Carney complex
Carney complex also called Carney syndrome, is a rare inherited multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac (heart) myxomas (tumors composed of mucous connective tissue), skin myxomas, multiple endocrine tumors or endocrine over-activity and schwannomas 1. Signs and symptoms of Carney complex commonly begin in the teens or early adulthood.
Individuals with Carney complex are at increased risk of developing noncancerous (benign) tumors called myxomas in the heart (cardiac myxoma) and other parts of the body. Cardiac myxomas may be found in any of the four chambers of the heart and can develop in more than one chamber. These tumors can block the flow of blood through the heart, causing serious complications or sudden death. Myxomas may also develop on the skin and in internal organs. Skin myxomas appear as small bumps on the surface of the skin or as lumps underneath the skin. In Carney complex, myxomas have a tendency to recur after they are removed.
Individuals with Carney complex also develop tumors in hormone-producing (endocrine) glands, such as the adrenal glands located on top of each kidney. People with Carney complex may develop a specific type of adrenal disease called primary pigmented nodular adrenocortical disease (PPNAD). PPNAD (primary pigmented nodular adrenocortical disease) causes the adrenal glands to produce an excess of the hormone cortisol. High levels of cortisol (hypercortisolism) can lead to the development of Cushing syndrome. Cushing’s syndrome causes weight gain in the face and upper body, slowed growth in children, fragile skin, fatigue, and other health problems.
People with Carney complex may also develop tumors of other endocrine tissues, including the thyroid, testes, and ovaries. A tumor called an adenoma may form in the pituitary gland, which is located at the base of the brain. A pituitary adenoma usually results in the production of too much growth hormone. Excess growth hormone leads to acromegaly, a condition characterized by large hands and feet, arthritis, and “coarse” facial features.
Some people with Carney complex develop a rare tumor called psammomatous melanotic schwannoma. This tumor occurs in specialized cells called Schwann cells, which wrap around and insulate nerves. This tumor is usually benign, but in some cases it can become cancerous (malignant).
Almost all people with Carney complex have areas of unusual skin pigmentation. Brown skin spots called lentigines may appear anywhere on the body but tend to occur around the lips, eyes, or genitalia. In addition, some affected individuals have at least one blue-black mole called a blue nevus.
Carney complex is a rare disorder with an unknown prevalence 2. In the largest genotyped series of patients, 63% were females and 37% were males 3. The NIH-Mayo clinic and other centers in the United States and the Cochin Hospital in France have collectively reported more than 750 Carney complex cases including Caucasians, African-Americans, and Asians from all continents [North and South America, Europe, Asia (Japan, China, India) 2.
Some families with Carney complex have been found to have mutations in the PRKAR1A gene located on the long arm (q) of chromosome 17 (17q22-q24) coding for the regulatory subunit type 1 alpha of protein kinase A (PKA) gene. Carney complex is believed to be inherited in an autosomal dominant manner, which means that one copy of the altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person inherits the mutation from one affected parent 4. Approximately 70% of Carney complex cases had an affected parent (67 families), whereas the remaining had no known affected relatives and carried de novo germline mutations 5. In all inherited cases, Carney complex was passed on as an autosomal dominant trait with an almost 100% penetrance.
Figure 1. Carney complex
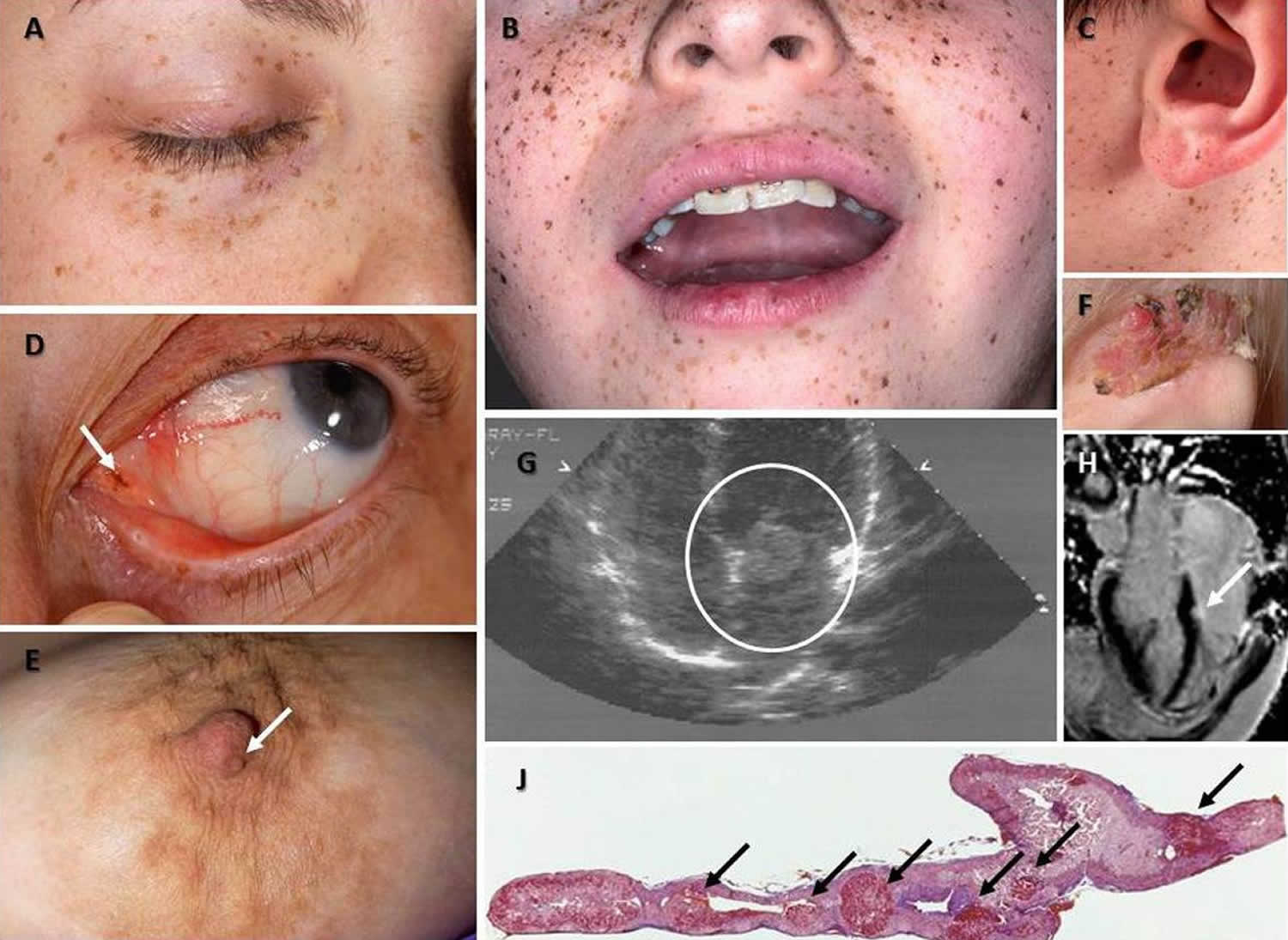
Footnote: (A) Characteristic distribution of the lentigines on the eyelids, (B) the vermillion border of the lips and the cheeks and the ears, including the ear canal (C) in patients with Carney complex; such typical pigmentation on the face is only present in less than one third of the patients but it is rather diagnostic when present. (D) A pigmented macule (arrow) on the outer canthus of a patient with Carney complex who had minimal other pigmentation; inner or outer canthal pigmentation such as the one shown here is only seen in Carney complex and Peutz-Jeghers syndrome making it diagnostic for these two conditions. (E) Nipple myxoma in a female patient with Carney complex. (F) Ear myxoma complicated by chronic infection and tissue overgrowth in a toddler with Carney complex. (G and H) Large myxoma (circled) between the left atrium and (G) ventricle detected by echocardiography in an adolescent with Carney complex who had surgery immediately thereafter and a much smaller myxoma (arrow) of the left ventricle originating from the cardiac diaphragm detected by cardiac MRI in an older patient with Carney complex (H); this myxoma was followed by serial echocardiogram and has yet to be operated, as it is not growing and poses no immediate risks. (J) 5x magnification hematoxylin and eosin staining of the adrenal gland of a patient with Carney complex: the characteristic nodules of PPNAD are shown by the arrows; the overall size of the gland is normal and the nodules may not be visible by imaging studies.
[Source 1 ]Carney complex causes
Mutations in the PRKAR1A gene cause most cases of Carney complex. The PRKAR1A gene is believed to be a tumor suppressor gene. A tumor suppressor is a gene that slows down cell division, repairs damage to the DNA of cells, and tells cells when to die, a normal process called apoptosis. PRKAR1A gene provides instructions for making one part (subunit) of an enzyme called protein kinase A, which promotes cell growth and division (proliferation). The subunit produced from the PRKAR1A gene, called type 1 alpha, helps control whether protein kinase A (PKA) is turned on or off.
Most mutations in the PRKAR1A gene that cause Carney complex result in an abnormal type 1 alpha subunit that is quickly broken down (degraded) by the cell. The lack of this subunit causes protein kinase A to be turned on more often than normal, which leads to uncontrolled cell proliferation. The signs and symptoms of Carney complex are related to the unregulated growth of cells in many parts of the body.
Some individuals with Carney complex do not have identified mutations in the PRKAR1A gene. Researchers believe that additional, as yet unidentified, genes may cause the disorder in these cases (genetic heterogeneity). Investigators have determined that an as yet unidentified gene on the short arm (p) of chromosome 2, designated as 2p16, is involved in some cases of Carney complex. These cases are sometimes referred to as Carney complex type 2. More research is necessary to determine the gene on this region of chromosome 2 that causes certain cases of Carney complex.
Carney complex inheritance pattern
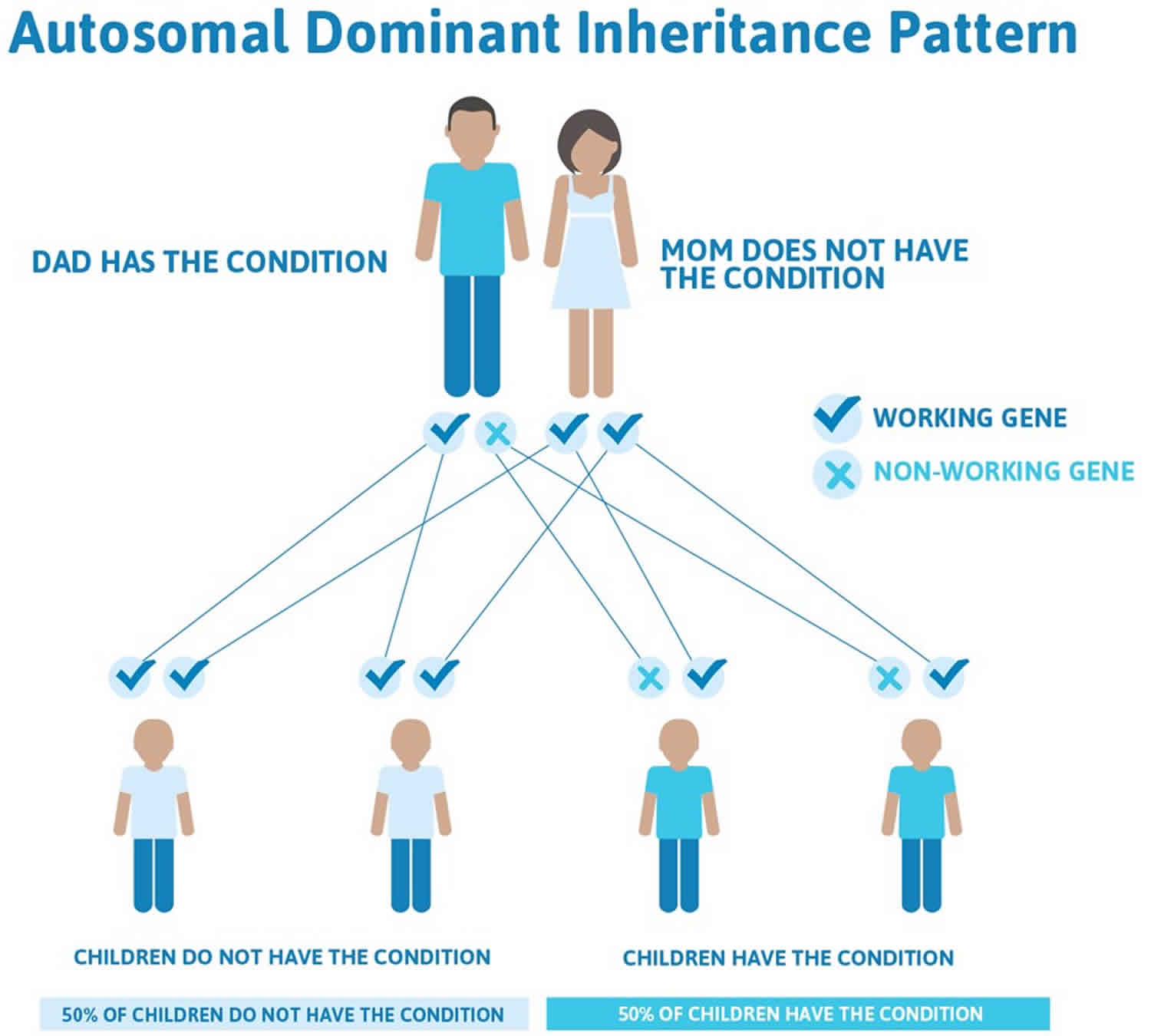
Carney complex is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In approximately 80 percent of cases, an affected person inherits the mutation from one affected parent. The remaining cases result from new mutations in the gene and occur in people with no history of Carney complex in their family. This is called a de novo mutation.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Figure 2 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 2. Carney complex autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Carney complex symptoms
The symptoms and severity of Carney complex can vary greatly from one person to another, even among members of the same family. The disorder may be evident at birth, but the median age of diagnosis is 20. Many of the signs and symptoms of Carney complex become apparent during the teen-age years or during early adulthood.
It is important to note that affected individuals may not have all of the symptoms discussed below. Affected individuals or parents of affected children should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
The presenting sign of Carney complex is often numerous tiny (freckle-like) black or brown spots on the skin (multiple lentigines). Although these tiny, flat discolorations resemble freckles, they tend to be darker and usually range between 2 and 10 millimeters in size. Lentigines are most often found around the upper and lower lips (pink part), on the eyelids, the membrane lining the eyes and the inside of the eyelids (conjunctiva), the ears and the genital area. Lentigines can be apparent at birth. In most cases, lentigines increase in number around puberty. Lentigines tend to fade in the 40s.
Another type of skin abnormality associated with Carney complex are blue nevi. Blue nevi are raised, small, blue or bluish-black spots on the skin. Less frequently, affected individuals may develop areas of light brown discoloration with irregular or jagged borders (café au lait spots) and white patches of skin due to loss of pigment (depigmented lesions).
Individuals with Carney complex are prone to developing a type of tumor known as a myxoma. Myxomas are small benign tumors consisting of connective tissue. Myxomas can affect any area of the body except the hands and feet and, in Carney complex, are most commonly seen in the heart (cardiac myxomas). One myxoma or multiple myxomas may be present. Myxomas may develop in any or all of the chambers of the heart. The normal heart has four chambers. The two upper chambers, known as atria, are separated from each other by a fibrous partition known as the atrial septum. The two lower chambers are known as ventricles and are separated from each other by the ventricular septum. Valves connect the atria (left and right) to their respective ventricles. The valves allow for blood to be pumped through the chambers. Blood travels from the right ventricle through the pulmonary artery to the lungs where it receives oxygen. The blood returns to the heart through pulmonary veins and enters the left ventricle. The left ventricle sends the now oxygen-filled blood into the main artery of the body (aorta).
Cardiac myxomas can potentially cause serious life-threatening complications usually due to the obstruction of blood flow. Specific complications can include stroke due to blockage of an artery (embolism) in the brain by a piece of detached cardiac myxoma or the inability of the heart to pump blood to the rest of the body, causing fluid buildup in the heart, lungs and various body tissues (congestive heart failure). Complete blockage (occlusion) of a valvular opening potentially can cause sudden death. Additional heart abnormalities that may occur in individuals with Carney complex include palpitations, diastolic heart murmurs and “tumor plop”, which is a distinctive sound related to the movement of a tumor within the heart. Cardiac myxomas may also cause general, nonspecific symptoms including fatigue, fever, muscle pain (myalgia), difficulty breathing (dyspnea) and unintended weight loss.
Less often, myxomas can be found in other areas of the body in addition to or instead of the heart. These areas include the eyelids, nipples and the external ear canal. Any area of the body can be affected except the hands and feet. Cutaneous myxomas may present as white, pink or flesh-colored papules or small nodules just under the surface of the skin. They generally do not cause any symptoms and can appear at any time from birth through the fourth decade. They are generally 1 cm or less in diameter. Myxomas may also occur in the oropharynx area, which encompasses the tongue, hard palate and the back wall of the throat (pharynx). In women, myxomas can also occur in the breasts after puberty. In addition, women may develop myxomas in the genital tract including the vagina, uterus and cervix. In rare cases, affected individuals may develop an osteochondromyxoma, a rare bone tumor predominantly affecting the nasal sinuses or the long bones of the arms and legs.
Individuals with Carney complex can develop a wide variety of abnormalities affecting the endocrine system including the development of multiple benign tumors. The endocrine system is the network of glands that secrete hormones into the bloodstream where they travel to various areas of the body. These hormones regulate the chemical processes (metabolism) that influence the function of various organs and activities within the body. Hormones are involved in numerous vital processes including regulating heart rate, body temperature and blood pressure as well as cell differentiation and growth and also in modulation of several metabolic processes.
The most common endocrine tumor associated with Carney complex is known as primary pigmented nodular adrenocortical disease (PPNAD). PPNAD affects approximately 25 percent of individuals with Carney complex. The condition is characterized by multiple tiny nodules affecting the adrenal glands. The adrenal glands are situated atop the kidneys and produce cortisol, which is a hormone that is involved in certain metabolic and cardiovascular processes and helps the body respond to stress. PPNAD is a rare disorder that predominantly occurs in individuals with Carney complex. Elevated cortisol levels due to PPNAD can cause a disorder known as Cushing’s syndrome.
Cushing’s syndrome is a disorder that occurs because of abnormally high levels of cortisol in the body. The symptoms develop slowly over time. Affected children may experience weight gain and growth delays. Adults may experience progressive weight gain resulting in extra fat in the midsection, between the shoulder blades, around the neck and in the face, giving the face a rounded appearance. Additional symptoms include high blood pressure (hypertension), fatigue, purple or red stretch marks (striae) on the abdomen, excessive thirst, weakness of the muscles closest to the body (proximal muscle weakness) and psychological disturbances. Some affected women may experience disturbances of their menstrual cycles and a male pattern of hair growth (hirsutism). Some affected individuals may have progressive thinning and loss of protein of bones (osteoporosis) because of prolonged mild elevation of cortisol.
Some individuals with Carney complex may have a benign tumor (adenoma) of the pituitary gland. The pituitary gland is a small gland located near the base of the skull that produces several hormones and releases them into the bloodstream as needed by the body. Infrequently, individuals with Carney complex can develop a condition known as acromegaly. Acromegaly occurs when a pituitary adenoma causes increased production of growth hormone. Symptoms include abnormal enlargement of the bones of the arms, legs and head. The bones in the jaws and in the front of the skull are typically most often affected. Consequently, affected individuals may exhibit abnormal enlargement of the hands, feet, jaws and face. Acromegaly may also cause thickening of the soft tissues of the body, particularly the heart and accelerated growth leading to tall stature. Acromegaly is a slowly progressive condition.
Some individuals with Carney complex may have multiple tumors (nodules) affecting the thyroid. The thyroid is a butterfly-shaped gland at the base of the neck that secretes hormones that help to regulate growth and development in the body. In most cases, these nodules are benign nonfunctioning adenomas. Nonfunctioning means that the adenoma does not produce excess hormones. Some affected individuals may have papillary or follicular thyroid carcinoma. In rare cases, thyroid carcinoma has developed in individuals with a longstanding history of multiple thyroid nodules.
In males, an endocrine tumor known as a large-cell calcifying Sertoli cell tumor (LCCSCT) may develop. This tumor is found in the testes as tiny areas of calcification and sometimes can be associated with early development of secondary sexual characteristics (precocious puberty). This tumor can potentially cause breast development in males (gynecomastia). LCCSCTs are almost always benign; only one case has ever been reported of malignant transformation. Approximately one-third of males with Carney complex have these tumors present when first diagnosed with the disorder, usually during the first decade of life. Virtually all adult males develop LCCSCTs at some point. Less frequently, two other testicular tumors can also occur in males with Carney complex, specifically Leydig cell tumors and pigmented nodular adrenocortical rest tumors. Leydig cell tumors potentially can become malignant. Pigmented nodular adrenocortical rest tumors are benign, but can cause Cushing’s syndrome.
In some cases, testicular tumors can affect fertility due to replacement and obstruction of the tiny tubes in which sperm is formed (seminiferous tubules) and decreased sperm motility (oligoasthenospermia). The presence of these tumors can cause the testes to become abnormally large (macroorchidism) as well.
Although not a frequent finding, some females with Carney complex have developed ovarian cysts. In approximately 10 percent of cases, individuals with Carney complex may develop a psammomatous melanotic schwannoma, which is a rare tumor of the peripheral nerve sheath. They can occur anywhere along the central and peripheral nervous system, but most often affect the gastrointestinal tract (including the esophagus) or the network of nerves adjacent to the spine (paraspinal sympathetic chain). Depending upon their location psammomatous melanotic schwannomas can cause pain or discomfort as well as damage to one or more nerves (radiculopathy). In rare cases, these tumors can become malignant.
Skin pigment abnormalities
- Pale brown to black lentigines are the most common presenting feature of Carney complex and may be present at birth. Typically, they increase in number and appear anywhere on the body including the face, the lips, and mucosa around puberty. These lentigines tend to fade after the fourth decade, but may still be evident in the eighth decade.
- Additional pigmentary abnormalities that develop over time are epithelioid-type blue nevi (small bluish domed papules with a smooth surface), combined nevi, café au lait macules, and depigmented lesions.
Myxomas
- Cutaneous myxomas are papules or subcutaneous nodules that usually have a smooth surface and are white, flesh-colored, opalescent, or pink. They appear between birth and the fourth decade. Most individuals with Carney complex have multiple lesions. Myxomas occur on any part of the body except the hands and feet and typically affect the eyelids, external ear canal, and nipples.
- Cardiac myxomas occur at a young age and may occur in any or all cardiac chambers. Cardiac myxomas present with symptoms related to intracardiac obstruction of blood flow, embolic phenomenon (into the systemic circulation), and/or heart failure. Myxomas that completely occlude a valvular orifice can cause sudden death.
- Breast myxomas, often bilateral, occur in females after puberty. Both males and females may develop nipple myxomas at any age.
- Other sites for myxomas include the oropharynx (tongue, hard palate, pharynx) and the female genital tract (uterus, cervix, vagina).
- Osteochondromyxoma is a rare myxomatous tumor of the bone that affects nasal sinuses and long bones.
Endocrine tumors
- Primary pigmented nodular adrenocortical disease (PPNAD) is associated with adrenocorticotropic hormone (ACTH)-independent overproduction of cortisol (hypercortisolism). PPNAD is the most frequently observed endocrine tumor in individuals with Carney complex, occurring in an estimated 25% of affected individuals. Among those with a PRKAR1A pathogenic variant, Cushing syndrome caused by PPNAD is seen in 70% of affected females before age 45 years but in only 45% of affected males, likely reflecting the generally higher frequency of Cushing syndrome in females. Histologic evidence of PPNAD has been found in almost every individual with Carney complex undergoing autopsy. Symptomatic individuals have Cushing syndrome. The hypercortisolism of PPNAD is usually insidious in onset. In children, hypercortisolism is manifest first as weight gain and growth arrest. In adults, long-standing hypercortisolism results in central obesity, “moon facies,” hirsutism, striae, hypertension, buffalo hump fat distribution, weakness, easy bruising, and psychological disturbance. In a minority of individuals, PPNAD presents in the first two to three years; in the majority, it presents in the second or third decade.
- Growth hormone (GH)-producing adenoma. Clinically evident acromegaly is a relatively frequent manifestation of Carney complex, occurring in approximately 10% of adults at the time of presentation. Gigantism, resulting from excess GH secretion prior to puberty, is rare. However, asymptomatic increased serum concentration of GH and insulin-like growth factor type-1 (IGF-1), as well as subtle hyperprolactinemia, may be present in up to 75% of individuals with Carney complex. Somatomammotroph hyperplasia, a putative precursor of GH-producing adenoma, may explain the protracted period of onset of clinical acromegaly in individuals with Carney complex.
- Testicular tumors. Large-cell calcifying Sertoli cell tumors (LCCSCT) are observed in one third of affected males at the time of presentation, which is often within the first decade. Most adult males with Carney complex have evidence of LCCSCT. The tumors are often multicentric and bilateral. LCCSCT is almost always benign; malignancy has been reported only once, in an individual age 62 years. LCCSCT may be hormone producing; gynecomastia in prepubertal and peripubertal boys may result from increased P-450 aromatase expression. Other testicular tumors observed in individuals with LCCSCT include Leydig cell tumors and (pigmented nodular) adrenocortical rest tumors.
- Thyroid adenoma or carcinoma. Up to 75% of individuals with Carney complex have multiple thyroid nodules, most of which are nonfunctioning thyroid follicular adenomas. Thyroid carcinomas, both papillary and follicular, can occur and occasionally may develop in a person with a long history of multiple thyroid adenomas.
Psammomatous melanotic schwannoma (PMS). This rare tumor of the nerve sheath occurs in approximately 10% of individuals with Carney complex. Malignant degeneration occurs in approximately 10% of those with Carney complex 6. Psammomatous melanotic schwannoma may occur anywhere in the central and peripheral nervous system; it is most frequently found in the nerves of the gastrointestinal tract (esophagus and stomach) and paraspinal sympathetic chain (28%). The spinal tumors present as pain and radiculopathy in adults (mean age 32 years).
Breast ductal adenoma is a benign tumor of the mammary gland ducts.
Fertility
Large-cell calcifying Sertoli cell tumors (LCCSCT) causes replacement and obstruction of seminiferous tubules, macroorchidism, oligoasthenospermia, and inappropriate hormone production or aromatization. Despite these findings, fertility is frequently preserved.
Carney complex diagnosis
A diagnosis of Carney complex is made based upon a detailed patient history, a thorough clinical evaluation, a variety of specialized tests and identification of characteristic symptoms. According to the medical literature, identification of two or more of the following symptoms in typical fashion is indicative of Carney complex: cardiac myxoma: skin myxoma; lentiginosis; multiple blue nevi; primary pigmented nodular adrenocortical disease (PPNAD); testicular tumors; acromegaly; thyroid tumors, melanotic schwannoma; or an osteochondromyxoma.
Tests that may performed to help obtain a diagnosis of Carney complex include surgical removal and microscopic study of affected skin (skin biopsy), urine analysis to detect elevated levels of cortisol (indicative of Cushing’s syndrome), an echocardiogram to detect the presence of cardiac myxomas, and blood tests to detect abnormal high levels of certain hormones such as insulin-like growth factor, cortisol and prolactin due to the presence of endocrine tumors.
Carney complex should be suspected in individuals with the following features 4
Major diagnostic criteria for Carney complex
- Spotty skin pigmentation with typical distribution (lips, conjunctiva and inner or outer canthi, vaginal and penile mucosa)
- Myxoma* (cutaneous and mucosal)
- Cardiac myxoma*
- Breast myxomatosis* or fat-suppressed MRI findings suggestive of this diagnosis
- Primary pigmented nodular adrenocortical disease (PPNAD)* or paradoxic positive response of urinary glucocorticosteroid excretion to dexamethasone administration during Liddle’s test
- Acromegaly as a result of growth hormone (GH)-producing adenoma*
- Large-cell calcifying Sertoli cell tumor (LCCSCT)* or characteristic calcification on testicular ultrasound
- Thyroid carcinoma* or multiple, hypoechoic nodules on thyroid ultrasound in a child younger than age 18 years
- Psammomatous melanotic schwannomas (PMS)*
- Blue nevus, epithelioid blue nevus*
- Breast ductal adenoma*
- Osteochondromyxoma*
Note: * denotes after histologic confirmation 7.
Supplementary criteria
- Affected first-degree relative
- Inactivating pathogenic variant in PRKAR1A
Findings suggestive of or possibly associated with Carney complex, but not diagnostic for the disease 8
- Intense freckling (without darkly pigmented spots or typical distribution)
- Blue nevus, common type (if multiple)
- Café au lait macules or other “birthmarks”
- Elevated IGF-I levels, abnormal glucose tolerance test (GTT), or paradoxic GH response to TRH (thyrotropin-releasing hormone) testing in the absence of clinical acromegaly
- Cardiomyopathy
- Pilonidal sinus
- History of Cushing’s syndrome, acromegaly, or sudden death in extended family
- Multiple skin tags or other skin lesions; lipomas
- Colonic polyps (usually in association with acromegaly)
- Hyperprolactinemia (usually mild and almost always combined with clinical or subclinical acromegaly)
- Single, benign thyroid nodule in a child younger than age 18 years; multiple thyroid nodules in an individual older than age 18 years (detected on ultrasound examination)
- Family history of carcinoma, in particular of the thyroid, colon, pancreas, and ovary; other multiple benign or malignant tumors
A diagnosis of Carney complex can be confirmed in some cases through molecular genetic testing, which can reveal the characteristic mutation of the PRKAR1A gene that causes the disorder in many cases. Molecular genetic testing is available on a clinical basis.
Carney complex treatment
The treatment of Carney complex is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, surgeons, cardiologists, cardiothoracic surgeons, endocrinologists, dermatologists and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment.
Affected individuals should receive regular screening for the various potential symptoms associated with Carney complex. No specific guidelines have been agreed upon in the medical literature, but most sources recommend yearly screening for cardiac myxoma.
The specific therapeutic procedures and interventions for individuals with Carney complex will vary, depending upon numerous factors including the specific symptoms present, the extent of the disorder, an individual’s age and overall health, tolerance of certain medications or procedures, personal preference and other factors. Decisions concerning the use of particular therapeutic interventions should be made by physicians and other members of the healthcare team in careful consultation with the patient and/or parents based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
Cardiac myxomas require open-heart surgical removal. Despite surgery, cardiac myxomas can recur. Cutaneous and mammary myxomas also require surgical removal. Surgical removal of the adrenal glands (adrenalectomy) may be required in individuals who develop Cushing’s syndrome.
A pituitary adenoma may be treated by transsphenoidal surgery; a procedure in which all or part of a pituitary tumor is removed. In some cases, surgery results in a rapid therapeutic response, and lowering growth hormone levels.
Although rare, cancerous tumors may occur in Carney complex including thyroid carcinoma and malignant psammomatous melanotic schwannoma. Surgical removal of the primary tumor and any metastatic lesions is necessary. Individuals who have their thyroid removed will require lifelong supplementation of the hormones normally produced by the thyroid.
Surgical removal of one or both testes (orchiectomy) may be recommended to avoid or cope with the adverse effects (e.g., gynecomastia) of excessive hormone production that can occur in males with large-cell calcifying Sertoli cell tumor (LCCSCT). However, because these tumors are benign, some physicians prefer surgery that spares fertility. In some cases, surgery that spares the testicles combined with strict monitoring of growth and puberty milestones has been performed. The administration of antiestrogen drugs may be necessary in the case of recurrence. Leydig cell tumors, because of the potential of malignant transformation, are usually treated by orchiectomy.
Genetic counseling should be offered to affected individuals and their families. Other treatment is symptomatic and supportive.
Monitoring
- For prepubertal children: echocardiography annually or biannually for those with a history of excised myxoma; testicular ultrasound with close monitoring of linear growth rate and annual pubertal staging.
- For postpubertal children and adults: echocardiogram annually or biannually for those with a history of excised myxoma; annual testicular ultrasound; baseline thyroid ultrasound with repeat as necessary; baseline transabdominal ultrasound of the ovaries with repeat as necessary; annual urinary free cortisol levels; annual serum IGF-1 levels.
Further evaluation in all age groups may include: diurnal cortisol levels, dexamethasome stimulation test, and adrenal computed tomography for primary pigmented nodular adrenocortical disease; pituitary MRI, 3-hour oral glucose tolerance test, and 90-minute thyroid releasing hormone testing for gigantism/acromegaly; MRI (brain, spine, chest, abdomen, retroperitoneum, pelvis) for psammamotous melanotic schwannoma.
Carney complex life expectancy
Most individuals with Carney complex have a normal life span. However, because some die at an early age, the average life expectancy for individuals with Carney complex is 50 years 4. Causes of death include complications of cardiac myxoma (myxoma emboli, cardiomyopathy, cardiac arrhythmia, surgical intervention), metastatic or intracranial psammomatous melanotic schwannomas, thyroid carcinoma, and metastatic pancreatic and testicular tumors.
References- Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol. 2015;173(4):M85-M97. doi:10.1530/EJE-15-0209 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4553126
- Espiard S, Bertherat J. Carney complex. Front Horm Res. 2013;41:50-62. doi: 10.1159/000345669
- Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, René-Corail F, Stergiopoulos S, Bourdeau I, Bei T, Clauser E, Calender A, Kirschner LS, Bertagna X, Carney JA, Stratakis CA. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009 Jun;94(6):2085-91. doi: 10.1210/jc.2008-2333.
- Stratakis CA, Raygada M. Carney Complex. 2003 Feb 5 [Updated 2018 Aug 16]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1286
- Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001 Sep;86(9):4041-6. doi: 10.1210/jcem.86.9.7903
- Watson JC, Stratakis CA, Bryant-Greenwood PK, Koch CA, Kirschner LS, Nguyen T, Carney JA, Oldfield EH. Neurosurgical implications of Carney complex. J Neurosurg. 2000;92:413–8.
- Mateus C, Palangié A, Franck N, Groussin L, Bertagna X, Avril MF, Bertherat J, Dupin N. Heterogeneity of skin manifestations in patients with Carney complex. J Am Acad Dermatol. 2008;59:801–10.
- Mateus C, Palangié A, Franck N, Groussin L, Bertagna X, Avril MF, Bertherat J, Dupin N. Heterogeneity of skin manifestations in patients with Carney complex. J Am Acad Dermatol. 2008 Nov;59(5):801-10. doi: 10.1016/j.jaad.2008.07.032





{kind=link}