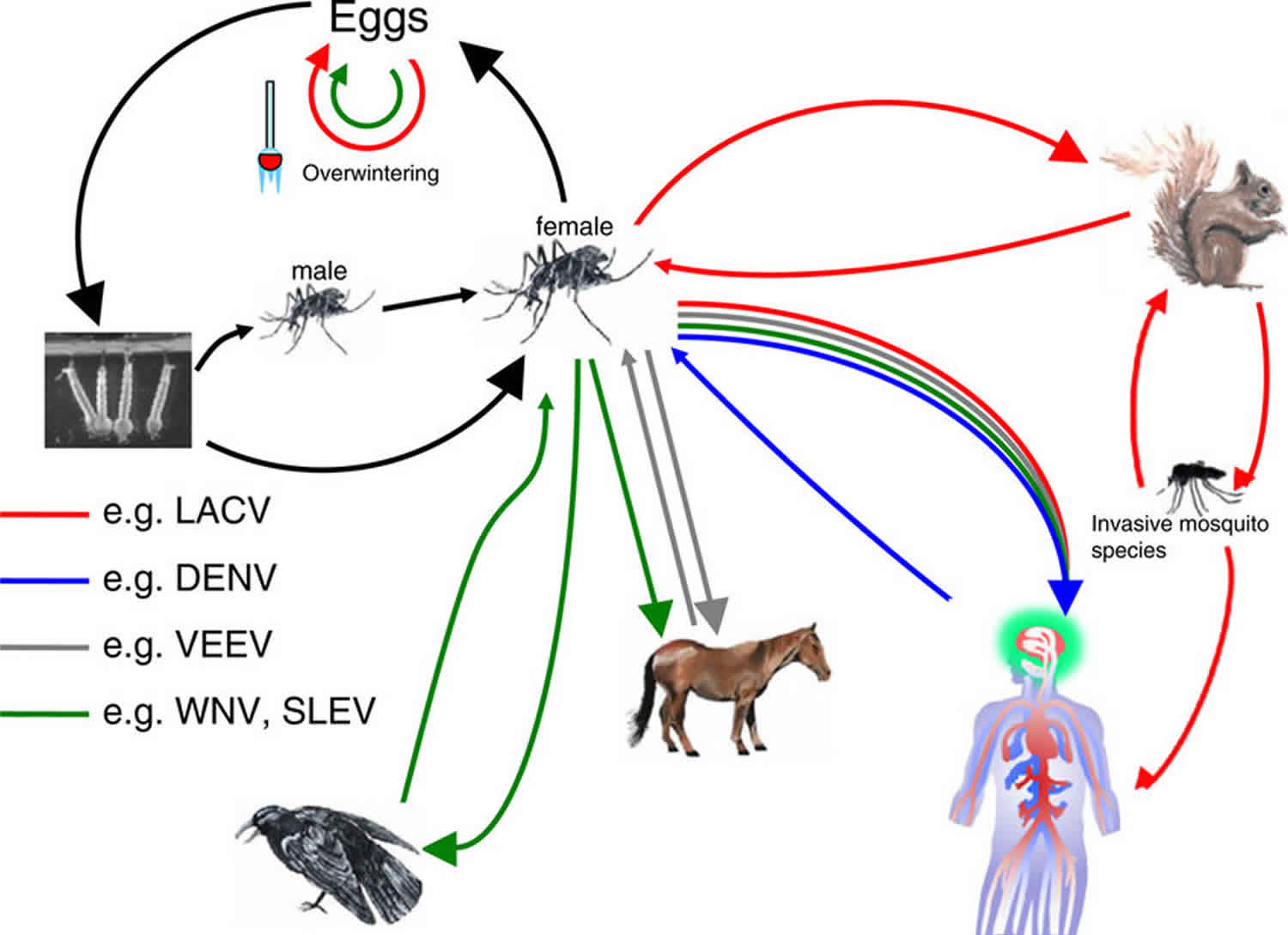
[Source 1 ]Arboviruses are maintained in complex life cycles involving nonhuman primate/vertebrate hosts and primary arthropod vectors (see Figure 2). Mosquitoes are the most important vectors that transmit zoonotic viruses. Different mosquito species (Culex spp., Aedes spp., etc.) may act as vectors for the same virus in different vertebrate hosts depending on different geographical and ecological locations. Ticks, sandflies (Phlebotomus spp.) and gnats (Culicoides spp.) are also important vectors of some arboviruses. Vertical transmission (transovarial and transstadial) occurs in some arthropod vectors as they transmit some arboviruses from parent arthropod to offspring arthropods (see Figure 3). This type of transmission mainly occurs in tick-borne encephalitis viruses (TBEVs) but it has been also reported in some mosquito-borne viruses 8. For example, La Crosse virus, one of the most important viruses among agents causing California encephalitis, is transmitted by its main vector, Aedes triseriatus, not only by transovarial and transstadial routes but also sexually 9.
Most known arboviruses were first isolated in tropical regions such as Africa and South America and in some Asian countries. However, the geographic distribution and frequency of epidemic outbreaks of arboviral diseases have expanded dramatically across the world in the past several decades 10. Several factors such as changes in viral genetics, host and/or vector population, and climate changes facilitated expansion and transmission of arboviruses resulting in emergence/reemergence of arboviral disease outbreaks in new regions in the world. Extensive tropical urbanization and faster and increased movement of humans and animals with modern transportation helped vectors to be in closer contact with vertebrate reservoir hosts raising transmission potential. Introduction of West Nile virus into the New World and the emergence of Japanese encephalitis virus in Australia are a few prominent examples of recent unexpected emerging/reemerging zoonotic diseases 11.
Epidemics/epizootics of humans and domestic animals usually occur when the enzootic virus is introduced into rural environments or comes to close contact with humans by a bridge vector (Figure 2). Usually, humans and domestic animals develop clinical disease but do not develop a sufficient level of viremia to infect arthropods, thus, they are considered dead-end hosts and do not contribute to the transmission cycle 12. However, some arboviruses such as dengue fever, yellow fever, and chikungunya viruses cause high levels of viremia in humans and can be transmitted from person to person by mosquitoes (urban cycle) (Figure 2) 13.
Figure 2. Arbovirus vertebrate host and vector transmission cycles
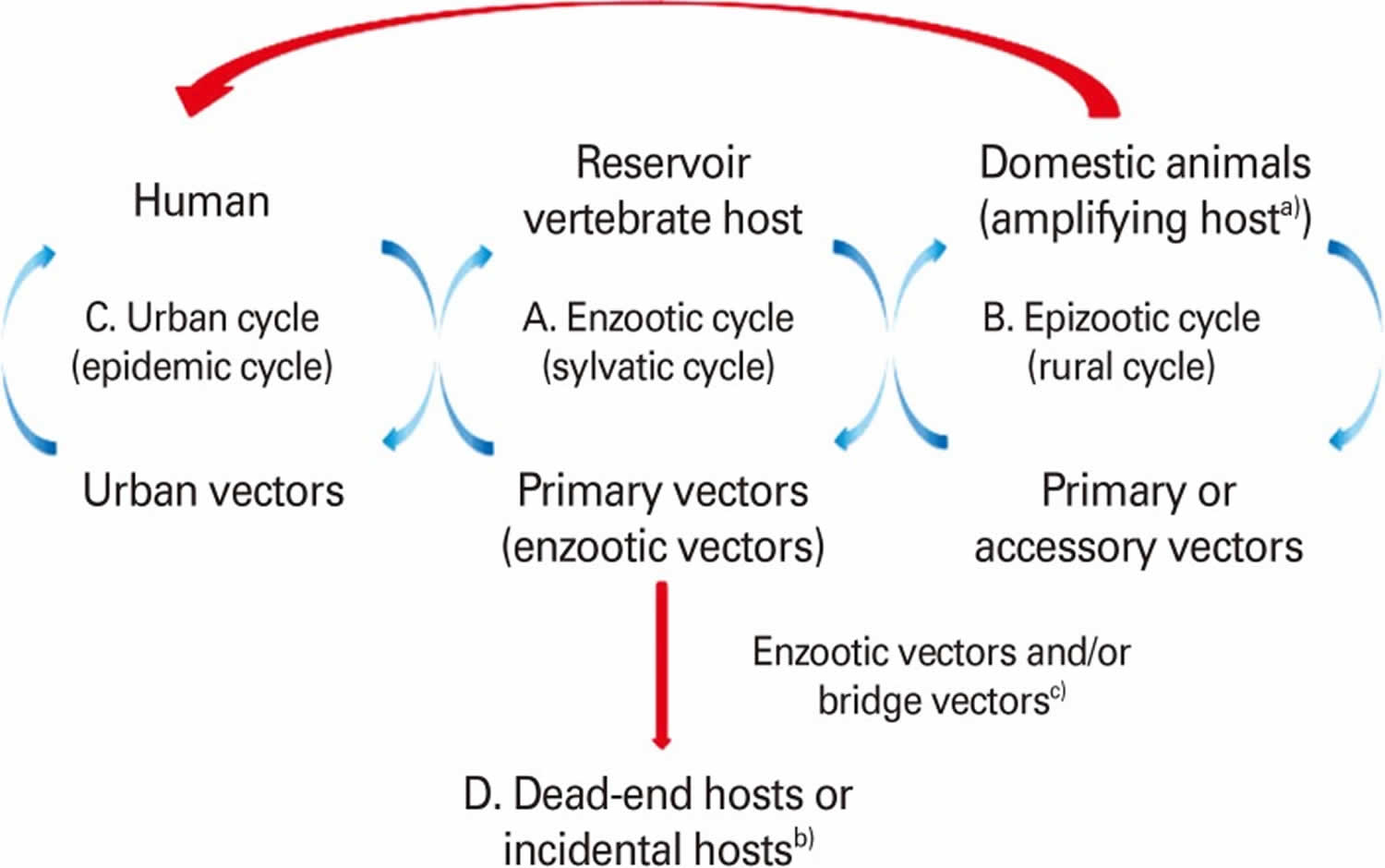
Footnote: (A) Enzootic cycle (sylvatic or jungle cycle). The natural transmission of virus between wild animals (vertebrate hosts) and primary or enzootic insect vectors and that leads to the amplification of the virus in the vector. The vertebrate host is the reservoir host that can harbor a virus indefinitely with no ill effects. Therefore, reservoir host is the primary host of a virus and may be re-infected several times during their life.
(B) Epizootic cycle (rural cycle). The virus is transmitted between non-wild or domestic animals and the primary or accessory insect vectors. This can lead to an epidemic outbreak of viral disease in a domestic animal population where the virus is amplified (amplifying host), often with the implication that it may extend to humans by insect vectors (e.g., Japanese encephalitis virus, Venezuelan equine encephalitis virus [VEEV]).
(C) Urban cycle. Humans are the source of infection for mosquitoes due to high level of viremia. The virus cycles between humans and insect vectors (urban vector e.g., A. aegypti) repeatedly, as reinfection occurs with every new insect bite (e.g.,dengue virus, yellow fever virus, St. Louis encephalitis virus, Venezuelan equine encephalitis virus (VEEV), chikungunya virus, Rift Valley fever virus).
(D) Humans are dead-end hosts in the infection chain and do not develop sufficient viremia and do not serve for amplification of the virus to be transmitted again to insect vector (e.g., Eastern equine encephalitis virus, Western equine encephalitis virus, West Nile virus, and Sindbis virus).
a) Amplifying host is in which the level of virus can become high enough that an insect vector such as a mosquito that feeds on it will probably become infectious.
b) Dead-end host or incidental host is an intermediate host that does generally not allow transmission of the virus to the definitive host. They do not develop sufficient viremia to be picked up by the insect vectors. c)Bridge vector is an arthropod that acquires virus from an infected wild animal and subsequently transmits the agent to human or secondary host.
[Source 1 ]Figure 3. Arbovirus vertical transmission cycle
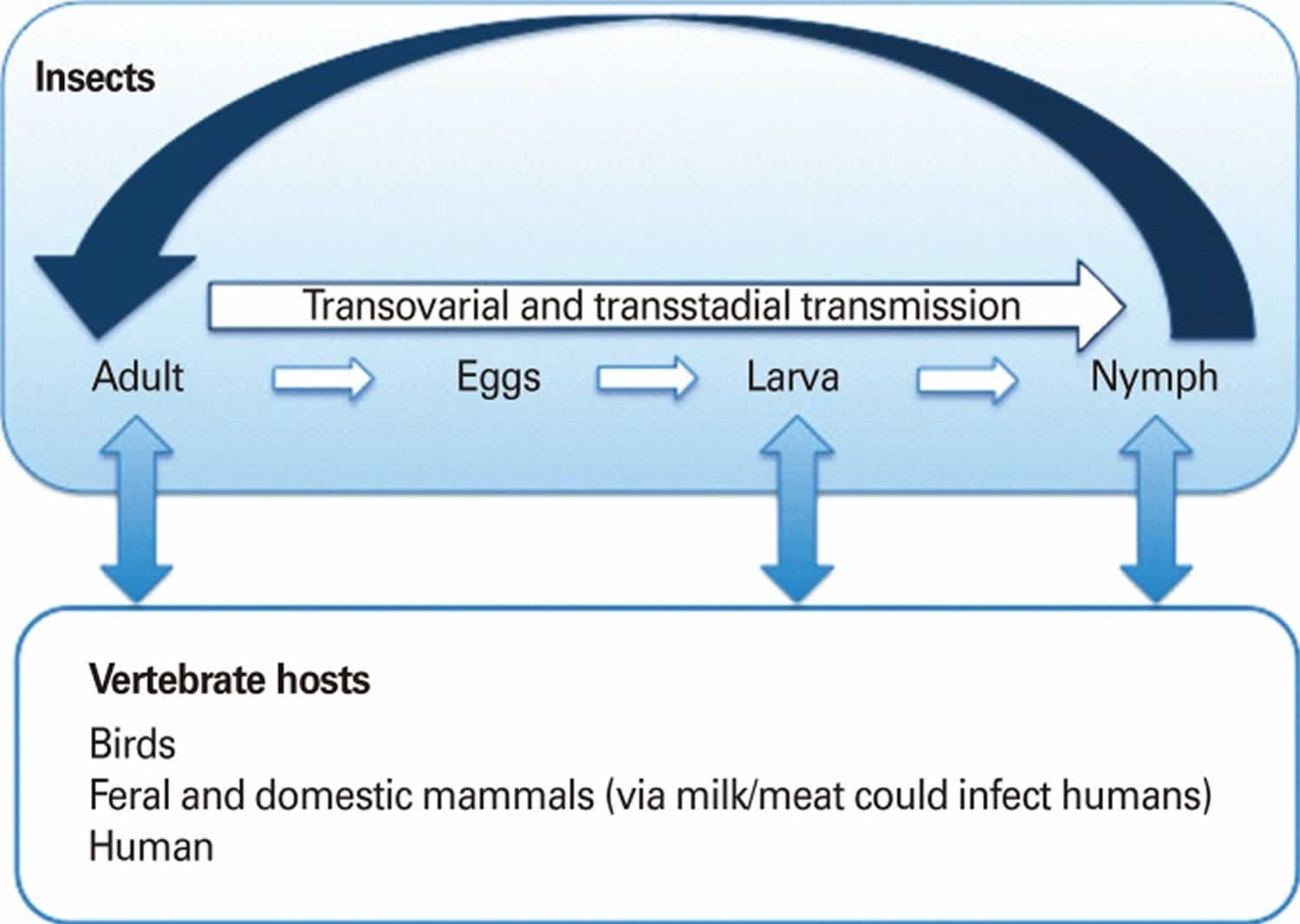
Footnote: Vertical transmission cycle (transovarial and transstadial). A vertical transmission exists in some arthropod vectors and is epidemiologically important. This type of transmission is found in viruses that belong to tick-borne encephalitis complex. However, it is also found in some of the mosquito-transmitted viruses (e.g., La Crosse encephalitis, Murray Valley encephalitis, St. Louis encephalitis, Japanese encephalitis,West Nile, and Western equine encephalitis).
[Source 1 ]Arbovirus causes
Arboviruses are spread to vertebrate hosts by hematophagous arthropod vectors. Transmission occurs via biological transfer, requiring successful replication in vector species as well as adequate
viremia in the host before transmission is achievable. As of 1992, 535 virus species belonging to 14 virus families are registered in the International Catalog of Arboviruses 14, and new viruses are being described on a regular basis 15. Of these species, greater than 100 are known to cause zoonotic diseases, mainly in four virus families: Togaviridae, Flaviviridae, Bunyaviridae, and Reoviridae 14. While the majority of arboviruses circulate in tropical and subtropical regions, many arboviruses also have been introduced and thrive within temperate regions. These viruses, along with their vector species, have spread exponentially in their geographical distributions in accordance with global trade routes and industrialization 16. This post targets arboviruses transmitted by mosquitoes that have high public health importance and risk, namely chikungunya virus (Togaviridae), dengue virus (Flaviviridae), Zika virus (Flaviviridae), yellow fever virus (Flaviviridae), Japanese encephalitis virus (Flaviviridae), and West Nile virus (Flaviviridae). Combined, these viruses account for hundreds of millions of clinical/symptomatic infections globally, resulting in tens of thousands of deaths per year 17. However, symptoms in humans and animals range from mild to subclinical infection all the way to encephalitic or hemorrhagic, so the total number of cases per year may be underestimated (Table 1). In addition, due to the paucity of data on nutrition and arbovirus infection, other viruses of concern will also be mentioned where literature is available, including La Crosse virus (Bunyaviridae), Sindbis virus (Togaviridae), Ross River virus (Togaviridae), Western equine encephalitis virus (Togaviridae), Rift Valley Fever virus (Bunyaviridae), and St. Louis encephalitis virus (Flaviviridae).
Table 1. Vectors, hosts, symptomology and estimated numbers of cases and deaths of selected arboviruses

Abbreviations: DENV = dengue virus; CHIKV = chikungunya virus; JEV = Japanese encephalitis virus; WNV = West Nile virus; YFV = yellow fever virus; ZIKV = Zika virus.
[Source 17 ]Table 2. Epidemiologic and clinical characteristics of arbovirus infections
| Family/Virus | Vector | Geographic Distribution | Clinical Presentation | At-Risk Populations | Diagnosis |
|---|---|---|---|---|---|
| Flaviviridae/West Nile virus | Mosquito: Culex pipiens | Continental United States, southern Europe | Fever, myalgia, meningismus, altered mental status | Elderly and immunocompromised, seasonal exposure | Serology and CSF IgM, PCR of nucleic acid in immunocompromised |
| Flaviviridae/dengue virus | Mosquito: Aedes aegypti | Worldwide in tropics and subtropics | Fever, myalgia, arthralgia | Children and travelers | Clinical presentation, acute and convalescent serology |
| Flaviviridae/St Louis encephalitis virus | Mosquito: C. pipiens | Southeastern United States | Fever, myalgia, meningismus, altered mental status | Elderly, seasonal exposure | Serology and CSF IgM |
| Flaviviridae/ Powassan virus | Tick: Ixodes species | Northern central and eastern United States | Fever, headache, vomiting, meningismus, altered mental status | Outdoor exposure, seasonal exposure to ticks | Serum and CSF IgM antibody production |
| Togaviridae/eastern equine encephalitis virus | Mosquito: Culex and Aedes species | Eastern seaboard of United States, swamplands | Fever, meningismus, altered mental status, cranial neuropathies | Geographic exposure during epidemic cycle | Acute and convalescent serology, IgM-positive serum, CSF IgM |
| Togaviridae/ Venezuelan equine encephalitis virus | Mosquito: Aedes and Culex species | Central America and South America | Fever, headache, sore throat, altered mental status | Geographic exposure during epidemic cycle | Acute and convalescent serology, IgM-positive serum, CSF-positive IgM, PCR for viral nucleic acid |
| Togaviridae/ Chikungunya virus | Mosquito: A. aegypti | Worldwide in tropics and subtropics | Fever, headache, gastrointestinal symptoms, myalgia, arthralgia, arthritis | Geographic exposure | Acute and convalescent serology, CSF IgM, PCR for viral nucleic acid in serum and CSF |
| Bunyaviridae/La Crosse virus | Mosquito: Aedes species | Midwestern United States | Fever, headache, vomiting, altered mental status, seizures | Children | Acute and convalescent serology, CSF IgM |
Abbreviations: CSF = cerebrospinal fluid; IgM = immunoglobulin M; PCR = polymerase chain reaction.
[Source 2 ]Arboviral diseases key points 2
- Less than 1% of West Nile virus–infected individuals develop neuroinvasive disease.
- West Nile virus neuroinvasive disease can manifest as meningitis, encephalitis, acute flaccid paralysis, or a combination of syndromes.
- West Nile virus neuroinvasive disease is more common in individuals older than 60 years of age.
- CSF from patients with West Nile virus neuroinvasive disease may exhibit a neutrophil-predominant pleocytosis.
- West Nile virus encephalitis most commonly causes MRI T2-signal abnormalities in the deep gray nuclei.
- West Nile virus neuroinvasive disease is often diagnosed by measurement of CSF IgM.
- Dengue virus infections are associated with encephalitis and peripheral nerve syndromes.
- Cases of eastern equine encephalitis virus and Venezuelan equine encephalitis virus encephalitis occur during localized epidemic transmission.
- Chikungunya virus is a rapidly emerging virus with rare associations with neurologic disease.
- Chikungunya virus is associated with meningitis and encephalitis during acute infection.
- La Crosse virus causes cases of encephalitis in children.
- Mortality following infection from the California encephalitis group of viruses is low, and outcomes are typically favorable.
Flaviviruses (Flaviviridae)
Flaviviruses formerly group B arboviruses, include agents pathogenic for humans and animals that are transmitted by mosquitoes or ticks 18. Flaviviruses are enveloped viruses containing a single-stranded positive-sense RNA genome of approximately 11 kb in length. The viral genome encodes three structural proteins (capsid [C], premembrane [prM], and envelope [E] proteins) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) 19. The genus Flavivirus is comprised of more than sixty-six virus species, many of which are arthropod-borne human pathogens (mainly mosquitoes and ticks) and are highly pathogenic for both humans and animals 20. Flaviviruses are further classified into antigenic complexes and subcomplexes 21 that are related serologically, genetically, and etiologically. The diseases caused by flaviviruses range from fevers and encephalitides to hemorrhagic fever. There are three complexes of flaviviruses that include important agents pathogenic for humans 20.
‘Virus complexes transmitted by mosquitoes’ (Japanese encephalitis virus and related encephalitis virus complex)—the mosquito-borne flaviviruses infect a variety of animal species and humans, with birds being most important to the enzootic transmission cycle of many of these viruses. Culex mosquitoes are the main vectors of these viruses.
The following flaviviruses are members of this group 20:
- Japanese encephalitis virus in Southeast Asia,
- West Nile virus (WNV; West Nile fever/encephalitis) in Africa, North America, Europe, and Asia,
- St. Louis encephalitis virus (SLEV; St. Louis encephalitis) in North and South America,
- Kunjin virus (KUN; Kunjin virus encephalitis) in Australia,
- Murray Valley encephalitis virus (MVEV; Murray Valley encephalitis) in Australia and New Guinea,
- Rocio virus (Rocio encephalitis) in South America, and
- Usutu virus in Africa and Europe.
‘Virus complexes transmitted by ticks’ (tick-borne encephalitis complex)—Rodents are the most important vertebrate hosts. Hedgehogs, deer, and livestock may also be inapparently infected. Ixodes, Dermacentor, and Haemaphysalis ticks are the principal vectors of these viruses.
The following flavivirus zoonoses are transmitted by ticks 21, 20:
- Tick-borne encephalitis, European subtype (Central European encephalitis),
- Tick-borne encephalitis, Eastern subtype (Russian spring-summer encephalitis),
- Powassan encephalitis and Modoc virus encephalitis in North America,
- Louping ill in Scotland and Negishi virus encephalitis in Japan,
- Omsk hemorrhagic fever in Siberia,
- Kyasanur Forest disease in India, and
- Alkhurma hemorrhagic fever in Saudi Arabia.
‘Agents causing yellow fever and dengue which form two closely related virus complexes’—Simians and humans are viremic hosts and Aedes mosquitoes are the vectors.
Following are the zoonotic diseases caused by viruses in this complex 20:
- Yellow fever virus (YFV; yellow fever) in Central Africa and South America,
- Dengue virus (DENV) serotypes 1-4 in Asia, Africa, and Central and South America (dengue hemorrhagic fever, dengue shock syndrome), and
- Wesselsbron fever in Africa.
Figure 4. Enzootic and epizootic transmission cycles of Japanese encephalitis virus and West Nile virus
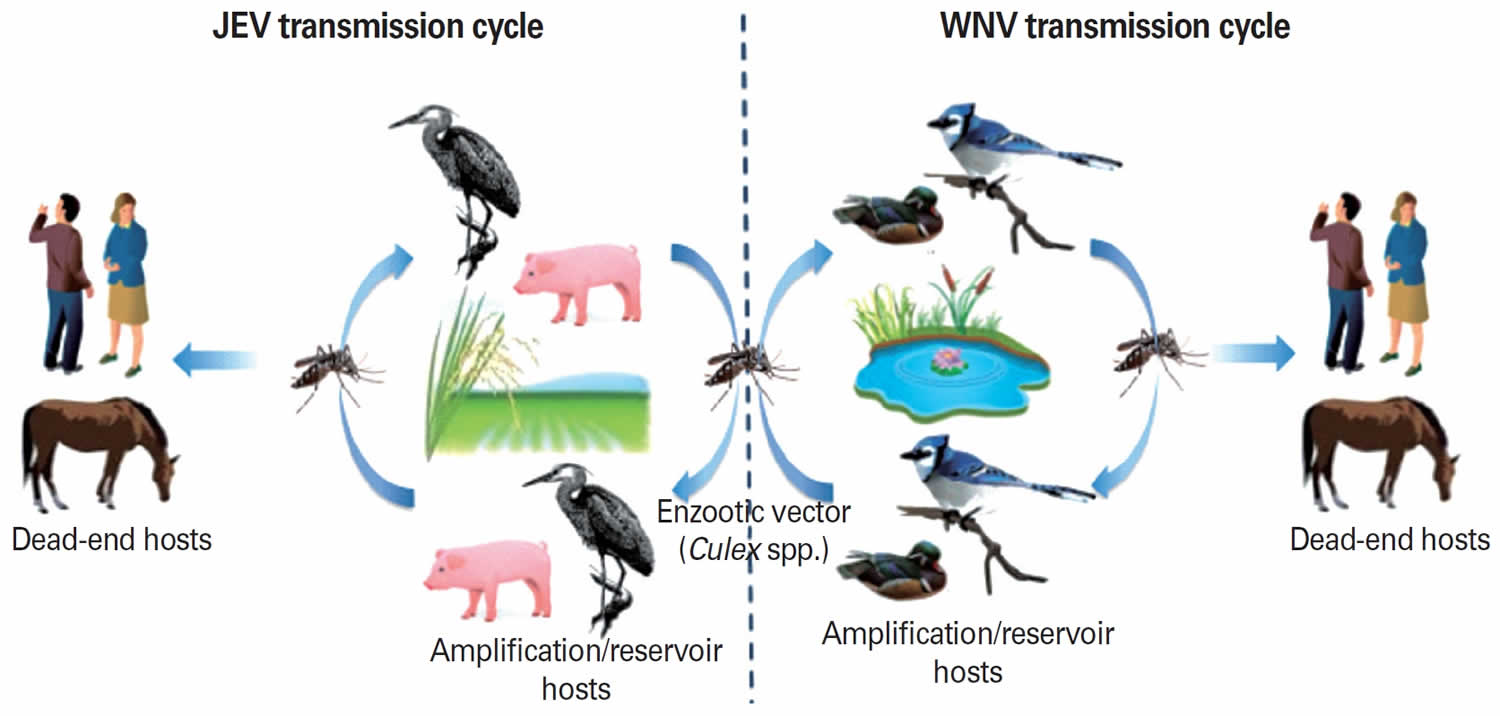
Footnote: Enzootic and epizootic/epidemic transmission cycles of Japanese encephalitis virus (JEV) and West Nile virus (WNV). Left: Japanese encephalitis virus (JEV) is transmitted by primarily Culex tritaeniorhynchus. Pigs and aquatic birds are the efficient amplification and reservoir hosts of JEV that develop high-titered viremia. Humans and horses are dead-end hosts since the level of viremia is insufficient for mosquito transmission. Right: West Nile virus (WNV) maintains an enzootic transmission cycle between Culex mosquitoes and birds (reservoir host). Horses, humans, and other mammals infected in a spillover transmission are “dead-end” hosts.
[Source 1 ]Japanese encephalitis virus
Japanese encephalitis formerly known as Japanese B encephalitis. is the most common mosquito-transmitted encephalitic disease in Asian countries where it is regarded as a major health threat 22. Japanese encephalitis virus was first documented as viral encephalitis in the 1870s and isolated in 1935 from the brain of a fatal human encephalitis case in Tokyo, Japan 23. Recently, cases of Japanese encephalitis virus have been reported in Pakistan, Papua New Guinea, and Australia indicating expansion of the virus into new geographic regions 24. Japanese encephalitis virus is classified into one single serotype with at least four distinct genotypes (I-IV) 25. Genotype I strains have been isolated in Australia, Cambodia, China, Thailand, Japan, Korea, Taiwan, and Vietnam since 1967. Japanese encephalitis virus strains isolated in Australia, Indonesia, Malaysia, Papua New Guinea, and Thailand between 1951 and 1999 were included in Genotype II. Genotype III isolates have been found in temperate areas of Asia including China, Japan, Taiwan, and the Philippines since 1935, and genotype IV was found only in Indonesia in 1980 and 1981. Recently, Genotype V has been suggested for an isolate from the Muar region of Malaysia 26. Genotypes I and III frequently occur in epidemic regions whereas genotypes II and IV are mostly associated with endemic transmission 27.
Up to 70% of adults in tropical regions of Asia have Japanese encephalitis virus antibodies. An estimated 50,000 cases occur every year where the most acutely infected patients are children or non-immune individuals 28. Clinical infections are severe with a case fatality rate of 30% or severe neurologic sequels in up to 50% of the patients 29. Japanese encephalitis virus infections occur all year long in South Asian countries while they occur during summer in temperate regions 30. The infection in animals is subclinical, however, it causes abortion in pregnant sows and death in newborn piglets 31. Japanese encephalitis virus causes inapparent infection in horses, but neurologic signs are observed occasionally with a high fatality rate. Seroepidemiological studies show that Japanese encephalitis virus can incidentally infect a variety of other vertebrates, including dogs, ducks, chickens, cattle, bats, snakes, and frogs 22.
Japanese encephalitis virus transmission cycle
Japanese encephalitis virus is maintained in a zoonotic cycle and transmitted by mosquitoes,primarily Culex tritaeniorhynchus 31. This mosquito vector mainly hatches in rice fields in India and other Asian countries, with multiplication increasing up to 50% in fertilized fields. Japanese encephalitis virus also can be transmitted by C. annulus, C. annulirostris, and Aedes mosquitoes 31. Pigs and aquatic birds are the efficient amplification and reservoir hosts of Japanese encephalitis virus that develop high-titered viremia providing a source of infection for mosquitoes 31. In enzootic regions, pigs, frogs, and waterbirds (e.g., egrets and herons) increase the risk of transmission to humans and equines, especially in agricultural settings such as rice cultivation areas 31. The combination of increased rice crops and pig farming gives Japanese encephalitis virus an epidemiological advantage. Humans and horses are dead-end hosts since the level of viremia is insufficient for mosquito transmission and they succumb to encephalitis after infection.
Japanese encephalitis virus vaccines
Despite the use of effective vaccines including both inactivated whole virus and live attenuated vaccines, Japanese encephalitis virus remains as an important cause of arthropod-transmitted viral encephalitis. The first Japanese encephalitis vaccines available were inactivated vaccines prepared in mouse brains or primary hamster kidney cells with protection efficacy of 76% to 95% 32. Due to safety concerns, the inactivated vaccines were replaced by the live-attenuated vaccine SA14-14-2. Recently, a new purified inactivated Japanese encephalitis vaccine derived from Vero cell-adapted SA14-14-2 strain (IXIARO, Intercell AG, Vienna, Austria) has been licensed in the US, Europe, Canada, and Australia 33. In addition, a live chimeric vaccine containing prM and E proteins of Japanese encephalitis virus in a backbone of attenuated YFV 17D strain was developed by Sanofi Pasteur (Chimerivax/IMOJapanese encephalitis virus, Lyon, France) 34. The Chimerivax/IMOJapanese encephalitis virus showed outstanding immunogenicity without concerning adverse effects, thus, it was recently licensed in Australia and is currently under review in Thailand 35.
West Nile Virus
Most arboviruses maintain an enzootic (animal) cycle that does not involve human infection. West Nile virus is maintained in an enzootic cycle between mosquitos such as Culex pipiens and passerine (perching) bird species such as jays and finches. After an infected mosquito bites a human, the virus replicates in dendritic cells and macrophages in local tissue and lymph nodes, resulting in viremia that eventually disseminates virus to end organs, including the central nervous system (CNS = brain and spinal cord) 36. Of note, West Nile viremia in humans is not high enough or sustained enough to support subsequent transmission to mosquitoes, so humans are dead-end hosts. The ability of an arbovirus to invade the central nervous system (neuroinvasiveness) is determined by multiple viral and host factors 37. Proposed routes of arboviral CNS entry include penetration of the cerebral microvasculature after infection of endothelial cells, diapedesis (movement of infected blood cells across brain capillary walls) of infected leukocytes, passage through the choroid plexus, or passage through fenestrated endothelial cells in the vasculature of specific regions in the CNS 36.
After crossing the blood-brain barrier, many arboviruses can directly infect and cause death of neurons.5 Several studies have shown that apoptosis is an important mechanism of West Nile virus–induced neuron cell death and CNS injury 38. Immune responses also contribute to clearance of virus but contribute to immune-mediated neuronal cell death as well 39.
West Nile virus infection is now the most common cause of epidemic viral encephalitis in the United States. Since its emergence in New York City in 1999, over 40,000 cases of West Nile virus infection have been reported in the United States, with over 17,000 cases (42%) of neuroinvasive disease and 1700 (4%) deaths 40. The epidemics of West Nile virus in 2003 and 2012 were the largest outbreaks of neuroinvasive viral infections ever reported in the Western Hemisphere 41.
Almost all cases of West Nile virus disease are caused by transmission through a mosquito bite. In most areas of the United States, the majority of cases occur between June and October, reflecting the peak activity period of biting mosquitoes. Occasional cases of West Nile virus disease also occur following transplantation of West Nile virus–infected organs 42, breast-feeding 43, and blood transfusions from asymptomatic West Nile virus–infected individuals 44. Currently, donor organs and blood products are screened for West Nile virus nucleic acids to prevent iatrogenic transmission.
Following transmission of West Nile virus, most infections (approximately 80%) are asymptomatic. Approximately 20% of infected individuals develop an acute febrile flulike illness (West Nile fever), characterized by fever, headache, fatigue, anorexia, nausea, myalgia, and lymphadenopathy. A maculopapular rash involving the trunk and limbs occurs in 25% to 50% of cases 45. Less than 1% of West Nile virus–infected individuals develop neuroinvasive disease, which can manifest as meningitis, encephalitis, acute flaccid paralysis, or a combination of these clinical syndromes 46. The symptomatology reflects the common predilection for the virus to injure the basal ganglia, thalamus, upper brainstem, and cerebellum as well as the anterior horns of the spinal cord. An estimated 30% to 40% of patients with neuroinvasive West Nile virus infection develop meningitis, 50% to 60% develop encephalitis, and 5% to 10% develop acute flaccid paralysis 47. Other reported syndromes include chorioretinitis,28 myositis,29 and autonomic nerve dysfunction 48.
Neuroinvasive disease most commonly occurs in older individuals (over 60 years of age). In one study, the odds ratio of developing encephalitis was 2.2 in individuals older than 64 years 49. Additional identified risk factors for encephalitis include hypertension and diabetes mellitus 50. Patients who are immunocompromised, including organ transplant recipients, are at high risk of developing severe West Nile virus disease. Specific genetic factors in humans shown to enhance susceptibility to serious West Nile virus disease include single nucleotide polymorphisms in the oligoadenylate synthetase gene, which encodes an interferon-inducible enzyme involved in antiviral innate immunity 51, and a genetic deficiency of the chemokine receptor CCR5, which may inhibit trafficking of West Nile virus–specific CD8+ T cells into the CNS 52.
West Nile virus meningitis is characterized by the abrupt onset of fever, headache, meningeal signs, photophobia, and phonophobia. Patients have a CSF pleocytosis with an average of 226 cells/μL, mildly elevated protein, and normal glucose. In one study, neutrophils, rather than lymphocytes, were found to predominate in the CSF in approximately 50% of patients with West Nile virus meningitis 53. In some cases of West Nile virus neuroinvasive disease, very atypical-appearing monocytes are noted in the CSF similar to Mollaret cells noted in cases of recurrent meningitis. In cases of West Nile virus meningitis, neuroimaging studies are unremarkable, and the EEG is usually normal.
West Nile virus encephalitis is distinguished from meningitis by the presence of signs and symptoms of brain parenchymal involvement on examination or diagnostic testing including neuroimaging or EEG. Results from multiple studies suggest that patients with West Nile virus encephalitis present with fever (70% to 100%), headache (50% to 100%), and altered mental status (45% to 100%) 54. Signs common in West Nile virus encephalitis but unusual in other forms of viral encephalitis include tremor, parkinsonism, and myoclonus (20% to 40%) 55. Weakness is common and may be of a lower motor neuron type associated with hypotonia and areflexia, with preserved sensation. Cranial neuropathies, most commonly involving unilateral or bilateral peripheral facial palsy, occur in approximately 20%.
The prevalence of tremors due to West Nile virus neuroinvasive disease ranges from 12% to nearly 100% 56. When present, tremors are described as coarse and isolated to the upper extremities and have postural and kinetic components 57. Parkinsonian features also occur with variable frequency and include signs of bradykinesia, cogwheel rigidity, hypomimia, and postural instability 56. Myoclonus can resemble that seen in prion diseases, and usually involves the upper extremities and face. Cerebellar abnormalities including incoordination and gait ataxia occur in a variable percentage of cases 56.
Patients with West Nile virus infection have a normal complete blood count or mild leukocytosis 58. The CSF findings in patients with West Nile virus encephalitis are almost identical to the findings with meningitis, including pleocytosis (mean 227 cells/μL), elevated protein, and normal glucose. Neutrophils predominate rather than lymphocytes in 37% of cases 52. MRI is abnormal in approximately 50% to 70% of West Nile virus encephalitis cases, with the frequency of positive MRI findings increasing with imaging later in the course of disease (more than 7 days after symptom onset) and increased sensitivity with use of T2-weighted fast spin echo and fluid-attenuated inversion recovery (FLAIR) sequences. When present, MRI abnormalities typically involve the thalamus, basal ganglia, and brainstem. CT is considerably less sensitive than MRI and is usually normal.
West Nile virus can also cause a poliomyelitislike acute flaccid paralysis that results from viral injury to motor neurons in the anterior horns of the spinal cord 59. Patients typically develop acute onset of asymmetric limb paralysis associated with decreased or absent reflexes and preserved sensation. Weakness may be associated with respiratory impairment from diaphragm or intercostal muscle paralysis. Electrophysiology studies obtained acutely show reduction in amplitude or absence of compound muscle action potentials with relatively preserved sensory nerve action potentials. EMG studies obtained 2 to 3 weeks after onset show characteristic features of denervation, including increased insertional activity and fasciculations. In contrast to Guillain-Barré syndrome, no evidence of significant demyelination (slowed conduction velocities or conduction block) is seen. In most, but not all, cases of West Nile virus, acute flaccid paralysis is associated with clinical signs and symptoms of systemic infection, and the syndrome may occur in association with meningitis or encephalitis. Patients typically have CSF features similar to the features seen in meningoencephalitis. MRI of the spinal cord may show increased signal in the anterior horns on T2 and FLAIR sequences.
West Nile virus neuroinvasive disease is usually diagnosed by demonstration of West Nile virus–specific IgM in CSF by enzyme-linked immunosorbent assay (ELISA) 60. In some patients, CSF West Nile virus IgM may persist for 1 year or longer, and it may be necessary to perform serial studies of serum and CSF IgG and IgM to definitively distinguish acute from remote infection. CSF polymerase chain reaction (PCR) for West Nile virus is highly specific, but less sensitive than serologic studies. CSF PCR may be particularly useful early in infection, before antibody responses have fully evolved, and in immunocompromised individuals who may have delayed or absent seroconversion 60. Antibodies reacting with West Nile virus antigens in ELISA tests may occur as a result of heterologous cross-reactions induced by infection with or vaccination against other flaviviruses, including St Louis encephalitis virus, yellow fever virus, and Japanese encephalitis virus. In some cases, it may be necessary to confirm ELISA results by plaque-reduction neutralization assays. Neutralization antibody titers are typically highest against the inciting virus compared with cross-reacting species.
No specific therapy of proven benefit for West Nile virus infection exists. Isolated case reports and small series describe both benefit and lack of effect from treatment with IVIg containing high-titer anti-West Nile virus antibodies and with interferon alfa. A multicenter randomized controlled trial of a high-titer anti–West Nile virus IVIg preparation was conducted by the Collaborative Antiviral Study Group and did not show evidence of a therapeutic benefit 61. A phase 2/3 trial to evaluate the safety and efficacy of a humanized monoclonal antibody directed against an epitope on the West Nile virus envelope glycoprotein was closed because of low enrollment (NCT00515385). Isolated reports of use of corticosteroids in patients with West Nile virus acute flaccid paralysis and brainstem disease do not permit any conclusions about efficacy 62.
Mortality from West Nile virus neuro-invasive disease is approximately 12% and occurs almost exclusively in the subsets of patients with severe encephalitis or severe acute flaccid paralysis. The frequency and severity of sequelae are still not well understood 63. Six months after the acute infection, 40% of patients with movement disorders such as myoclonus, parkinsonism, or tremors have residual symptoms, and 20% have ongoing symptoms at 18 months of follow-up 63. Up to 50% of West Nile virus encephalitis survivors report cognitive problems, decreased motor speed, and diminished dexterity 3 months after the initial infection 63.
Dengue virus
According to the World Health Organization (WHO), dengue virus causes approximately 50 million cases of disease annually throughout the world and continues to expand its geographic distribution throughout tropical and subtropical regions. Dengue virus isolates are divided into four different serotypes and are maintained in endemic cycles between humans and the Aedes aegypti mosquito. Dengue virus infection causes an acute self-limited febrile syndrome characterized by headache, retroorbital pain, rash, nausea, vomiting, diarrhea, myalgia, and arthralgia. In individuals with prior exposure to dengue virus, reexposure to another serotype places the individual at increased risk for dengue hemorrhagic fever, characterized by increased vascular permeability, thrombocytopenia, hypotension, and hemorrhagic manifestations.
Acute dengue virus infections are rarely associated with CNS involvement, and direct dengue virus infection of the nervous system remains controversial 64. A 2014 study in India and Nepal found that 9.2% of dengue infections were associated with a neurologic complication 64. Of the 45 patients in a prospective cohort study with neurologic manifestations associated with dengue virus infection, encephalitis was the most common presentation at 33% 64. Other associated neurologic abnormalities include encephalopathy (22%), myelitis, Guillain-Barré syndrome, myositis, and neuralgic amyotrophy 64. The diagnosis of dengue is often clinical in endemic locations. Diagnosis can be supported with serology using acute and convalescent serum for antibody to the dengue virus envelope protein. No known therapy exists for acute dengue virus infection. Among other problems, the development of dengue hemorrhagic fever with recurrent infection from new serotypes has hampered the development of efficacious vaccination strategies.
Powassan virus
Powassan virus is a rare tick-borne flavivirus maintained in an enzootic cycle between Ixodes ticks and small mammals 65. Powassan virus causes rare sporadic cases of neurologic disease in humans. In the United States, Powassan virus has been reported in the northeastern and north central states 65. Symptoms of infection vary from mild fever and myalgia to acute flaccid paralysis, encephalitis, and death 66.
St Louis encephalitis virus
St Louis encephalitis virus is another member of the Flaviviridae family, spread by mosquito vectors in southern Midwest states. Cases of St Louis encephalitis have rapidly decreased with the expansion of West Nile virus. According to Centers for Disease Control and Prevention (CDC) data, two cases of neuroinvasive St Louis encephalitis were reported in the District of Columbia in 2010. St Louis encephalitis causes neuroinvasive disease in a minority of patients, with very similar clinical patterns compared to West Nile virus.
Alphaviruses (Togaviridae)
The family Togaviridae is composed of enveloped single-stranded positive-sense RNA viruses. In North America, eastern equine encephalitis virus is the most important member of this group causing encephalitis and is, fortunately, rare. Chikungunya virus has recently emerged as the cause of massive epidemics of a febrile arthralgic illness that, in rare cases, is associated with neurologic manifestations. However, since some epidemics can involve millions of infected cases, even low-incidence involvement of the nervous system can result in significant numbers of affected individuals.
Eastern Equine Encephalitis virus
Eastern equine encephalitis virus is an alphavirus that causes a sporadic mosquito-borne viral infection endemic in the eastern United States and Caribbean. Four lineages of eastern equine encephalitis virus exist; group I causes most disease in humans, whereas groups IIA, IIB, and III cause primarily equine disease in Central America and South America 67. Eastern equine encephalitis virus is maintained in an enzootic cycle with avian species after a bite from a mosquito vector, Culiseta melanura 68. Birds serve as the primary reservoir hosts and amplifying hosts, and humans are incidentally infected by various mosquito bridging vectors, including Culex, Culiseta, and Aedes species 69. In the United States, most cases are along the eastern seaboard, and cases are found sporadically along the Gulf Coast, typically within 5 miles of swamplands or marshlands. Approximately 1 in 30 individuals exposed to eastern equine encephalitis virus develops disease 70. Eastern equine encephalitis virus causes sporadic infections in human populations during the summer months and occasional larger epidemic outbreaks.
Figure 5. Enzootic and epizootic transmission cycles of Eastern equine encephalitis virus
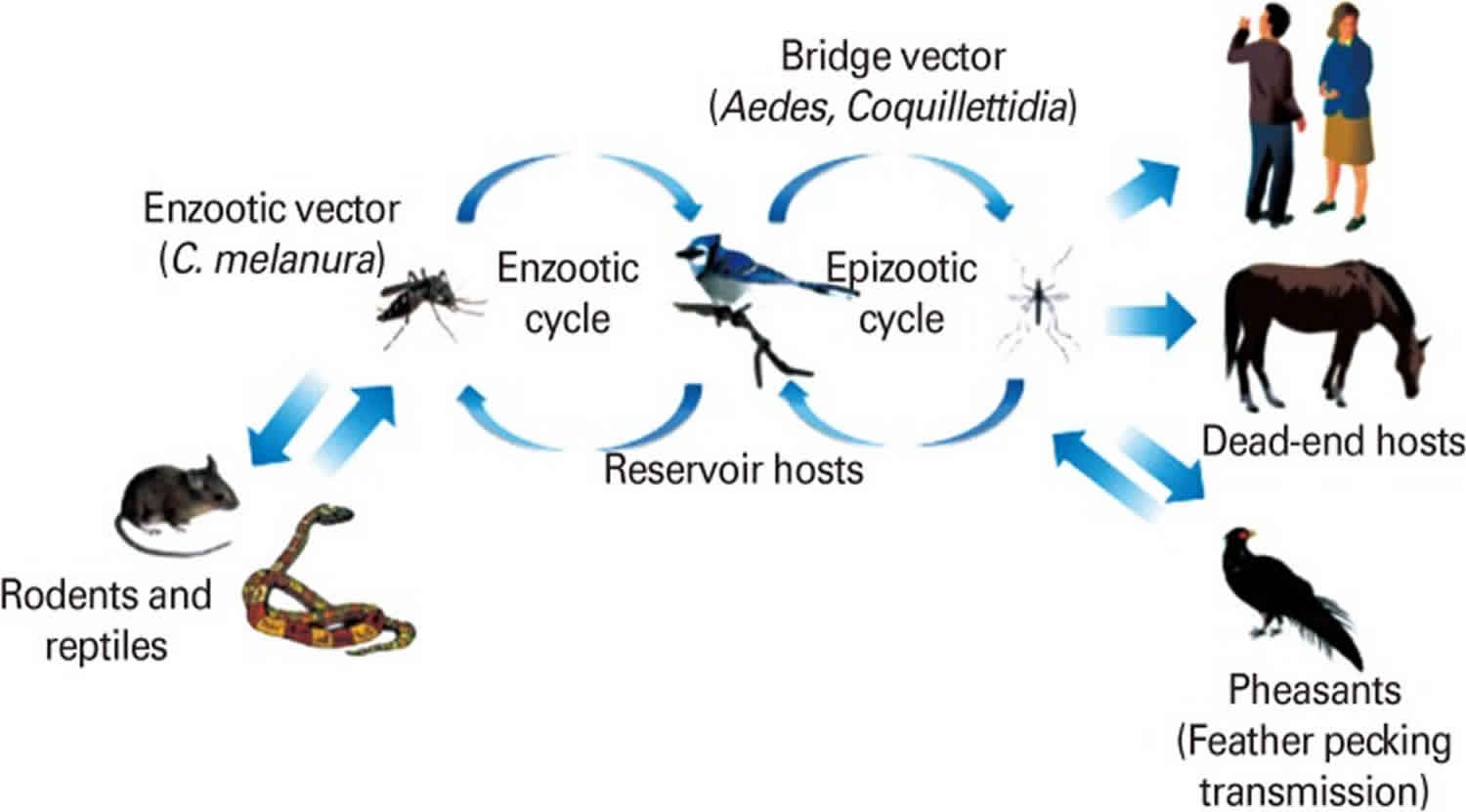
Footnote: Enzootic and epizootic/epidemic transmission cycles of Eastern equine encephalitis virus (EEEV). The enzootic Eastern equine encephalitis virus transmission cycle is maintained between passerine birds as reservoir/amplification hosts and Culiseta melanura, as the main enzootic vector in swamp habitats. Rodents/marsupials may serve as principal enzootic vectors and reservoirs in South America. Passerine birds develop extremely high levels of viremia, enough to infect both enzootic vectors as well as a variety of bridge vectors. Humans and equids are dead-end hosts since they do not develop sufficient viremia to transmit the virus.
[Source 1 ]Patients infected with eastern equine encephalitis virus develop nonspecific symptoms common to other causes of viral encephalitis, including fever, chills, malaise, and myalgia. The prodrome is followed by either recovery without neurologic illness or the onset of encephalitis characterized by severe headache, confusion, nausea, and vomiting. Seizures, focal neurologic deficits such as cranial nerve palsies or focal weakness, and meningismus are common findings 71. Brainstem involvement is common and is associated with gaze palsies, nystagmus, and pupillary abnormalities. Mortality is about 33% but increases to 50% in patients older than 60 years of age. Moderate to severe sequelae occur in one-third of survivors 71.
Similar to the flaviviruses above, MRI abnormalities occur predominantly in the thalamus, basal ganglia, and brainstem 71. EEG is typically diffusely slow, with some severely ill patients having burst suppression or diffuse high-voltage delta-wave slowing 72. Laboratory studies may show a peripheral leukocytosis with a neutrophil predominance in 69% and hyponatremia in 60% of patients 71. CSF typically shows a significant pleocytosis with a mean cell count of 370 leukocytes/μL in one study53 and 940 leukocytes/μL in another 72. Approximately 60% of eastern equine encephalitis virus cases will display a neutrophil predominance in the CSF with a median neutrophil proportion of 70% of nucleated cells 71. CSF protein is often elevated (median 97 mg/dL), and 90% of patients have CSF glucose concentrations less than 60% of coincident serum values. CSF red blood cells are common, reflecting the necrotic and hemorrhagic features of the encephalitis pathologically.
Diagnosis is typically made by demonstration of IgM antibodies in CSF by capture ELISA, demonstration of serum IgM antibodies, or a fourfold increase in IgG antibodies between acute and convalescent sera. No proven antiviral therapy exists for eastern equine encephalitis virus, and treatment is focused on supportive care and managing complications such as seizures and increased intracranial pressure. No commercial vaccine for eastern equine encephalitis virus exists, but standard precautions to prevent mosquito bites may help to prevent infection. In evaluating laboratory and imaging studies for prognostic value, one study found that CSF leukocytosis greater than 500 cells/μL and hyponatremia less than 130 mEq/L were predictive of a poor outcome 71, but a longer prodromal period was associated with a better prognosis. Sequelae may be more common and generally more severe in children 71.
Western equine encephalitis virus
Western equine encephalitis virus is genetically diverse and both epizootic and enzootic strains have been identified. Epizootic North American strains are more virulent than strains that are enzootic in South America (sporadic cases of Western equine encephalitis). Western equine encephalitis was the first equine encephalitic arbovirus identified in North America. It is closely related to Sindbis and Semliki Forest virus since it emerged from a recombination of viruses in the Eastern equine encephalitis and the Sindbis lineages 73. Western equine encephalitis virus was first isolated from brains of affected horses during an equine epizootic outbreak in the San Joaquin Valley of California in 1930 74. In 1938, the first lethal human infection of Western equine encephalitis virus was confirmed 75 and since then it spread to the west of North America and the American Midwest with periodic equine epizootics and epidemics 76. Epidemiological studies have shown that Western equine encephalitis virus occurs throughout most of the Americas from the western half of North to South America, including Guyana, Ecuador, Brazil, Uruguay, and Argentina 73. In South America, with the exception of Argentina, only small equine epizootics, but no human Western equine encephalitis cases have been reported 77.
Western equine encephalitis virus continues to cause equine encephalitis in northern South America and Central America with occasional outbreaks in Florida and the southwestern US, but only a few human cases of Western equine encephalitis have been reported, with low fatality rate, in the past several decades in North America 78. Most Western equine encephalitis virus infections in humans and equines occur in summer, June and July, and slightly later in temperate regions like Canada. Although most human cases of Western equine encephalitis are asymptomatic, infants and children are highly susceptible to Western equine encephalitis virus infection and are most likely to develop severe encephalitis. Clinical manifestations develop after 2 to 10 days of incubation and are characterized by nonspecific febrile viremia, malaise, and headache often in association with meningismus. The case fatality rate in humans is about 3% to 4% 79. The case fatality rate in horses is 20% to 30%, but can be up to 50% in some epidemics 80. For horses, Western equine encephalitis virus is less virulent than Eastern equine encephalitis virus.
In addition, Highland J virus, Fort Morgan virus, and related Buggy Creek virus, distinct but closely related to Western equine encephalitis virus, were also isolated in North America 81. Highland J virus has been identified in the eastern US (Florida) and is transmitted from C. melanura mosquitoes to songbirds in freshwater swamps 82. It has a low pathogenicity in mammals and is rarely seen in humans or horses. Exposure to Highland J virus has not been directly associated with human illness. However, Highland J virus can cause sporadic encephalitis in horses, and is also pathogenic to turkeys and partridges 83. Similar to Western equine encephalitis virus, Buggy Creek virus is a natural recombinant virus derived from Old World Sindbis virus and New World Eastern equine encephalitis virus 73. Buggy Creek virus (and the closely related Fort Morgan virus) is apparently widely distributed in North America, having been found in Texas, Oklahoma, Nebraska, ColoradoColorado, South Dakota, and Washington State. It was first isolated in 1980 at Buggy Creek in Grady County, Oklahoma. However, the ecologically very similar Fort Morgan virus was discovered in the 1970s in Colorado 84. Buggy Creek virus is commonly associated with the cimicid swallow bug (Oeciacus vicarius) 84. The bug is an ectoparasite of the colonially nesting cliff swallow (Petrochelidon pyrrhonota) and, to a lesser extent, the house sparrow (Passer domesticus), with both birds serving as hosts to Buggy Creek virus. Fort Morgan virus is also associated with swallow bugs, cliff swallows, and house sparrows. These two viruses are pathogenic to swallows but not to humans or horses. These four viruses in North America (Western equine encephalitis virus, Buggy Creek virus, Fort Morgan virus, and Highland J virus), the Aura virus in South America, and Sindbis virus with its four subtypes found in Africa, Asia, Australia, and Europe, are regarded as members of the Western equine encephalitis complex 85.
Western equine encephalitis virus transmission cycle
Western equine encephalitis virus is maintained in an enzootic cycle between passerine birds as reservoirs and its specific mosquito vector, Culex tarsalis, abundant in agricultural settings in the western US. Domestic and wild birds are considered important reservoir and epizootic amplifying hosts (Figure 6). It has been also suggested that lagomorphs and rodents can serve as amplification hosts when they are infected with Western equine encephalitis virus by Aedes mosquitoes 84.
Epizootic transmission to horses and humans is mediated by bridge vectors, such as Ochlerotatus melanimon in California, Aedes dorsalis in Utah and New Mexico and A. campestris in New Mexico. The seasonal continuation of the natural Western equine encephalitis virus transmission cycle in temperate regions is not clear. However, the annual reintroduction of migratory birds and vertical transmission among A. dorsalis mosquitoes are suspected for the maintenance mechanism in temperate regions 86.
Figure 6. Enzootic and epizootic transmission cycles of Western equine encephalitis virus
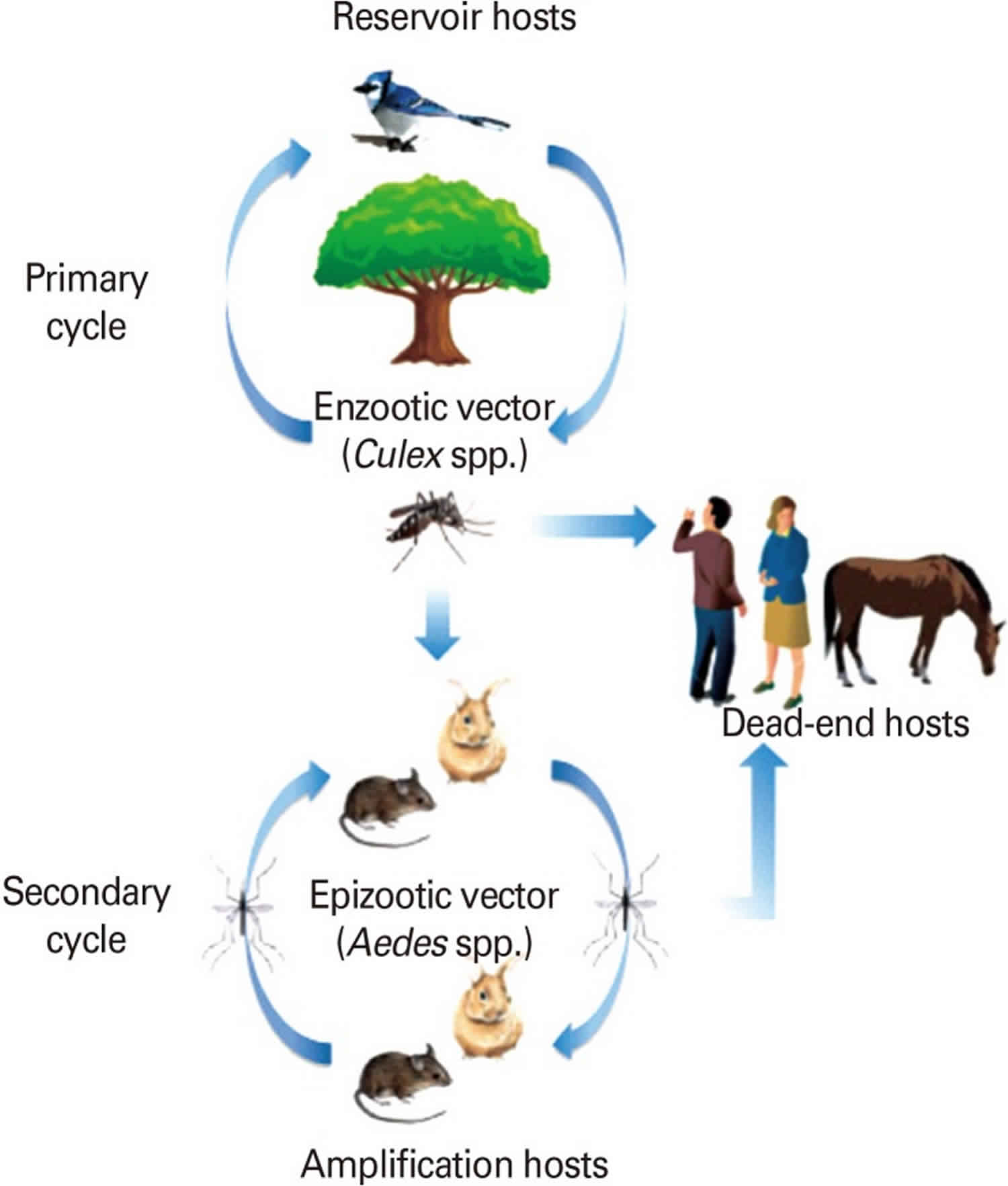
Footnote: Enzootic and epizootic/epidemic transmission cycles of Western equine encephalitis virus (WEEV). Western equine encephalitis virus is maintained in an enzootic cycle between passerine birds as reservoirs and its specific mosquito vector, C. tarsalis. Domestic and wild birds are considered important reservoir and epizootic amplifying hosts. It has also been suggested that lagomorphs and rodents can serve as amplification hosts when they are infected with Western equine encephalitis virus by Aedes mosquitos.
[Source 1 ]Western equine encephalitis virus vaccine
Formalin-inactivated vaccines have been developed experimentally for the protection of laboratory workers and other persons at high risk 87. There is a formalin-inactivated vaccine that is available as a double vaccine in combination with Eastern equine encephalitis virus only for veterinary use (horses) 88. Horses are vaccinated twice a year due to low immunogenicity of the inactivated vaccine. Presence of neutralizing antibodies is used as a correlate of protection and to monitor the success of immunization.
Venezuelan Equine Encephalitis virus
Venezuelan equine encephalitis virus is an Alphavirus that was originally isolated from the brains of dead horses 89. Venezuelan equine encephalitis virus circulates between a mosquito vector, Culex melanoconion, and forest-dwelling small mammals and birds in Central America and South America. It emerges during epizootic outbreaks to infect horses and humans via bridge vectors such as Aedes taeniorhynchus. Epidemics typically occur in northern South America, but have extended as far north as Mexico and Texas 90. In areas of sylvatic (forest) activity, human seroprevalence can be 50%. In contrast to many encephalitic arbovirus infections, Venezuelan equine encephalitis viremia in humans is sufficient to transmit virus to mosquitos for approximately 72 hours 91. However, humans are not likely to play an important role in spread; instead, epizootic strains of Venezuelan equine encephalitis virus are dependent on equine amplification for epidemic spread. Forty percent of patients with Venezuelan equine encephalitis virus have virus in the pharynx, suggesting that direct spread between humans may be possible, although human-to-human spread has never been proven 92.
Symptomatic Venezuelan equine encephalitis virus infection results in neurologic disease in a minority of cases following a viral prodrome of fever, headache, photophobia, conjunctival injection, myalgia, arthralgia, nausea, and dizziness. Pharyngeal inflammation, painful cervical lymphadenopathy, somnolence, and tremulousness may occur 93. CSF analysis reveals lymphocytic pleocytosis, elevated protein, and a normal CSF glucose. Few reports of neuroimaging studies exist. CT scans are usually normal. EEG typically shows diffuse slowing, although some cases have focal temporal slowing similar to that seen in herpes simplex virus encephalitis.
Venezuelan equine encephalitis virus infection is diagnosed by detection of specific IgM antibody in the CSF or serum, and fatality rates range from 0.2% to 1% of symptomatic Venezuelan equine virus–infected patients without symptoms of encephalitis and increases to 10% to 25% in patients with encephalitis. No antiviral therapy or human vaccine has shown proven benefit.
Figure 7. Enzootic and epizootic transmission cycles of Venezuelan equine encephalitis virus
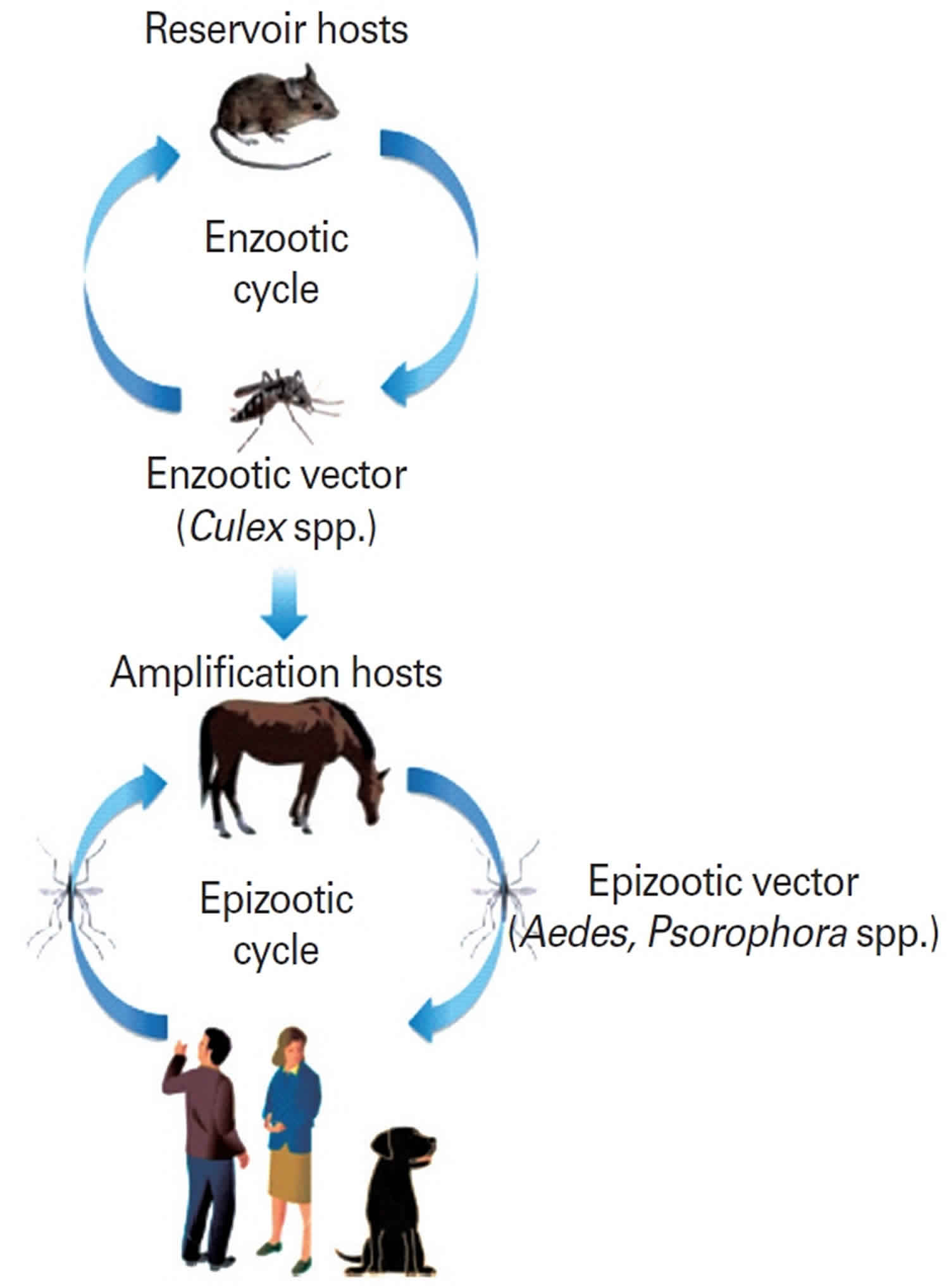
Footnote: Enzootic and epizootic/epidemic transmission cycles of Venezuelan equine encephalitis virus (VEEV). The enzootic transmission cycle of Venezuelan equine encephalitis virus is maintained among rodents and other vertebrates (e.g., cotton rats, spiny rats, bats, and opossums) as reservoirs and mosquitoes in the subgenus Culex (Melanoconion) as primary vectors. In contrast, epizootic Venezuelan equine encephalitis virus strains are transmitted by several mosquito vectors (e.g., Aedes and Psorophora spp.) to susceptible amplification hosts, horses. During epizootic or epidemic cycles, horses are efficient amplification hosts with high titered viremia for mosquito transmission.
[Source 1 ]Chikungunya virus
Recently, an emerging alphavirus called chikungunya virus has rapidly spread across the tropical and subtropical regions around the globe. The name “chikungunya” is derived from the Makonde language of Tanzania and means “that which bends up.” Over 1 million cases of chikungunya virus disease were reported in recent epidemics in India 94. The virus rapidly spread west and now is endemic in the Caribbean islands, South America, Central America, and recently the southeastern United States 95. Chikungunya virus has rapidly adapted to multiple mosquito-mammal transmission cycles depending on the geographic region.63 An urban cycle between the mosquito vector, A. aegypti, and humans is thought to support much of the recent epidemic spread. Following the bite of an infected mosquito, chikungunya virus causes a characteristic febrile syndrome manifested by headache, rash, and severe arthralgia. Patients can develop a poly-articular large-joint arthritis during acute infection, and symptoms of joint pain can linger for months following infection.
Chikungunya virus infection of the CNS is rare but has been reported. Associated neurologic clinical manifestations of chikungunya virus have included meningitis, encephalitis, and encephalomyeloradiculitis 96. Diagnosis of chikungunya virus infection can be made with serology using acute and convalescent serum, and cases of chikungunya virus–associated CNS disease have been confirmed following detection of anti–chikungunya virus IgM in the CSF 97. No known efficacious therapy or vaccine exists for chikungunya virus.
Bunyaviridae
California Encephalitis group
California encephalitis virus, La Crosse virus, Jamestown Canyon virus, and Tahyna virus are the major causes of encephalitis in the California encephalitis group within the family of Bunyaviridae and genus Bunyavirus. Of these viruses, La Crosse virus, California encephalitis virus, and Jamestown Canyon virus are causes of disease in the United States, and Tahyna virus is predominantly a cause of encephalitis in Russia. La Crosse virus is the most common cause of disease in the California encephalitis group. It was originally described in 1965 after a postmortem examination of a child who died of encephalitis in La Crosse, Wisconsin 98. La Crosse virus is transmitted in an enzootic pattern between squirrels and chipmunks by the mosquito Aedes triseriatus in areas of the Mississippi and Ohio River basins 99. Recent outbreaks of La Crosse virus have demonstrated a shift in incidence to the Appalachian region and West Virginia 100. Human exposure is often associated with camping or other recreational activities in wooded areas in endemic regions.
California encephalitis virus was originally isolated in 1941, but it is rare. Most human cases occur in the western United States and Canada. The ratio of asymptomatic to symptomatic infections is 1000:1 101.
Although La Crosse and California encephalitis viruses mostly cause disease in children, with a mean age of La Crosse virus infection in children of 7.5 years, Jamestown Canyon virus affects predominantly elderly individuals in regions of the northern United States, with seroprevalence in some areas reaching 10% 102. A recent report of a Jamestown Canyon virus infection in Montana underscores continued low-level transmission in the northern continental United States 103.
Symptoms of La Crosse virus encephalitis include fever, headache, vomiting in 70%, seizures in 46%, and altered mental status in 42% of cases 101. Focal neurologic signs include hemiparesis, aphasia, dysarthria, and chorea. About 10% of patients develop increased intracranial pressure, and, rarely, cerebral herniation can occur 101. Jamestown Canyon virus has similar clinical features.
CSF analysis for the California encephalitis virus group reveals a lymphocytic pleocytosis of approximately 600 cells/μL and normal glucose. An increased CSF protein is found in 30% of patients with encephalitis 101. Peripheral leukocytosis and hyponatremia secondary to syndrome of inappropriate secretion of antidiuretic hormone (SIADH) are common. IgM detection in the CSF or a fourfold increase in paired sera for IgG is considered diagnostic for infection.
No antiviral therapy currently exists for the California encephalitis group of viruses, and no vaccine is available. Mortality from La Crosse encephalitis is approximately 1% to 3%, and most survivors return to normal function. Ribavirin treatment for La Crosse encephalitis in children is not recommended because of problems with pharmacokinetics, toxicity at higher doses, and penetration into the CNS 104.
References- Go, Yun & Balasuriya, Udeni & Lee, Chong-Kyo. (2014). Zoonotic encephalitides caused by arboviruses: Transmission and epidemiology of alphaviruses and flaviviruses. Clinical and experimental vaccine research. 3. 58-77. 10.7774/cevr.2014.3.1.58
- Beckham JD, Tyler KL. Arbovirus Infections. Continuum (Minneap Minn). 2015;21(6 Neuroinfectious Disease):1599–1611. doi:10.1212/CON.0000000000000240 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5089063
- Hollidge BS, Gonzalez-Scarano F, Soldan SS. Arboviral encephalitides: transmission, emergence, and pathogenesis. J Neuroimmune Pharmacol 2010;5:428-42.
- Soldan SS, Gonzalez-Scarano F. Emerging infectious diseases: the Bunyaviridae. J Neurovirol 2005;11:412-23.
- Elliott R. Bunyaviruses: general features. In: Mahy BW, van Regenmortel MH, editors. Encyclopedia of virology. London: Elsevier-Academic Press; 2008. p.390-9.
- Romero JR, Simonsen KA. Powassan encephalitis and Colorado tick fever. Infect Dis Clin North Am 2008;22: 545-59.
- JOINT WHO/FAO expert committee on zoonoses. World Health Organ Tech Rep Ser 1959;58:1-84.
- Pfeffer M, Dobler G. Emergence of zoonotic arboviruses by animal trade and migration. Parasit Vectors 2010;3:35.
- Watts DM, Thompson WH, Yuill TM, DeFoliart GR, Hanson RP. Overwintering of La Crosse virus in Aedes triseriatus. Am J Trop Med Hyg 1974;23:694-700.
- Gubler DJ. The global resurgence of arboviral diseases. Trans R Soc Trop Med Hyg 1996;90:449-51.
- Hanna JN, Ritchie SA, Phillips DA, et al. An outbreak of Japanese encephalitis in the Torres Strait, Australia, 1995. Med J Aust 1996;165:256-60.
- Gubler DJ. Human arbovirus infections worldwide. Ann N Y Acad Sci 2001;951:13-24.
- Weaver SC, Barrett AD. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol 2004;2:789-801.
- Karabatsos NE. International catalogue of arboviruses, including certain other viruses of vertebrates. 3rd ed. ed. Karabatsos N, editor. San Antonio, Texas:: American Society of Tropical Medicine and Hygiene for The Subcommittee on Information Exchange of the American Committee on Arthropodborne Viruses; 1985.
- Junglen S, Drosten C. Virus discovery and recent insights into virus diversity in arthropods. Curr Opin Microbiol. 2013; 16(4):507±13. https://doi.org/10.1016/j.mib.2013.06.005
- Liang G, Gao X, Gould EA. Factors responsible for the emergence of arboviruses; strategies, challenges and limitations for their control. Emerg Microbes Infect. 2015; 4:e18. https://doi.org/10.1038/emi.2015.18
- Weger, James & Auerswald, Heidi & Vignuzzi, Marco & Dussart, Philippe & Karlsson, Erik. (2018). Taking a bite out of nutrition and arbovirus infection. PLOS Neglected Tropical Diseases. 12. e0006247. 10.1371/journal.pntd.0006247
- Gould EA, Solomon T. Pathogenic flaviviruses. Lancet 2008;371:500-9.
- Gubler DJ, Kuno G, Markoff L. Flaviviruses. In: Knipe DM, Howley PM, Griffin DE, et al., editors. Fields virology. Philadelphia: Lippincott William & Wilkins; 2007. p.1153-253.
- Simmonds P, Becher P, Collet MS, et al. Family Flaviviridae. In: King AM, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Virus taxonomy classification and nomenclature of viruses. Ninth report of the International Commitee on Taxonomy of Viruses. San Diego: Elsevier Academic Press; 2011. p.1003-20.
- Calisher CH. Antigenic classification and taxonomy of flaviviruses (family Flaviviridae) emphasizing a universal system for the taxonomy of viruses causing tick-borne encephalitis. Acta Virol 1988;32:469-78.
- van den Hurk AF, Ritchie SA, Mackenzie JS. Ecology and geographical expansion of Japanese encephalitis virus. Annu Rev Entomol 2009;54:17-35.
- Lewis L, Taylor HG, Sorem MB, Norcross JW, Kindsvatter VH. Japanese B encephalitis: clinical observations in an outbreak on Okinawa Shima. Arch Neurol Psychiatry 1947;57:430-63.
- Hanna JN, Ritchie SA, Phillips DA, et al. Japanese encephalitis in north Queensland, Australia, 1998. Med J Aust 1999;170:533-6.
- Tsarev SA, Sanders ML, Vaughn DW, Innis BL. Phylogenetic analysis suggests only one serotype of Japanese encephalitis virus. Vaccine 2000;18 Suppl 2:36-43.
- Mohammed MA, Galbraith SE, Radford AD, et al. Molecular phylogenetic and evolutionary analyses of Muar strain of Japanese encephalitis virus reveal it is the missing fifth genotype. Infect Genet Evol 2011;11:855-62.
- Chen WR, Tesh RB, Rico-Hesse R. Genetic variation of Japanese encephalitis virus in nature. J Gen Virol 1990; 71(Pt 12):2915-22.
- Tamada K, Kasai H, Hara M, Aoyama T, Takeuchi Y, Yamashita R. Japanese encephalitis in a 7-month-old Japanese boy. Acta Paediatr Jpn 1990;32:98-103.
- Kumar R, Mathur A, Singh KB, et al. Clinical sequelae of Japanese encephalitis in children. Indian J Med Res 1993; 97:9-13.
- Mackenzie JS. Emerging zoonotic encephalitis viruses: lessons from Southeast Asia and Oceania. J Neurovirol 2005;11:434-40.
- Rosen L. The natural history of Japanese encephalitis virus. Annu Rev Microbiol 1986;40:395-414.
- Halstead SB, Thomas SJ. Japanese encephalitis: new options for active immunization. Clin Infect Dis 2010;50: 1155-64.
- Tauber E, Kollaritsch H, Korinek M, et al. Safety and immunogenicity of a Vero-cell-derived, inactivated Japanese encephalitis vaccine: a non-inferiority, phase III, randomised controlled trial. Lancet 2007;370:1847-53.
- Guy B, Guirakhoo F, Barban V, Higgs S, Monath TP, Lang J. Preclinical and clinical development of YFV 17D-based chimeric vaccines against dengue, West Nile and Japanese encephalitis viruses. Vaccine 2010;28:632-49.
- Halstead SB, Thomas SJ. New Japanese encephalitis vaccines: alternatives to production in mouse brain. Expert Rev Vaccines 2011;10:355-64.
- Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA. 2013;310(3):308–315. doi: 10.1001/jama.2013.8042
- Wang P, Bai F, Zenewicz LA, et al. IL-22 signaling contributes to West Nile encephalitis pathogenesis. PLoS One. 2012;7(8):e44153. doi: 10.1371/journal.pone.0044153
- Melian EB, Edmonds JH, Nagasaki TK, et al. West Nile virus NS2A protein facilitates virus-induced apoptosis independently of interferon response. J Gen Virol. 2013;94(pt 2):308–313. doi: 10.1099/vir.0.047076-0
- Clarke P, Leser JS, Quick ED, et al. Death receptor-mediated apoptotic signaling is activated in the brain following infection with West Nile virus in the absence of a peripheral immune response. J Virol. 2014;88(2):1080–1089. doi: 10.1128/JVI.02944-13
- West Nile virus. https://www.cdc.gov/westnile/index.html
- Racsa L, Gander R, Chung W, et al. Clinical features of West Nile virus epidemic in Dallas, Texas, 2012. Diagn Microbiol Infect Dis. 2014;78(2):132–136. doi: 10.1016/j.diagmicrobio.2013.11.006
- Iwamoto M, Jernigan DB, Guasch A, et al. Transmission of West Nile virus from an organ donor to four transplant recipients. N Engl J Med. 2003;348(22):2196–2203. doi: 10.1056/NEJMoa022987
- Centers for Disease Control and Prevention (CDC) Possible West Nile virus transmission to an infant through breast-feeding—Michigan, 2002. MMWR Morb Mortal Wkly Rep. 2002;51(39):877–878.
- Biggerstaff BJ, Petersen LR. Estimated risk of West Nile virus transmission through blood transfusion during an epidemic in Queens, New York City. Transfusion. 2002;42(8):1019–1026. doi: 10.1046/j.1537-2995.2002.00167.x
- Watson JT, Pertel PE, Jones RC, et al. Clinical characteristics and functional outcomes of West Nile Fever. Ann Intern Med. 2004;141(5):360–365. doi: 10.7326/0003-4819-141-5-200409070-00010
- Bode AV, Sejvar JJ, Pape WJ, et al. West Nile virus disease: a descriptive study of 228 patients hospitalized in a 4-county region of Colorado in 2003. Clin Infect Dis. 2006;42(9):1234–1240. doi: 10.1086/503038
- Sejvar JJ, Haddad MB, Tierney BC, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290(4):511–515. doi: 10.1001/jama.290.4.511
- Fratkin JD, Leis AA, Stokic DS, et al. Spinal cord neuropathology in human West Nile virus infection. Arch Pathol Lab Med. 2004;128(5):533–537.
- Jean CM, Honarmand S, Louie JK, Glaser CA. Risk factors for West Nile virus neuroinvasive disease, California, 2005. Emerg Infect Dis. 2007;13(12):1918–1920. doi: 10.3201/eid1312.061265
- Nash D, Mostashari F, Fine A, et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344(24):1807–1814. doi: 10.1056/NEJM200106143442401
- Yakub I, Lillibridge KM, Moran A, et al. Single nucleotide polymorphisms in genes for 2′-5′-oligoadenylate synthetase and RNase L in patients hospitalized with West Nile virus infection. J Infect Dis. 2005;192(10):1741–1748. doi: 10.1086/497340
- Lim JK, Louie CY, Glaser C, et al. Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J Infect Dis. 2008;197(2):262–265. doi: 10.1086/524691
- Tyler KL, Pape J, Goody RJ, et al. CSF findings in 250 patients with serologically confirmed West Nile virus meningitis and encephalitis. Neurology. 2006;66(3):361–365. doi: 10.1212/01.wnl.0000195890.70898.1f
- Beckham JD, Tyler KL. Neuro-intensive care of patients with acute CNS infections. Neurotherapeutics. 2012;9(1):124–138. doi: 10.1007/s13311-011-0086-5
- Debiasi RL, Tyler KL. West Nile virus meningoencephalitis. Nat Clin Pract Neurol. 2006;2(5):264–275. doi: 10.1038/ncpneuro0176
- Davis LE, DeBiasi R, Goade DE, et al. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60(3):286–300. doi: 10.1002/ana.20959
- Sejvar JJ, Marfin AA. Manifestations of West Nile neuroinvasive disease. Rev Med Virol. 2006;16(4):209–224. doi: 10.1002/rmv.501
- Davis LE, Beckham JD, Tyler KL. North American encephalitic arboviruses. Neurol Clin. 2008;26(3):727–757. ix. doi: 10.1016/j.ncl.2008.03.012
- Sejvar JJ. West Nile virus and “poliomyelitis” Neurology. 2004;63(2):206–207. doi: 10.1212/01.WNL.0000130361.62281.69
- Shi PY, Wong SJ. Serologic diagnosis of West Nile virus infection. Expert Rev Mol Diagn. 2003;3(6):733–741. doi: 10.1586/14737159.3.6.733
- Hart J, Jr, Tillman G, Kraut MA, et al. West Nile virus neuroinvasive disease: neurological manifestations and prospective longitudinal outcomes. BMC Infect Dis. 2014;14:248. doi: 10.1186/1471-2334-14-248
- Pyrgos V, Younus F. High-dose steroids in the management of acute flaccid paralysis due to West Nile virus infection. Scand J Infect Dis. 2004;36(6–7):509–512. doi: 10.1080/00365540410020659
- Sejvar JJ. The long-term outcomes of human West Nile virus infection. Clin Infect Dis. 2007;44(12):1617–1624. doi: 10.1086/518281
- Sahu R, Verma R, Jain A, et al. Neurologic complications in dengue virus infection: a prospective cohort study. Neurology. 2014;83(18):1601–1609. doi: 10.1212/WNL.0000000000000935
- Sung S, Wurcel AG, Whittier S, et al. Powassan meningoencephalitis, New York, New York, USA. Emerg Infect Dis. 2013;19(9) doi: 10.3201/eid1909.121846
- Tavakoli NP, Wang H, Dupuis M, et al. Fatal case of deer tick virus encephalitis. N Engl J Med. 2009;360(20):2099–2107. doi: 10.1056/NEJMoa0806326
- Centers for Disease Control and Prevention (CDC) Eastern equine encephalitis—New Hampshire and Massachusetts, August-September 2005. MMWR Morb Mortal Wkly Rep. 2006;55(25):697–700.
- Loftin KC, Diallo AA, Herbert MW, et al. Five-year surveillance of West Nile and eastern equine encephalitis viruses in Southeastern Virginia. J Environ Health. 2006;68(9):33–40.
- Mitchell CJ, Niebylski ML, Smith GC, et al. Isolation of eastern equine encephalitis virus from Aedes albopictus in Florida. Science. 1992;257(5069):526–527. doi: 10.1126/science.1321985
- Goldfield M, Taylor BF, Welsh JN, et al. The persistence of eastern encephalitis serologic reactivity following overt and inapparent human infection—an eight year follow-up. Am J Epidemiol. 1968;87(1):50–57.
- Deresiewicz RL, Thaler SJ, Hsu L, Zamani AA. Clinical and neuroradiographic manifestations of eastern equine encephalitis. N Engl J Med. 1997;336(26):1867–1874.
- Przelomski MM, O’Rourke E, Grady GF, et al. Eastern equine encephalitis in Massachusetts: a report of 16 cases, 1970–1984. Neurology. 1988;38(5):736–739.
- Weaver SC, Kang W, Shirako Y, Rumenapf T, Strauss EG, Strauss JH. Recombinational history and molecular evolution of western equine encephalomyelitis complex alphaviruses. J Virol 1997;71:613-23.
- Meyer KF, Haring CM, Howitt B. The etiology of epizootic encephalomyelitis of horses in the San Joaquin Valley, 1930. Science 1931;74:227-8.
- Howitt B. Recovery of the virus of equine encephalomyelitis from the brain of a child. Science 1938;88:455-6.
- Reisen WK, Chiles RE. Prevalence of antibodies to western equine encephalomyelitis and St. Louis encephalitis viruses in residents of California exposed to sporadic and consistent enzootic transmission. Am J Trop Med Hyg 1997;57:526-9.
- Sabattini MS, Monath TP, Mitchell CJ, et al. Arbovirus investigations in Argentina, 1977-1980. I. Historical aspects and description of study sites. Am J Trop Med Hyg 1985; 34:937-44.
- Reimann CA, Hayes EB, DiGuiseppi C, et al. Epidemiology of neuroinvasive arboviral disease in the United States, 1999-2007. Am J Trop Med Hyg 2008;79:974-9.
- Longshore WA Jr, Stevens IM, Hollister AC Jr, Gittelsohn A, Lennette EH. Epidemiologic observations on acute infectious encephalitis in California, with special reference to the 1952 outbreak. Am J Hyg 1956;63:69-86.
- Doby PB, Schnurrenberger PR, Martin RJ, Hanson LE, Sherrick GW, Schoenholz WK. Western encephalitis in Illinois horses and ponies. J Am Vet Med Assoc 1966;148: 422-7.
- Weaver SC, Powers AM, Brault AC, Barrett AD. Molecular epidemiological studies of veterinary arboviral encephalitides. Vet J 1999;157:123-38.
- Karabatsos N, Lewis AL, Calisher CH, Hunt AR, Roehrig JT. Identification of Highlands J virus from a Florida horse. Am J Trop Med Hyg 1988;39:603-6.
- Wages DP, Ficken MD, Guy JS, Cummings TS, Jennings SR. Egg-production drop in turkeys associated with alphaviruses: eastern equine encephalitis virus and Highlands J virus. Avian Dis 1993;37:1163-6.
- Reisen WK, Monath TP. Western equine encephalomyelitis. In: Monath TP, editor. The arboviruses: epidemiology and ecology. Boca Raton: CRC Press; 1988. p.89-138.
- Calisher CH, Karabatsos N, Lazuick JS, Monath TP, Wolff KL. Reevaluation of the western equine encephalitis antigenic complex of alphaviruses (family Togaviridae) as determined by neutralization tests. Am J Trop Med Hyg 1988;38:447-52.
- Fulhorst CF, Hardy JL, Eldridge BF, Presser SB, Reeves WC. Natural vertical transmission of western equine encephalomyelitis virus in mosquitoes. Science 1994;263: 676-8.
- Pittman P, Plotkin SA. Biodefense and special pathogen vaccines. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccines. Philadelphia: Elsevier; 2008. p.1123-33.
- Barber TL, Walton TE, Lewis KJ. Efficacy of trivalent inactivated encephalomyelitis virus vaccine in horses. Am J Vet Res 1978;39:621-5.
- Beck CE, Wyckoff RW. Venezuelan equine encephalomyelitis. Science. 1938;88(2292):530.
- Griffin DE, Levine B, Tyor WR, Irani DN. The immune response in viral encephalitis. Semin Immunol. 1992;4(2):111–119.
- Wang E, Bowen RA, Medina G, et al. Virulence and viremia characteristics of 1992 epizootic subtype IC Venezuelan equine encephalitis viruses and closely related enzootic subtype ID strains. Am J Trop Med Hyg. 2001;65(1):64–69.
- Weaver SC, Salas R, Rico-Hesse R, et al. Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America. VEE Study Group. Lancet. 1996;348(9025):436–440. doi: 10.1016/S0140-6736(96)02275-1
- Molina OM, Morales MC, Soto ID, et al. Venezuelan equine encephalitis. 1995 outbreak: clinical profile of the case with neurologic involvement. Rev Neurol. 1999;29(2):296–298.
- Pialoux G, Gauzere BA, Jaureguiberry S, Strobel M. Chikungunya, an epidemic arbovirosis. Lancet Infect Dis. 2007;7(5):319–327. doi: 10.1016/S1473-3099(07)70107-X
- Prince HE, Seaton BL, Matud JL, Batterman HJ. Chikungunya virus RNA and antibody testing at a national reference laboratory since the emergence of Chikungunya in the Americas. Clin Vaccine Immunol. 2015;22(3):291–297. doi: 10.1128/CVI.00720-14
- Nelson J, Waggoner JJ, Sahoo MK, et al. Encephalitis caused by Chikungunya virus in a traveler from the Kingdom of Tonga. J Clin Microbiol. 2014;52(9):3459–3461. doi: 10.1128/JCM.01288-14
- Robin S, Ramful D, Le Seach’ F, et al. Neurologic manifestations of pediatric chikungunya infection. J Child Neurol. 2008;23(9):1028–1035. doi: 10.1177/0883073808314151
- Thompson WH, Kalfayan B, Anslow RO. Isolation of California encephalitis group virus from a fatal human illness. Am J Epidemiol. 1965;81:245–253.
- Rust RS, Thompson WH, Matthews CG, et al. La Crosse and other forms of California encephalitis. J Child Neurol. 1999;14(1):1–14. doi: 10.1177/088307389901400101
- Haddow AD, Bixler D, Odoi A. The spatial epidemiology and clinical features of reported cases of La Crosse virus infection in West Virginia from 2003 to 2007. BMC Infect Dis. 2011;11:29. doi: 10.1186/1471-2334-11-29.
- McJunkin JE, de los Reyes EC, Irazuzta JE, et al. La Crosse encephalitis in children. N Engl J Med. 2001;344(11):801–807. doi: 10.1056/NEJM200103153441103
- Mayo D, Karabatsos N, Scarano FJ, et al. Jamestown Canyon virus: seroprevalence in Connecticut. Emerg Infect Dis. 2001;7(5):911–912.
- Centers for Disease Control and Prevention (CDC) Human Jamestown canyon virus infection—Montana, 2009. MMWR Morb Mortal Wkly Rep. 2011;60(20):652–655.
- McJunkin JE, Nahata MC, De Los Reyes EC, et al. Safety and pharmacokinetics of ribavirin for the treatment of la crosse encephalitis. Pediatr Infect Dis J. 2011;30(10):860–865. doi: 10.1097/INF.0b013e31821c922c





{kind=link}