Barth syndrome
Barth syndrome is a rare genetic metabolic and neuromuscular disorder characterized by an enlarged and weakened heart (dilated cardiomyopathy), weakness in muscles used for movement (skeletal myopathy), recurrent infections due to small numbers of white blood cells (neutropenia), and growth retardation, potentially leading to short stature 1. Barth syndrome occurs almost exclusively in males.
In males with Barth syndrome, dilated cardiomyopathy is often present at birth or develops within the first months of life. Over time, the heart muscle becomes increasingly weakened and is less able to pump blood. Individuals with Barth syndrome may have elastic fibers in place of muscle fibers in some areas of the heart muscle, which contributes to the cardiomyopathy. This condition is called endocardial fibroelastosis; it results in thickening of the muscle and impairs its ability to pump blood. In people with Barth syndrome, the heart problems can lead to heart failure. In rare cases, the cardiomyopathy gets better over time and affected individuals eventually have no symptoms of heart disease.
In Barth syndrome, skeletal myopathy, particularly of the muscles closest to the center of the body (proximal muscles), is usually noticeable from birth and causes low muscle tone (hypotonia). The muscle weakness often causes delay of motor skills such as crawling and walking. Additionally, affected individuals tend to experience extreme tiredness (fatigue) during strenuous physical activity.
Most males with Barth syndrome have neutropenia. The levels of white blood cells can be consistently low (persistent), can vary from normal to low (intermittent), or can cycle between regular episodes of normal and low (cyclical). Neutropenia makes it more difficult for the body to fight off foreign invaders such as bacteria and viruses, so affected individuals have an increased risk of recurrent infections.
Newborns with Barth syndrome are often smaller than normal, and their growth continues to be slow throughout life. Some boys with this condition experience a growth spurt in puberty and are of average height as adults, but many men with Barth syndrome continue to have short stature in adulthood.
Males with Barth syndrome often have distinctive facial features including prominent cheeks. Affected individuals typically have normal intelligence but often have difficulty performing tasks involving math or visual-spatial skills such as puzzles.
Males with Barth syndrome have increased levels of a substance called 3-methylglutaconic acid in their blood and urine. The amount of the acid does not appear to influence the signs and symptoms of the condition. Barth syndrome is one of a group of metabolic disorders that can be diagnosed by the presence of increased levels of 3-methylglutaconic acid in urine (3-methylglutaconic aciduria).
Even though most features of Barth syndrome are present at birth or in infancy, affected individuals may not experience health problems until later in life. The age at which individuals with Barth syndrome display symptoms or are diagnosed varies greatly. The severity of signs and symptoms among affected individuals is also highly variable.
Males with Barth syndrome have a reduced life expectancy. Many affected children die of heart failure or infection in infancy or early childhood, but those who live into adulthood can survive into their late forties.
Barth syndrome is estimated to affect 1 in 300,000 to 400,000 individuals worldwide 2. More than 150 cases have been described in the scientific literature.
Barth syndrome is caused by mutations in the TAZ gene and is inherited in an X-linked recessive manner 3. Treatment is directed toward the specific symptoms that are apparent in each individual 3. Treatment may require the coordinated efforts of a team of medical professionals which includes a pediatrician, pediatric cardiologist, hematologist, specialist in the treatment of bacterial infections, physical therapist, occupational therapist, and/or other health care professionals. Many infants and children with Barth syndrome require therapy with diuretic and digitalis medications to treat heart failure. Some affected children are gradually removed from such cardiac therapy during later childhood due to improvement of heart functioning. For affected individuals with confirmed neutropenia, complications due to bacterial infection are often preventable by ongoing monitoring and early therapy of suspected infections with antibiotics. For example, antibiotics may be provided as a preventive (prophylactic) therapy during neutropenia to prevent the onset of infection. Other treatment for this disorder is typically symptomatic and supportive 3.
Barth syndrome causes
Mutations in the TAZ gene cause Barth syndrome. The TAZ gene provides instructions for making a protein called tafazzin. Tafazzin is located in structures called mitochondria, which are the energy-producing centers of cells. Tafazzin is involved in altering a fat (lipid) called cardiolipin, which plays critical roles in the mitochondrial inner membrane. Once altered by tafazzin, cardiolipin is key in maintaining mitochondrial shape, energy production, and protein transport within cells.
TAZ gene mutations result in the production of tafazzin proteins with little or no function. As a result, tafazzin cannot alter cardiolipin. A lack of functional cardiolipin impairs normal mitochondrial shape and functions. Tissues with high energy demands, such as the heart and skeletal muscles, are most susceptible to cell death due to reduced energy production in mitochondria. Additionally, abnormally shaped mitochondria are found in affected white blood cells, which could affect their ability to grow (proliferate) and mature (differentiate), leading to neutropenia. Dysfunctional mitochondria likely lead to other signs and symptoms of Barth syndrome.
Barth syndrome inheritance pattern
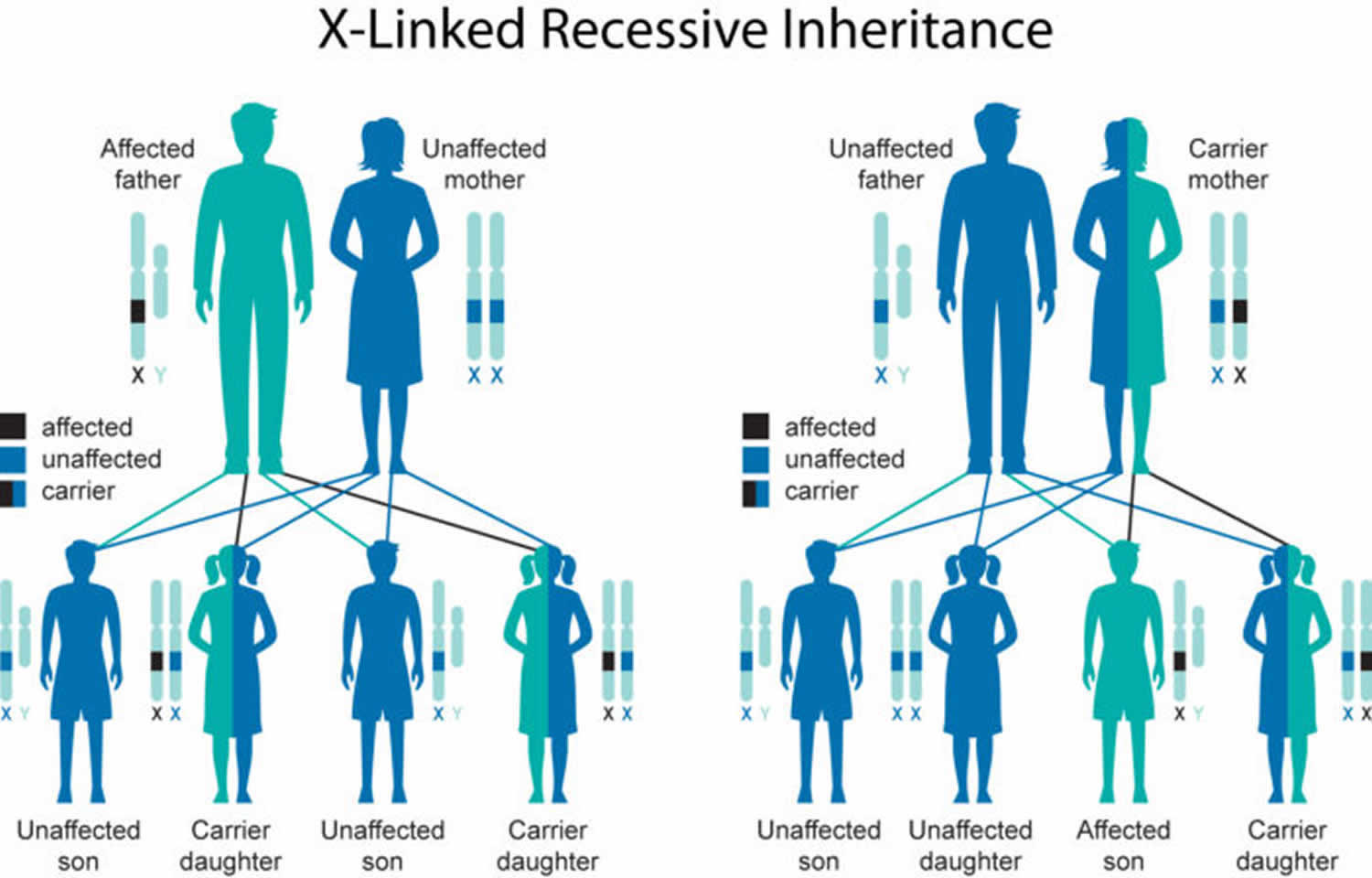
Barth syndrome is inherited in an X-linked recessive pattern. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
X-linked recessive genetic disorders are conditions caused by an abnormal gene on the X chromosome. Females have two X chromosomes but one of the X chromosomes is “turned off” and all of the genes on that chromosome are inactivated. Females who have a disease gene present on one of their X chromosomes are considered carriers for that disorder. Carrier females usually do not display symptoms of the disorder because it is usually the X chromosome with the abnormal gene that is “turned off”, and they have another X chromosome with a working copy of the gene. A male has only one X chromosome. Therefore, if he inherits an X chromosome that contains a non-working gene, he will develop the disease that is associated with that gene.[1] This is why Barth syndrome occurs exclusively in males.
Males with X-linked disorders pass the disease gene to all of their daughters, who will be carriers. A male cannot pass an X-linked gene to his sons, because males always pass their Y chromosome instead of their X chromosome to male offspring (which is what makes the offspring male). A female carrier of an X-linked disorder has two X chromosomes and will always pass one of them onto her offspring (whether it is male or female). Female carriers of and X-linked disorder have a 25 percent chance with each pregnancy to have a carrier daughter like themselves, a 25 percent chance to have a non-carrier daughter, a 25 percent chance to have a son affected with the disease, and a 25 percent chance to have an unaffected son. In some instances, the mother of an affected male may not be a carrier for Barth syndrome and there is no apparent family history of the disease. In such cases, the disorder appears to result from a new mutation of the gene on the X chromosome of the affected individual that occurred randomly for unknown reasons (sporadically).
Figure 1. Barth syndrome X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Barth syndrome symptoms
Barth syndrome is mainly found in early infancy or childhood. However, in some patients, symptoms appear in adulthood. Symptoms can present differently and can vary from one person to another.
Males with Barth syndrome could have various heart problems like dilated cardiomyopathy, hypertrophic cardiomyopathy, endocardial fibroelastosis and left ventricular non-compaction. Dilated cardiomyopathy is when the left ventricle muscle becomes enlarged and weak which decreases the heart’s ability to pump blood. In some people with Barth syndrome, the heart muscles become very thick making it difficult to pump blood (hypertrophic cardiomyopathy). Sometimes, this thickening may be due to the build-up of connective tissues and elastin fibres (endocardial fibroelastosis). In other patients, the left ventricles do not develop properly (left ventricular noncompaction) so instead of the muscle being smooth, it becomes thick and spongy making it difficult to pump blood. These heart findings are almost always present before the age of 5. Sometimes the heart problems can be seen on an ultrasound exam in the last trimester in pregnancy. In addition to structural differences to the heart, in some adolescents and young adults, there could be an irregular heartbeat identified (arrhythmia). The heart problems might lead to decrease in blood circulation from in the body and to the lungs (heart failure). Symptoms of heart failure may include shortness of breath, tiredness and nausea, but the symptoms depend on the child and other factors.
People with Barth syndrome have a low level of white blood cells (neutropenia). The white blood cells in our body help us fight infections. Due to neutropenia, people have mouth ulcers, pneumonia or blood infections. Males with Barth syndrome have weak muscles (hypotonia) especially in the hands and feet. Due to the hypotonia, children take longer to develop gross motor skills like crawling, sitting or walking. Due to the heart issues and weak muscles, these boys do not tolerate exercise well. Males with the condition have growth delay during childhood, but there is a significant growth spurt in puberty. Other symptoms include curvature of the spine (scoliosis) and delayed bone age.
Males with Barth syndrome have distinct facial features. They have a round face with prominent chin and full cheeks. The ears are large, and they have deep set eyes. The facial features become less noticeable with age. The striking feature in adolescence and adulthood is the fat distribution in the hips, thighs and chest.
People diagnosed with the condition have some form of learning disability. They have age appropriate reading skills and vocabulary. However, they may need extra help with mathematics. Their first words or forming sentences can be delayed in comparison to other people. They have delay in developing skills like reading a map, recognizing shapes and finding objects in a picture. The boys have feeding difficulties. The Barth syndrome registry data suggest that a third of males with this condition would need a tube put through the nose or directly to the stomach for feeding. Boys with this condition are picky eaters. Salty, cheesy and spicy food are some of the foods they prefer.
In addition to the cardiomyopathy, neutropenia and growth delay, people with this condition have increased levels of biochemical markers. Increased levels of 3-methyglutaconic acid and 2-ethyl hydracrylic acid in the urine or blood is the common marker used to reach a diagnosis. However, there have been no symptoms associated with the increased levels of these chemicals.
Barth syndrome diagnosis
Consider Barth syndrome if someone has:
- Heart findings like dilated cardiomyopathy, hypertrophic cardiomyopathy and noncompaction of left ventricle
- Increased levels of 3-methylglutaconic acid in blood and/or urine
- Neutropenia
- Hypotonia
- Growth delay
- Characteristic facial features
- Multiple pregnancy losses involving a male fetus have been observed in some families with Barth syndrome.
Barth syndrome may be diagnosed during infancy or early childhood (or, in some cases, at a later age), based upon a thorough clinical evaluation, identification of characteristic physical findings, a complete patient and family history, and a variety of specialized tests. Experts indicate that a diagnosis of Barth syndrome should be considered for any male infant or child with dilated cardiomyopathy of unknown cause (idiopathic); low levels of circulating neutrophils (neutropenia); elevated urinary levels of 3-methylglutaconic acid (aciduria); abnormal mitochondria within heart muscle; and/or muscle abnormalities (myopathy) of unknown cause that occur in association with growth retardation. For infants and children with signs of cardiomyopathy, metabolic screening tests should be conducted, including studies to measure levels of 3-methylglutaconic acid and other organic acids in the urine and blood. An elevated urinary level of 3-methylglutaconic acid (3-methylglutaconic aciduria) has been recognized as a diagnostic sign of Barth syndrome. Persistent low levels of neutrophils in the blood help to confirm the diagnosis in combination with these other signs. Diagnosis may also be confirmed via genetic testing 3.
Molecular genetic testing for mutations in the TAZ gene confirms the diagnosis of Barth syndrome. The TAZ gene testing can be done individually or as a part of a multigene panel.
Barth syndrome treatment
The treatment of Barth syndrome is for specific symptoms. Such treatments may need the efforts of a team of medical professionals, such as pediatricians; physicians who specialize in childhood heart disease (pediatric cardiologists); specialists in the study of the blood and blood-forming tissues (hematologists); specialists in the treatment of bacterial infections, physical therapists; occupational therapists; and/or other health care professionals.
Heart failure and/or bacterial infections are the threats to a patient with Barth syndrome. This is one of the main reasons for a reduced life expectancy. Standard heart failure medications like beta blockers, ACE inhibitors and digoxin are used. This helps in improving the heart function and reduces symptoms of heart failure. Aspirin is used for reducing clot formation. Heart transplant is considered when there is severe heart failure. The heart functioning tends to improve after infancy, so heart transplant should be carefully considered.
For affected people with confirmed neutropenia, complications due to bacterial infection can be prevented by monitoring and starting early therapy of suspected infections with antibiotics. For example, antibiotics may be provided as a preventive (prophylactic) therapy during neutropenia to prevent the onset of infection. Giving uncooked cornstarch before bedtime is recommended to prevent muscle loss. Early intervention like physical therapy is recommended for increasing muscle tone and helps children to attain various developmental milestones.
Genetic counseling is recommended for affected individuals and their families. Other treatment for this disorder is symptomatic and supportive.
Barth syndrome prognosis
One of the most frequent causes of death in Barth syndrome is intractable heart failure 4. Heart failure is thus a significant cause not only of morbidity but also of mortality. Cardiac function usually steadily decreases with disease progression, as has been shown in a study of 73 males 5. In a study of 22 males, 54 hospitalizations were due to heart failure 6. In this cohort, nine patients died from heart failure and two patients from sepsis 6. Median age at death in this cohort was 5.1 months 6. In cases where intractable heart failure requires management by heart transplantation, there is the risk that patients will develop malignancy from chronic immunosuppression 7. The prognosis of cardiac involvement significantly improves if patients survive the first 5 years of life 8. In a study of 27 patients, most of them had recovered near normal cardiac function when assessed by conventional echocardiography 8. However, when analyzing strain, abnormal myocardial deformation and abnormal rotational mechanics were still evident 8. Another factor determining the outcome of Barth syndrome patients is neutropenia. Since neutropenia is associated with an increased risk of infectious disease, affected patients also have an increased risk of dying from an intractable infection or sepsis. Factors identified as influencing survival include severe neutropenia at diagnosis and birth before or after the year 2000 9. Patients with a leukocyte count of <500 cells/μL have a 1-year survival rate of only 25% compared to 68% among those with a leukocyte count of >500 cells//μL 6. The 5-year survival rate among 22 patients was 50%, with no death of a patient older than 3 years 6.
Barth syndrome life expectancy
In general, life expectancy is reduced in patients with Barth syndrome but single patients exceptionally survive into their forties, fifties, or even sixties 10.
References- Barth PG, Wanders RJA, Vreken P. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) – MIM 302060. J Pediatr. 1999;135(3):273–276.
- Barth syndrome. https://ghr.nlm.nih.gov/condition/barth-syndrome
- Barth Syndrome. https://rarediseases.org/rare-diseases/barth-syndrome
- D’Adamo P, Fassone L, Gedeon A, et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet. 1997;61:862–867. doi:10.1086/514886
- Roberts AE, Nixon C, Steward CG, et al. The Barth syndrome registry: distinguishing disease characteristics and growth data from a longitudinal study. Am J Med Genet A. 2012;158A:2726–2732. doi:10.1002/ajmg.a.35609
- Rigaud C, Lebre AS, Touraine R, et al. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis. 2013;8:70. doi:10.1186/1750-1172-8-70
- Ronghe MD, Foot AB, Martin R, Ashworth M, Steward CG. Non-Epstein-Barr virus-associated T-cell lymphoma following cardiac transplantation for Barth syndrome. Acta Paediatr. 2001;90:584–586.
- Kang SL, Forsey J, Dudley D, Steward CG, Tsai-Goodman B. Clinical characteristics and outcomes of cardiomyopathy in Barth syndrome: the UK experience. Pediatr Cardiol. 2016;37:167–176. doi:10.1007/s00246-015-1260-z
- Spencer CT, Bryant RM, Day J, et al. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 2006;118:e337–e346. doi:10.1542/peds.2005-2667
- Ronvelia D, Greenwood J, Platt J, Hakim S, Zaragoza MV. Intrafamilial variability for novel TAZ gene mutation: barth syndrome with dilated cardiomyopathy and heart failure in an infant and left ventricular noncompaction in his great-uncle. Mol Genet Metab. 2012;107:428–432. doi:10.1016/j.ymgme.2012.09.013





{kind=link}