
What is cat scratch disease
Cat scratch disease also called cat scratch fever or subacute regional lymphadenitis is a bacterial infection caused by the Bartonella henselae bacteria. As it’s name suggests, cat scratch disease is contracted through the scratch or bite of an infected domestic or feral cats, especially kittens 1, 2. Research suggests a cat may get Bartonella henselae bacteria from fleas. These bacteria can be transmitted from a cat to a person during a scratch that is contaminated with flea stool. The Bartonella henselae bacteria are passed from an infected cat to a human after the infected cat licks an open wound or bites or scratches human skin hard enough to break the surface of the skin. Kittens younger than one year of age are more likely to scratch, increasing the likelihood of infection. Stray cats are more likely than pets to be infected with Bartonella henselae. Some evidence suggests that Bartonella henselae bacteria may spread directly to people by the bite of infected cat fleas, but this has not been proven 1. In a study conducted in the United States, seropositivity was detected in 10-40% of domestic cats 3. The Centers for Disease Control and Prevention (CDC) estimates more than 12,500 diagnosed cases of cat scratch disease annually in the United States, although the disease is prevalent worldwide 4, 5, 6, 7. The incidence of cat scratch disease was reported to be 9.3 per 100,000 in the United States 8. In two studies conducted in Turkey, seropositivity rates were 3.3% and 6% among healthy individuals. This rate was reported as 12.5% in veterinarians and 26.5% in cat owners 9, 10. In the United States, most cases of cat scratch disease occur in the fall and winter. Cat scratch disease is most common in children under the age of 15 11, 12. However, Oray et al. 13 reported in a Turkish study that adults accounted for 50% of the patients in their series of cat scratch disease with eye involvement. Curi et al. 14 reported that 71% of cat scratch disease patients with eye involvement were over the age of 18.
Cat scratch disease most common symptoms include:
- Low-grade fever
- Headache
- Fatigue
- Poor appetite
- Enlarged, tender lymph nodes (lymphadenitis) that develop 1–3 weeks after exposure to a cat, especially around the head, neck, and upper limbs
- A scab or pustule at the scratch site after 3-10 days
Lymph nodes involvement is most commonly observed in cat scratch disease 15.
For people with weak immune systems, cat scratch disease may cause more serious problems. Rarely, infections of the eye, liver, spleen, brain, bones, or heart valves can occur. Some of these infections occur primarily in people with weakened immune systems, such as those with advanced human immunodeficiency virus (HIV) infection.
The best way to avoid cat scratch disease is to avoid rough play with cats that could lead to scratches or bites. If you do get a scratch or bite, wash it well with soap and water. If the bite or scratch gets infected or if you have symptoms of cat scratch disease, see your doctor.
The use of antibiotics to shorten the course of cat scratch disease is debated. In most people, cat-scratch disease clears up without treatment, although some patients may develop complications from disseminated disease where the infection can travel to your bones, liver, or other organs. This requires more intensive treatment. You can take an over-the-counter pain reliever to help relieve pain and discomfort. Ibuprofen (two brands: Motrin, Advil) or naproxen (one brand: Aleve) can help. Applying heat compresses to the affected area may also help. If a lymph node is very large or painful, your doctor may drain it to help relieve the pain. Antibiotics may be needed if your symptoms don’t go away in a month or two. Azithromycin has been shown to decrease lymph node volume more rapidly compared to no treatment. The suggested dose of azithromycin for cat scratch disease is 1:
- Adults and children > 45.5 kg: 500 mg on day 1, followed by 250 mg for 4 days
- Children ≤ 45.5 kg: 10 mg/kg on day 1, followed by 5 mg/kg for 4 days
Cat scratch disease infection appears to confer lifelong immunity, as reports of recurrences of clinical cat scratch disease are rare 16.
Figure 1. Swollen lymph node from cat scratch disease
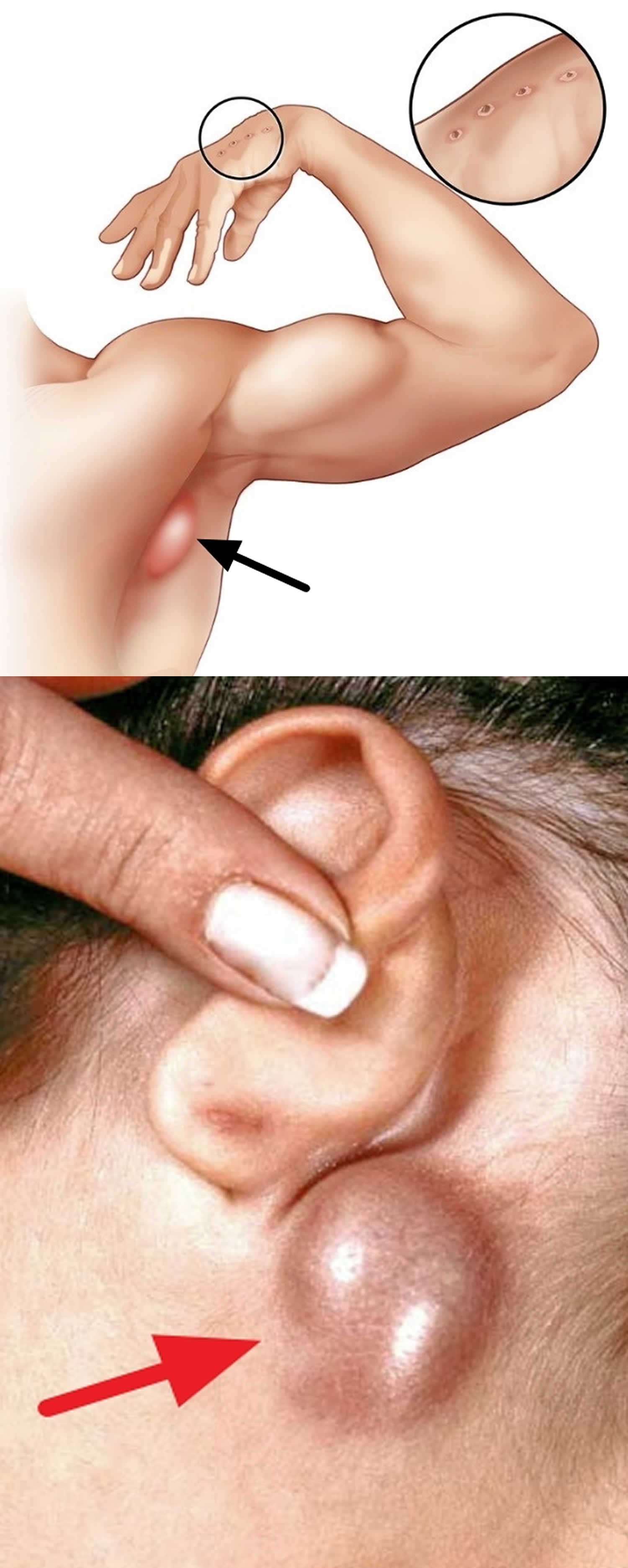
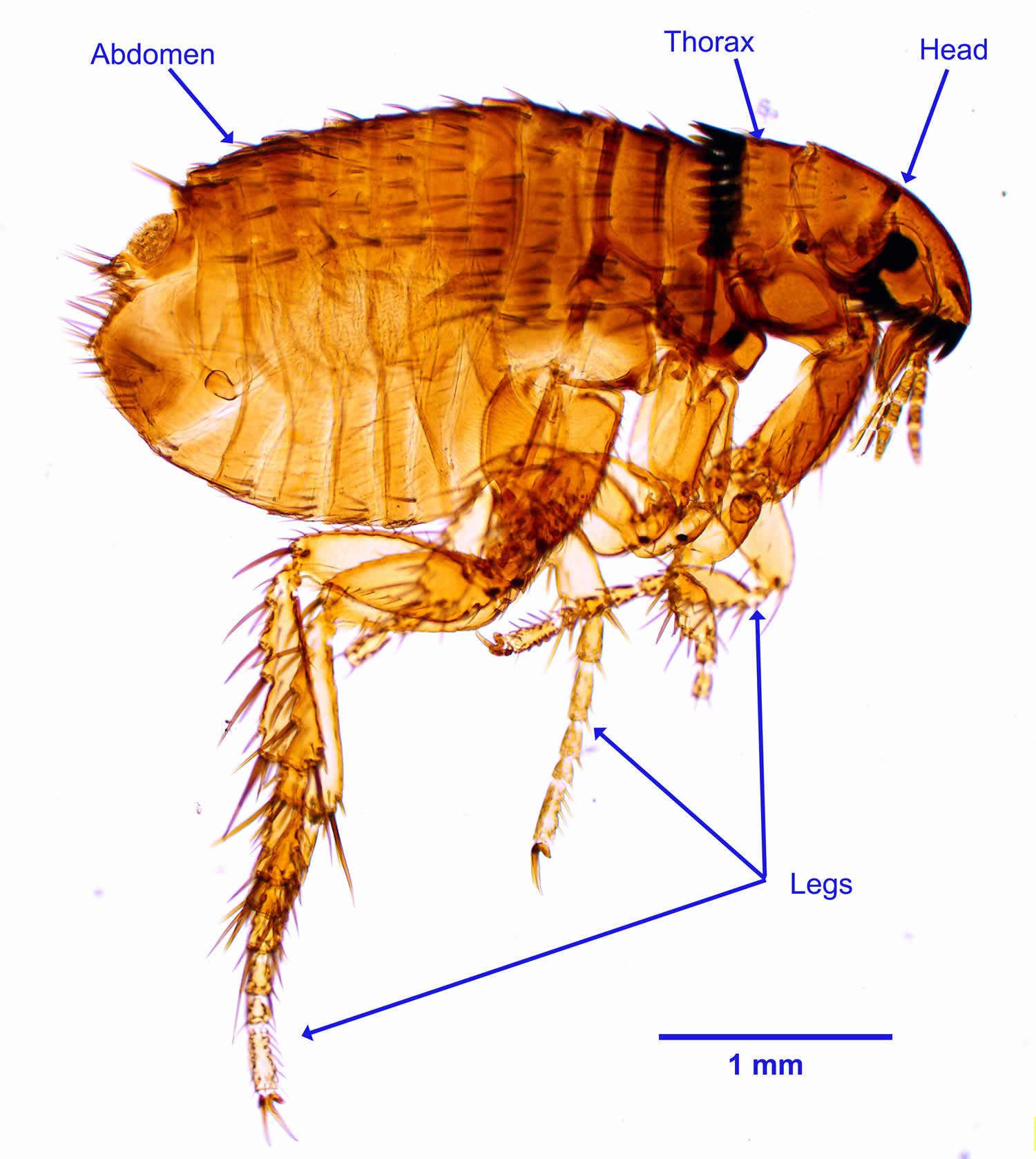
Figure 2. Cat flea (Ctenocephalides felis)
My cat is completely healthy. How can it have cat scratch disease?
Cats most commonly become infected with Bartonella henselae, the bacteria that cause cat scratch disease, through flea bites. They can less commonly become infected during fights with other infected cats or through feline blood transfusions. While some cats become ill, most simply have Bartonella henselae bacteria in their blood without getting sick. Some studies have found the Bartonella bacteria in the blood of up to 1 in 3 healthy cats, particularly kittens.
Should I get my pet tested and treated for Bartonella, just to be safe?
Testing and treatment is not recommended, unless your pet is sick. For cats that do become sick, the illness usually consists of fever for 2 to 3 days. Symptoms requiring veterinary care include fever lasting more than 3 days, vomiting, lethargy, red eyes, swollen lymph nodes, or decreased appetite. Treating Bartonella henselae with antibiotics can take a long time.
My child got scratched by the neighbor’s kitten and now my husband is worried about “cat scratch disease.” How worried should we be?
Most cat scratches do not result in cat scratch disease. Clean scratch wounds with soap and water. If your child develops a fever; enlarged, tender lymph nodes that develop 1 to 3 weeks after being scratched; or a pustule at the scratch site, see your family doctor. Treatment with antibiotics is usually not necessary, but may help reduce lymph node swelling.
I have a weakened immune system. Should I give my cat away?
If you are living with human immunodeficiency virus (HIV), are being treated for cancer, or have any other condition that might suppress your immune system, you can keep your cat. You do not need to test or treat a healthy cat for Bartonella bacteria.
To reduce your risk of getting cat scratch disease:
- Keep your cat indoors.
- Avoid cat scratches, bites, and licks, especially from kittens or stray cats. This is especially important for people who have weakened immune systems. Declawing is not recommended to prevent cat scratch disease.
- Promptly wash any cat scratches or bites with soap and water.
- Wash your hands promptly after handling cats.
- Avoid contact with fleas.
- Treat your cat with a flea control product recommended by your veterinarian. Never use products that contain permethrin on cats.
- If you are getting a cat, make sure it’s at least a year old, in good health, and free of fleas. Avoid and do not adopt stray cats or cats with flea infestations.
- Keep cats indoors and away from stray cats. People who have weakened immune systems should avoid owning cats less than one-year-old.
What causes cat-scratch disease?
People with cat scratch disease usually have suffered a scratch or bite from a cat or kitten that is infected with the Bartonella henselae bacteria previously known as Rochalimaea henselae bacteria. Bartonella henselae bacteria is a facultative intracellular Gram-negative bacteria that commonly infects cats and less commonly dogs 18, 11. The bacteria is passed from cat to cat via fleas 12, 3. In the absence of fleas, an infected cat cannot pass the infection to uninfected cats. The bacteria are present in saliva deposited in cat’s fur and claws.
Cat scratch disease is transmitted by cats through scratching or biting. Approximately 90% of patients have a history of cat contact 3. Children and adolescents, veterinarians, and cat owners are at increased risk 11, 12, 19. Human immunodeficiency virus (HIV) infection is also a risk factor for Bartonella infection 11, 20.
People at risk of getting cat scratch fever include those:
- Owning a cat younger than 12 months
- Licked, bitten or scratched by an infected kitten
- Petting an infected cat
- Owning at least one kitten or cat with fleas.
Cat scratch fever can occur in people of all ages but is most common in children and adolescents. 80% of patients with cat scratch disease are less than 21 years old.
Cat scratch disease prevention
To reduce your risk of getting cat scratch disease:
- Keep your cat indoors.
- Avoid cat scratches, bites, and licks, especially from kittens or stray cats. This is especially important for people who have weakened immune systems. Declawing is not recommended to prevent cat scratch disease.
- Promptly wash any cat scratches or bites with soap and water.
- Wash your hands promptly after handling cats.
- Avoid contact with fleas.
- Treat your cat with a flea control product recommended by your veterinarian. Never use products that contain permethrin on cats.
- If you are getting a cat, make sure it’s at least a year old, in good health, and free of fleas. Avoid and do not adopt stray cats or cats with flea infestations.
- Keep cats indoors and away from stray cats. People who have weakened immune systems should avoid owning cats less than one-year-old.
Cat scratch disease signs and symptoms
Cat scratch disease signs and symptoms may include:
- Low-grade fever
- Headache
- Malaise or fatigue
- Poor appetite
- Stomach pains
- Nausea and vomiting
- Sore throat
- Rash
- Enlarged, tender lymph nodes (lymphadenitis) that develop 1–3 weeks after exposure to a cat, especially around the head, neck, and upper limbs. The nearest lymph glands become swollen and tender, most often those on the head, neck and armpit. This is known as regional lymphadenopathy. The lymph glands may swell to 10 to 12 cm in the first two weeks of cat scratch disease. On questioning, patients often recall being licked, bitten or scratched by a cat in the previous one to eight weeks.
- A scab or pustule at the scratch site after 3-10 days. A small red raised spot develops at the site of contact with an infected cat’s saliva. This changes into a fluid-filled blister that later forms a crusty sore. This usually heals without scarring in several days or months. It is often mistaken for an insect bite. Most are found on the hands, arms, face or neck as people often hold kittens close to their chest and face.
Less common problems occur in about 10% of patients with cat scratch disease. These include:
- Parinaud’s oculoglandular syndrome characterized by an inflamed lump in the eye (granulomatous conjunctivitis), swollen lymph glands in front of the nearby ear (preauricular lymphadenopathy) and fever 15
- Bacillary angiomatosis (infection of the blood vessels)
- Bacillary hepatitis and splenitis (infection of the liver and spleen)
- Erythema nodosum (red lumps on the shins)
- Sepsis (infection disseminated through the blood stream)
- Encephalitis (brain infection). Encephalitis is an uncommon complication of cat scratch disease that occurs one to six weeks after the lymph glands swell up. The abrupt symptoms of fever, seizures and coma can be frightening. With hospitalization and high doses of antibiotics, most patients recover completely.
- Bacterial endocarditis (heart valve infection)
Rarely disseminated infection may cause serious clinical infections of the eye (conjunctivitis), lungs (pneumonia), liver (bacillary hepatitis), spleen (bacillary splenitis), brain (encephalitis & meningitis), bones (osteomyelitis) or heart valves (endocarditis) can occur 15, 21, 22. Some of these infections occur primarily in people with weakened immune systems (immunocompromised individuals), such as those with advanced HIV infection.
Bartonella henselae can sometimes cause infection of the heart valves called endocarditis. In many cases, blood cultures might be negative (culture-negative endocarditis), which can make the diagnosis more challenging.
Eye involvement (conjunctivitis) occurs in 5-10% of cat scratch disease patients 23. Eye involvement can manifest as granulomatous conjunctivitis and preauricular lymphadenopathy, as in Parinaud’s oculoglandular syndrome, but can also present with different clinical signs and symptoms, such as neuroretinitis, anterior uveitis, intermediate uveitis, focal/multifocal chorioretinitis, choroidal mass, retinal infiltrate, branch retinal vein or artery occlusion, serous retinal detachment, or acute endophthalmitis 22, 24. Cat scratch disease is the most common known cause of neuroretinitis.15,16 B. henselae seropositivity is detected in two-thirds of neuroretinitis patients 25, 26. Although Bartonella henselae is the most common cause, neuroretinitis is detected in only 1-2% of infected individuals 23.
Cat scratch disease diagnosis
Cat scratch disease diagnosis is based on the presence of three of the four following criteria:
- Contact with a cat and the presence of a scratch or lesion on the skin, eye or mucous membranes
- Enlarged regional lymph glands (lymphadenopathy), after excluding all other possible causes
- Positive skin test for cat scratch disease
- Suggestive biopsy of skin or lymph node
Bartonella henselae infection may be diagnosed clinically in patients with typical signs and symptoms and a compatible history of being licked, bitten or scratched by a cat.
Bartonella henselae bacteria is a fastidious, slow-growing bacterium. Cultures should be held for a minimum of 21 days. It is often helpful for doctors to alert the microbiology laboratory that Bartonella henselae is suspected to optimize conditions for growth.
Serology can aid the diagnosis of Bartonella henselae, although cross-reactivity with other Bartonella species may limit interpretation. Doctors should be aware that serological tests do not reliably differentiate among Bartonella species and positive results may persist for years even after effective treatment.
Bartonella henselae DNA may be detected by molecular assay of lymph node aspirates or blood, though sensitivity of these methods is not optimal for blood samples. However, lymph node aspiration is not generally recommended except to relieve severe pain and swelling or in cases where the diagnosis is unclear.
Patients with infectious endocarditis sometimes have damaged heart valves that need to be surgically replaced. Excised heart valve tissue can be tested by molecular assay to confirm infection with Bartonella henselae.
In addition to the role of history (cat contact) and clinical findings in the diagnosis of ocular cat scratch disease, serology and imaging such as fundus fluorescein angiography (FFA) and optical coherence tomography (OCT) are also important for a definitive diagnosis 27.
Cat scratch disease treatment
Treatment of uncomplicated cat scratch disease remains controversial. Because it is a benign and self-limiting condition, with most cases of enlarged regional lymph glands (lymphadenopathy) resolving spontaneously in two to four months, no specific treatment is usually necessary. However, some studies suggest that certain antibiotics including doxycycline, erythromycin, gentamicin, rifampicin, trimethoprim + sulphamethoxazole and ciprofloxacin may significantly shorten the duration of lymphadenopathy. Antibiotics are warranted in patients with severe or persistent symptoms of cat scratch disease.
Systemic antibiotic therapy is recommended for patients with immunodeficiency, severe systemic disease, and vision-threatening eye involvement (e.g., neuroretinitis, papillitis, retinitis) 28. Doxycycline, tetracycline, and erythromycin are first-line antibiotics in systemic therapy, while ciprofloxacin, rifampin, and trimethoprim-sulfamethoxazole can also be used as alternatives 29. Doxycycline and azithromycin and trimethoprim + sulfamethoxazole are the most commonly used systemic antibiotics. There are publications in the literature indicating that the use of oral or intravenous steroids in combination with systemic antibiotic therapy positively affects visual outcomes 30. In their retrospective case series, Habot-Wilner et al. 31 emphasized that combined antibiotic and corticosteroid treatment yielded better final visual acuity compared to antibiotic treatment alone, especially in patients with optic disc involvement and low initial visual acuity.
In rare cases, large pus-filled lymph nodes may persist for one to three years. The pus may need to be repeatedly drained through a needle. Pain and fever can be managed by increasing fluid intake and paracetamol. Warm moist compresses to affected lymph glands may decrease swelling and tenderness.
Cat scratch disease prognosis
The prognosis for healthy patients with cat scratch disease is excellent. Complete recovery without complications occurs in nearly all patients. Significant illness occurs in 5 to 10% of cat scratch disease cases, usually because of involvement of the central or peripheral nervous system or because of multi-system widespread cat scratch disease. Even in patients with central nervous system (brain and spinal cord) involvement, recovery without neurologic complications within weeks to months can be expected. Death caused by cat scratch disease in patients who are healthy is extremely rare 32, 33.
Tender and inflamed swollen lymph nodes (lymphadenitis) usually resolves spontaneously over 2-4 months, but 1 to 2 years may be required. One episode of cat scratch disease confers lifelong immunity to children and adolescents. Recurrent lymphadenopathy 6 to 13 months after the initial diagnosis has been reported in 3 adults with cat scratch disease 34.
People with weak immune system (immunocompromised patients) may experience a dramatic and potentially life-threatening course of cat scratch disease. However, with appropriate antibiotic use and management of complications, these patients also typically experience full resolution from cat scratch disease 34.
- Bartonella henselae infection or cat scratch disease (CSD). https://www.cdc.gov/bartonella/bartonella-henselae[↩][↩][↩]
- Bartonella henselae or cat scratch disease (CSD) FAQs. https://www.cdc.gov/bartonella/bartonella-henselae/faq.html[↩]
- Zangwill KM, Hamilton DH, Perkins BA, Regnery RL, Plikaytis BD, Hadler JL, Cartter ML, Wenger JD. Cat scratch disease in Connecticut. Epidemiology, risk factors, and evaluation of a new diagnostic test. N Engl J Med. 1993 Jul 1;329(1):8-13. doi: 10.1056/NEJM199307013290102[↩][↩][↩]
- Nelson C.A., Saha S., Mead P.S. Cat-Scratch Disease in the United States, 2005–2013. Emerg. Infect. Dis. 2016;22:1741–1746. doi: 10.3201/eid2210.160115[↩]
- Fournier P.-E., Robson J., Zeaiter Z., McDougall R. Improved culture from lymph nodes of patients with cat scratch disease and genotypic characterization of Bartonella henselae isolates in Australia. J. Clin. Microbiol. 2002;40:3620–3624. doi: 10.1128/JCM.40.10.3620-3624.2002[↩]
- Kelly P.J., Meads N., Theobald A., Fournier P.-E., Raoult D. Rickettsia felis, Bartonella henselae, and B. clarridgeiae, New Zealand. Emerg. Infect. Dis. 2004;10:967–968. doi: 10.3201/eid1005.030986[↩]
- Maruyama S., Izumikawa K., Miyashita M., Kabeya H., Mikami T., Yamanouchi H., Sasaki E., Yoshida H., Izumikawa K. First isolation of Bartonella henselae type I from a cat-scratch disease patient in Japan and its molecular analysis. Microbiol. Immunol. 2004;48:103–109. doi: 10.1111/j.1348-0421.2004.tb03495.x[↩]
- Jackson LA, Perkins BA, Wenger JD. Cat scratch disease in the United States: an analysis of three national databases. Am J Public Health. 1993 Dec;83(12):1707-11. doi: 10.2105/ajph.83.12.1707[↩]
- Aydin N, Bülbül R, Tellı M, Gültekın B. Aydın ili kan donörlerinde Bartonella henselae ve Bartonella quintana seroprevalansı [Seroprevalence of Bartonella henselae and Bartonella quintana in blood donors in Aydin province, Turkey]. Mikrobiyol Bul. 2014 Jul;48(3):477-83. Turkish.[↩]
- Yilmaz C, Ergin C, Kaleli I. Pamukkale Universitesi Kan Merkezine başvuran donörlerde Bartonella henselae seroprevalansinin araştirilmasi ve risk faktörlerinin irdelenmesi [Investigation of Bartonella henselae seroprevalence and related risk factors in blood donors admitted to Pamukkale University Blood Center]. Mikrobiyol Bul. 2009 Jul;43(3):391-401. Turkish.[↩]
- Biancardi AL, Curi AL. Cat-scratch disease. Ocul Immunol Inflamm. 2014 Apr;22(2):148-54. doi: 10.3109/09273948.2013.833631[↩][↩][↩][↩]
- Koehler JE, Glaser CA, Tappero JW. Rochalimaea henselae infection. A new zoonosis with the domestic cat as reservoir. JAMA. 1994 Feb 16;271(7):531-5. doi: 10.1001/jama.271.7.531[↩][↩][↩]
- Oray M, Önal S, Koç Akbay A, Tuğal Tutkun İ. Diverse Clinical Signs of Ocular Involvement in Cat Scratch Disease. Turk J Ophthalmol. 2017 Jan;47(1):9-17. doi: 10.4274/tjo.28009[↩]
- Curi AL, Machado D, Heringer G, Campos WR, Lamas C, Rozental T, Gutierres A, Orefice F, Lemos E. Cat-scratch disease: ocular manifestations and visual outcome. Int Ophthalmol. 2010 Oct;30(5):553-8. doi: 10.1007/s10792-010-9389-5[↩]
- Spach DH, Koehler JE. Bartonella-associated infections. Infect Dis Clin North Am. 1998 Mar;12(1):137-55. doi: 10.1016/s0891-5520(05)70414-1[↩][↩][↩]
- Dolan MJ, Wong MT, Regnery RL, Jorgensen JH, Garcia M, Peters J, Drehner D. Syndrome of Rochalimaea henselae adenitis suggesting cat scratch disease. Ann Intern Med. 1993 Mar 1;118(5):331-6. doi: 10.7326/0003-4819-118-5-199303010-00002[↩]
- Ctenocephalides (felis) felis (cat flea). https://wcvm.usask.ca/learnaboutparasites/parasites/ctenocephalides.php[↩]
- Cassady JV, Culbertson CS. Cat-Scratch disease and Parinaud`s Oculoglandular syndrome. AMA Arch Ophthalmol. 1953;50:68–74. doi: 10.1001/archopht.1953.00920030071010[↩]
- Noah DL, Kramer CM, Verbsky MP, Rooney JA, Smith KA, Childs JE. Survey of veterinary professionals and other veterinary conference attendees for antibodies to Bartonella henselae and B quintana. J Am Vet Med Assoc. 1997 Feb 1;210(3):342-4.[↩]
- Trataris AN, Rossouw J, Arntzen L, Karstaedt A, Frean J. Bartonella spp. in human and animal populations in Gauteng, South Africa, from 2007 to 2009. Onderstepoort J Vet Res. 2012 Jun 20;79(2):452. doi: 10.4102/ojvr.v79i2.452[↩]
- Midani S, Ayoub EM, Anderson B. Cat-scratch disease. Adv Pediatr. 1996;43:397-422.[↩]
- Ormerod LD, Dailey JP. Ocular manifestations of cat-scratch disease. Curr Opin Ophthalmol. 1999 Jun;10(3):209-16. doi: 10.1097/00055735-199906000-00010[↩][↩]
- Carithers HA. Cat-scratch disease. An overview based on a study of 1,200 patients. Am J Dis Child. 1985 Nov;139(11):1124-33. doi: 10.1001/archpedi.1985.02140130062031[↩][↩]
- Saatci AO, Oner FH, Kargi A, Kavukcu S. Unilateral neuroretinitis and periparillary serous retinal detachment in cat-scratch disease. Korean J Ophthalmol. 2002 Jun;16(1):43-6. doi: 10.3341/kjo.2002.16.1.43[↩]
- Dreyer RF, Hopen G, Gass JD, Smith JL. Leber’s idiopathic stellate neuroretinitis. Arch Ophthalmol. 1984 Aug;102(8):1140-5. doi: 10.1001/archopht.1984.01040030918013[↩]
- Weiss AH, Beck RW. Neuroretinitis in childhood. J Pediatr Ophthalmol Strabismus. 1989 Jul-Aug;26(4):198-203. doi: 10.3928/0191-3913-19890701-10[↩]
- Acar A, Çakar Özdal P, Başarır B, Özdemir Yalçınsoy K, Altan Ç, Budakoğlu Ö. A Case Series of Cat-Scratch Disease with Ocular Manifestations: Clinical Findings and Treatment Approach. Turk J Ophthalmol. 2023 Aug 19;53(4):226-233. doi: 10.4274/tjo.galenos.2022.44692[↩]
- Wade NK, Levi L, Jones MR, Bhisitkul R, Fine L, Cunningham ET Jr. Optic disk edema associated with peripapillary serous retinal detachment: an early sign of systemic Bartonella henselae infection. Am J Ophthalmol. 2000 Sep;130(3):327-34. doi: 10.1016/s0002-9394(00)00599-7[↩]
- Jerris RC, Regnery RL. Will the real agent of cat-scratch disease please stand up? Annu Rev Microbiol. 1996;50:707-25. doi: 10.1146/annurev.micro.50.1.707[↩]
- Gulati A, Yalamanchili S, Golnik KC, Lee AG. Cat scratch neuroretinitis: the role of acute and convalescent titers for diagnosis. J Neuroophthalmol. 2012 Sep;32(3):243-5. doi: 10.1097/WNO.0b013e318233a0a6[↩]
- Habot-Wilner Z, Trivizki O, Goldstein M, Kesler A, Shulman S, Horowitz J, Amer R, David R, Ben-Arie-Weintrob Y, Bakshi E, Almog Y, Sartani G, Vishnevskia-Dai V, Kramer M, Bar A, Kehat R, Ephros M, Giladi M. Cat-scratch disease: ocular manifestations and treatment outcome. Acta Ophthalmol. 2018 Jun;96(4):e524-e532. doi: 10.1111/aos.13684[↩]
- Fouch B, Coventry S. A case of fatal disseminated Bartonella henselae infection (cat-scratch disease) with encephalitis. Arch Pathol Lab Med. 2007 Oct;131(10):1591-4. doi: 10.5858/2007-131-1591-ACOFDB[↩]
- Gerber JE, Johnson JE, Scott MA, Madhusudhan KT. Fatal meningitis and encephalitis due to Bartonella henselae bacteria. J Forensic Sci. 2002 May;47(3):640-4.[↩]
- Cat Scratch Disease (Cat Scratch Fever). https://emedicine.medscape.com/article/214100-overview#a6[↩][↩]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}