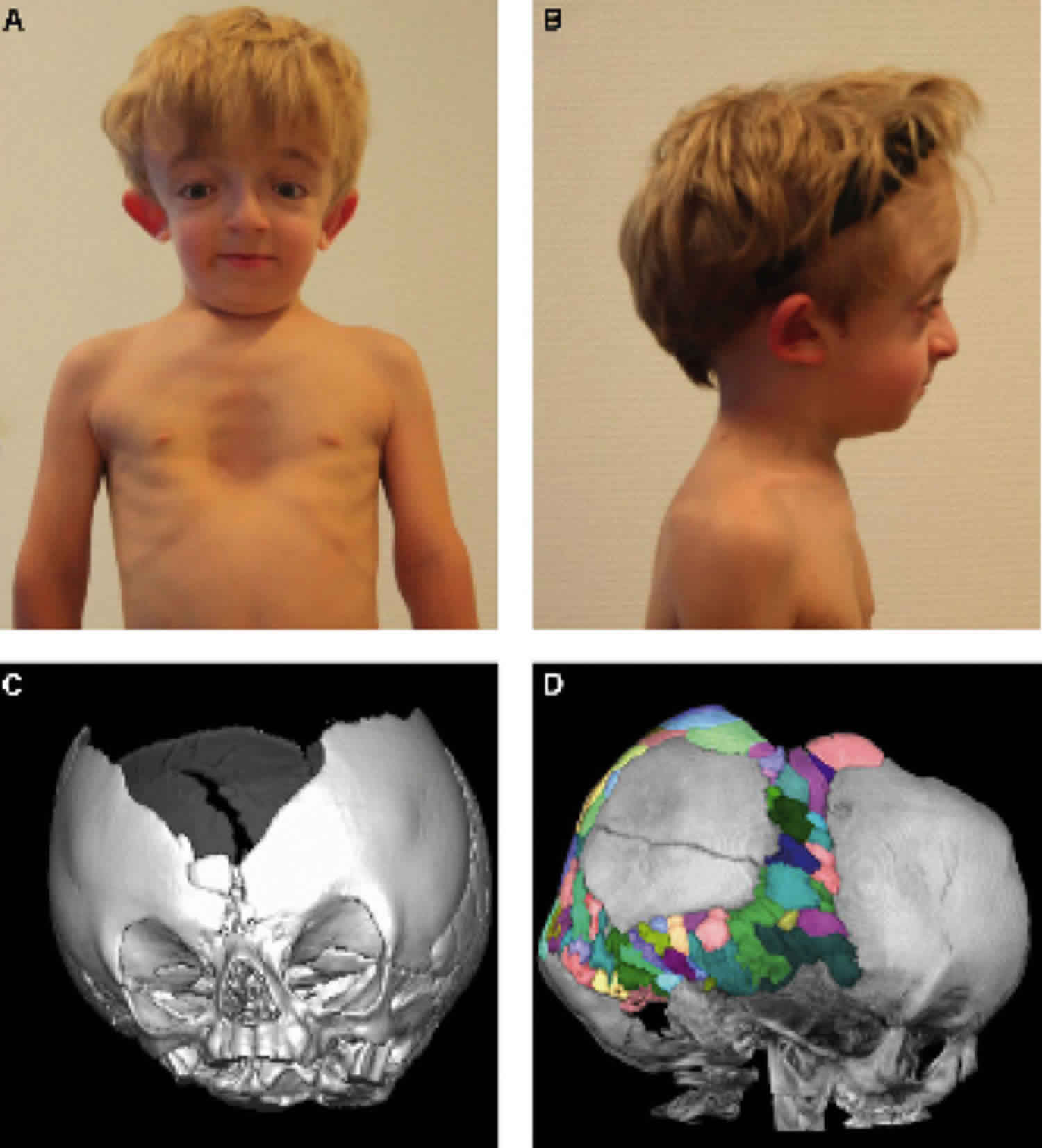
Carpenter syndrome
Carpenter syndrome also called acrocephalopolysyndactyly type 2, is a ultra-rare genetic condition characterized by the premature fusion of certain skull bones (craniosynostosis), abnormalities of the fingers and toes, and other developmental problems. Craniosynostosis prevents the skull from growing normally, frequently giving the head a pointed appearance (acrocephaly). Carpenter syndrome belongs to a group of rare genetic disorders known as “acrocephalopolysyndactyly” disorders 1. In severely affected individuals, the abnormal fusion of the skull bones results in a deformity called a cloverleaf skull (also known as Kleeblattschädel syndrome). Craniosynostosis can cause differences between the two sides of the head and face (craniofacial asymmetry). Early fusion of the skull bones can affect the development of the brain and lead to increased pressure within the skull (intracranial pressure). Premature fusion of the skull bones can cause several characteristic facial features in people with Carpenter syndrome. Distinctive facial features may include a flat nasal bridge, outside corners of the eyes that point downward (down-slanting palpebral fissures), low-set and abnormally shaped ears, underdeveloped upper and lower jaws, and abnormal eye shape. Some affected individuals also have dental abnormalities including small primary (baby) teeth. Vision problems also frequently occur.
Abnormalities of the fingers and toes include fusion of the skin between two or more fingers or toes (syndactyly), unusually short fingers or toes (brachydactyly), or extra fingers or toes (polydactyly). In Carpenter syndrome, cutaneous syndactyly is most common between the third (middle) and fourth (ring) fingers, and polydactyly frequently occurs next to the big or second toe or the fifth (pinky) finger.
People with Carpenter syndrome often have intellectual disability, which can range from mild to profound. However, some individuals with this condition have normal intelligence. The cause of intellectual disability is unknown, as the severity of craniosynostosis does not appear to be related to the severity of intellectual disability.
Other features of Carpenter syndrome include obesity that begins in childhood, a soft out-pouching around the belly-button (umbilical hernia), hearing loss, and heart defects. Additional skeletal abnormalities such as deformed hips, a rounded upper back that also curves to the side (kyphoscoliosis), and knees that are angled inward (genu valgum) frequently occur. Nearly all affected males have genital abnormalities, most frequently undescended testes (cryptorchidism).
A few people with Carpenter syndrome have organs or tissues within their chest and abdomen that are in mirror-image reversed positions. This abnormal placement may affect several internal organs (situs inversus); just the heart (dextrocardia), placing the heart on the right side of the body instead of on the left; or only the major (great) arteries of the heart, altering blood flow.
The signs and symptoms of Carpenter syndrome vary considerably, even within the same family. The life expectancy for individuals with Carpenter syndrome is shortened but extremely variable.
The signs and symptoms of Carpenter syndrome are similar to another genetic condition called Greig cephalopolysyndactyly syndrome. The overlapping features, which include craniosynostosis, polydactyly, and heart abnormalities, can cause these two conditions to be misdiagnosed; genetic testing is often required for an accurate diagnosis.
Most Carpenter syndrome cases are caused by mutations in the RAB23 gene. In several affected individuals, Carpenter syndrome was caused by mutations in the MEGF8 gene; these individuals are referred to as having Carpenter syndrome type 2. Both types of Carpenter syndrome are inherited in an autosomal recessive manner.
Carpenter syndrome is thought to be a rare condition; approximately 70 cases have been described in the scientific literature 2. Carpenter syndrome appears to affect males and females in relatively equal numbers. In 10 patients that had sequence analysis for the disease causing gene, homozygosity (two copies) for the same nonsense mutation, (a change in the DNA that causes a change in the protein) was found. This is indicative of a founder effect in patients of northern European descent, which means that a high prevalence of a genetic disorder in an isolated or inbred population is due to the fact that many members of the population are derived from a common ancestor who had the disease causing mutation.
Figure 1. Carpenter syndrome
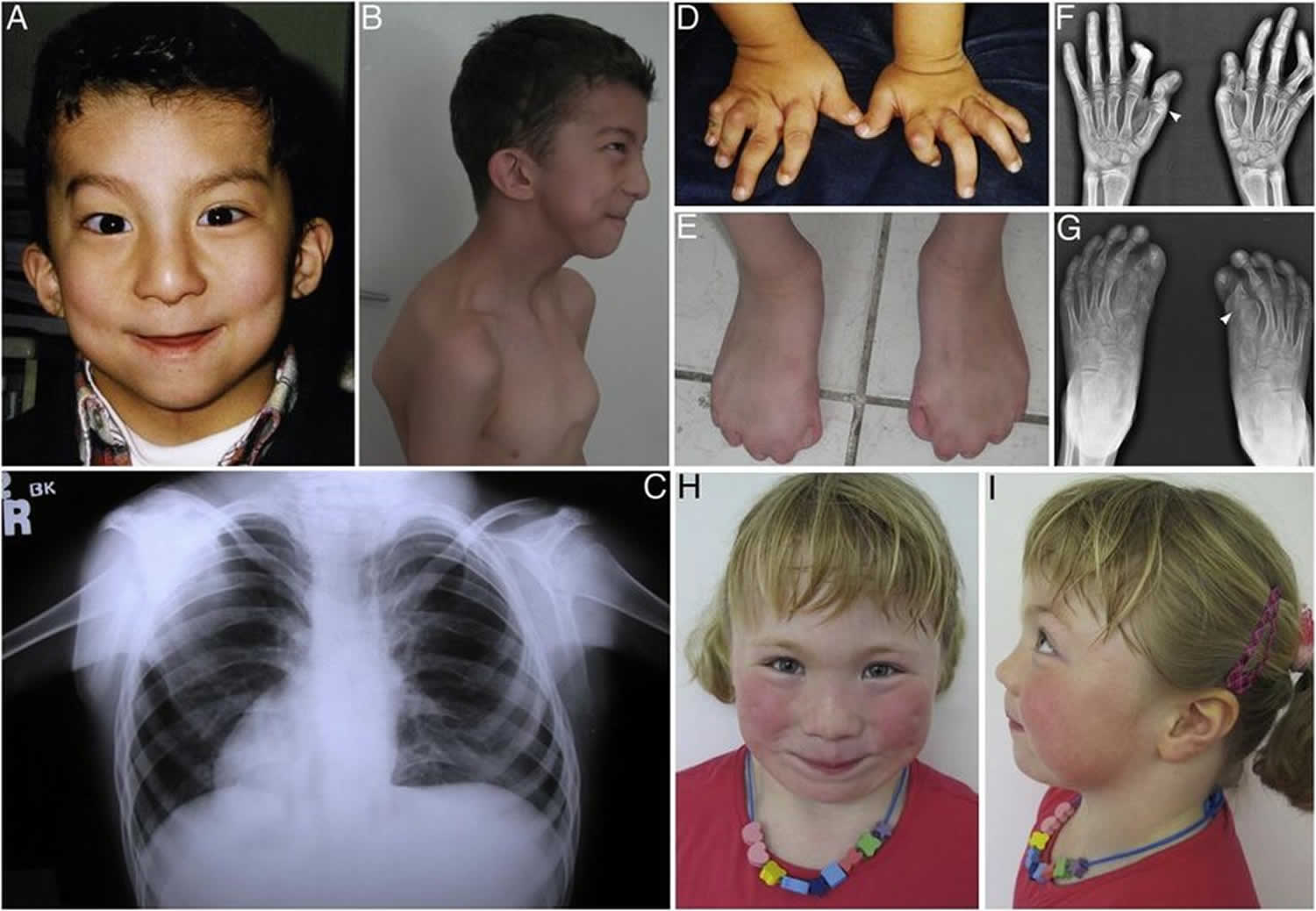
Footnote: Pictures of children with carpenter syndrome with a variety of related disorders.
[Source 3 ]Carpenter syndrome causes
Carpenter syndrome is caused by mutations in the RAB23 or MEGF8 gene. In most instances Carpenter syndrome are caused by a mutation in the RAB23 gene. In a small subset of people, Carpenter syndrome is caused by a mutation in the MEGF8 gene. The RAB23 gene provides instructions for making a protein that is involved in a process called vesicle trafficking, which moves proteins and other molecules within cells in sac-like structures called vesicles. The Rab23 protein transports vesicles from the cell membrane to their proper location inside the cell. Vesicle trafficking is important for the transport of materials that are needed to trigger signaling during development. For example, the Rab23 protein regulates a developmental pathway called the hedgehog signaling pathway that is critical in cell growth (proliferation), cell specialization, and the normal shaping (patterning) of many parts of the body.
The MEGF8 gene provides instructions for making a protein whose function is unclear. Based on its structure, the Megf8 protein may be involved in cell processes such as sticking cells together (cell adhesion) and helping proteins interact with each other. Researchers also suspect that the Megf8 protein plays a role in normal body patterning.
Mutations in the RAB23 or MEGF8 gene lead to the production of proteins with little or no function. It is unclear how disruptions in protein function lead to the features of Carpenter syndrome, but it is likely that interference with normal body patterning plays a role. For reasons that are unknown, people with MEGF8 gene mutations are more likely to have dextrocardia and other organ positioning abnormalities and less severe craniosynostosis than individuals with RAB23 gene mutations.
The RAB23 gene is located on the short arm of the chromosome 6 (6p12.1) and different mutations in the gene have been reported in the studied families. It is possible to read (sequence) this gene in some diagnostic and research laboratories to confirm the diagnosis if it is not clear clinically. Affected individuals in the same family with the same mutations may have different symptoms and variable severity (intrafamilial variability).
Carpenter syndrome inheritance pattern
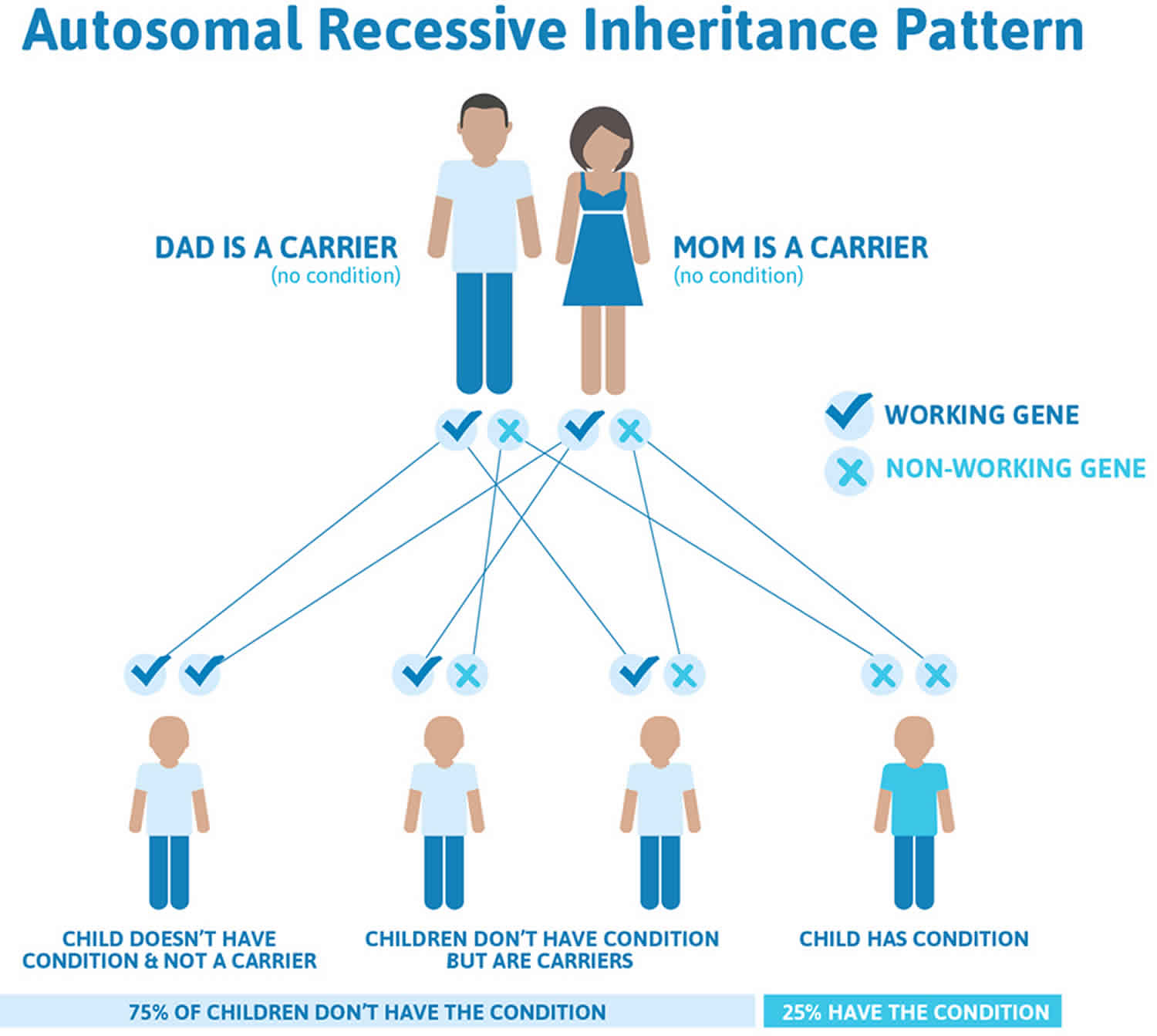
Carpenter syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Carpenter syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Carpenter syndrome signs and symptoms
Carpenter syndrome is typically evident at or shortly after birth. Primary findings associated with Carpenter syndrome include premature closure of the fibrous joints (cranial sutures) between particular bones in the skull (craniosynostosis), characteristic facial abnormalities, and/or malformations of the fingers and toes (digits). However, associated features may vary in range and severity from one person to another, even among affected members of the same family.
Due to craniosynostosis, the top of the head may appear unusually conical (acrocephaly) or the head may seem short and broad (brachycephaly). In addition, the cranial sutures often fuse unevenly, causing the head and face to appear dissimilar from one side to the other (craniofacial asymmetry). Additional malformations of the skull and facial (craniofacial) region may include downslanting eyelid folds (palpebral fissures); a flat nasal bridge; malformed (dysplastic), low-set ears; and a small, underdeveloped (hypoplastic) upper and/or lower jaw (maxilla and/or mandible).Individuals may also have unusually short fingers and toes (brachydactyly); partial fusion of the soft tissues (cutaneous syndactyly) between certain digits; and the presence of extra (supernumerary) toes or, less commonly, additional fingers (polydactyly). In some instances, additional physical abnormalities are present, such as short stature, structural heart malformations (congenital heart defects), mild to moderate obesity, weakening in the abdominal wall near the navel through which the intestine may protrude (umbilical hernia), or failure of the testes to descend into the scrotum (cryptorchidism) in affected males. In addition, many individuals with the disorder are affected by mild to moderate intellectual disability. However, intelligence is normal in some instances.
Although researchers have been able to establish a clear syndrome with characteristic or “core” symptoms, much about the disorder is not fully understood. Several factors including the small number of identified cases, the lack of large clinical studies, and the possibility of other genes influencing the disorder prevent physicians from developing a complete picture of associated symptoms and prognosis. Therefore, it is important to note that affected individuals may not have all of the symptoms discussed below. Parents should talk to their children’s physician and medical team about their child, potential associated symptoms and overall prognosis.
The symptoms of Carpenter syndrome types 1 and 2 are extremely similar and have significant overlap. Because only several individuals have been reported with Carpenter syndrome type 2, researchers are unable to determine whether the different associated genes lead to different symptoms or a different severity of symptoms.
Craniosynostosis is almost always present. The severity and degree of skull (cranial) malformation may be variable, depending on the cranial sutures involved as well as the rate and order of progression. In many affected infants and children, craniosynostosis initially involves the sutures between bones forming the upper sides and the back of the skull (i.e., sagittal and lambdoidal sutures); this is often followed by early closure of the sutures (i.e., coronal sutures) between bones forming the forehead (frontal bone) and the upper sides of the cranium (parietal bones). Such abnormalities may cause the upper portion of the skull or “skullcap” (calvaria) to appear variable in shape and, in some cases, may result in severe malformation. In many cases, the top of the head appears pointed (acrocephaly) or the head seems unusually short and broad (brachycephaly). In addition, involvement of certain sutures on one side of the skull (e.g., unilateral involvement of lambdoidal and/or coronal sutures) may cause the head and face to appear dissimilar from one side to the other (cranial asymmetry). Rarely, due to involvement of multiple cranial sutures (Kleeblattschadel type craniosynostosis), the skull appears to be abnormally divided into three lobes (cloverleaf skull deformity). In some instances, early closure of certain cranial sutures may lead to abnormally increased pressure within the skull (intracranial pressure).
Many infants and children have additional malformations of the skull and facial (craniofacial) area, resulting in a distinctive facial appearance. Such abnormalities include unusually small, underdeveloped ridges above the eyes (hypoplastic supraorbital ridges); downslanting eyelid folds (palpebral fissures); vertical skin folds (epicanthal folds) that may cover the eyes’ inner corners; and broad cheeks. Additional craniofacial malformations are also often present, such as a flat nasal bridge; an unusually narrow or highly arched roof of the mouth (palate); an underdeveloped lower and/or upper jaw (hypoplastic mandible and/or maxilla); relatively low-set, malformed ears; and a short neck. Carpenter syndrome may also be associated with eye (ocular) abnormalities. These may include smallness, improper development, and/or clouding of the front, normally transparent regions of the eyes (corneas); degeneration of the nerves that transmit impulses from the nerve-rich innermost membranes of the eyes (retinas) to the brain (optic atrophy); and/or other ocular defects.
Carpenter syndrome is also typically characterized by distinctive malformations of the fingers and toes (digits). Affected individuals have unusually short fingers and toes (brachydactyly) due to shortness or absence of the middle bones of the digits (middle phalanges). There may be partial fusion of the soft tissues (cutaneous syndactyly) between certain fingers, particularly the third and fourth digits. Some individuals may also have partial fusion of soft tissues between certain toes.
Some affected individuals may also have more than the normal number of digits (polydactyly). In most cases, affected individuals have additional (supernumerary) great toes (halluces) or second toes (preaxial polydactyly) that may also be webbed or partially fused. Less commonly, there may be additional fingers, such as duplication of the fifth fingers or “pinkies” (postaxial polydactyly). Those with the disorder may also have abnormal flexion (camptodactyly) and deviation (clinodactyly) of certain fingers and/or a deformity in which the heels of the feet are turned inward (talipes varus). Additional abnormalities may also be present, including abnormal skin ridge patterns of the hands; deformity of the hip (coxa valga); it also may be present a malformation in which the knees are abnormally close together and the ankles are unusually far apart (genu valgum); and/or an abnormal curvature of the spine (kyphoscoliosis).
Mild short stature and mild to moderate obesity of the face, neck, trunk, forearms, and thighs are common findings. Many individuals are affected by mild intellectual disability. However, normal intelligence has also been reported. In some individuals, hearing loss may occur due to improper conduction of sound from the outer or middle ear to the inner ear (conductive hearing loss); abnormalities of the nerves (i.e., acoustic nerves) that transmit sound impulses to the brain (sensorineural hearing loss); or both (mixed hearing loss).
Some individuals with Carpenter syndrome may also have structural heart malformations at birth (congenital heart defects). Such defects commonly include an abnormal opening in the fibrous partition (septum) that separates the lower or upper chambers of the heart (ventricular or atrial septal defects). In some cases, there may be an abnormal opening (i.e., patent ductus arteriosus) between the artery that transports oxygen-rich blood to most of the body (aorta) and the pulmonary artery, which carries oxygen-deficient blood to the lungs. Additional congenital heart defects may include abnormal narrowing of the opening between the pulmonary artery and the lower right chamber of the heart (i.e., pulmonary stenosis) or a deformity known as tetralogy of Fallot. The latter describes a combination of heart defects, including pulmonary stenosis; an abnormal opening in the partition between the lower chambers of the heart (ventricular septal defect); displacement of the aorta, enabling oxygen-deficient blood to flow from the right ventricle to the aorta; and enlargement of the right ventricle (hypertrophy). In some instances, other congenital heart defects may also be present.
Additional findings may include failure of the testes to descend into the scrotum (cryptorchidism); deficient functioning of the testes (hypogonadism); and/or protrusion (herniation) of portions of the intestine through an abnormal opening in the abdominal wall into the inguinal canal or the passageway through which the testes normally descend into the scrotum. In addition, in some cases, there may be bulging of the intestine through an abdominal wall defect near the navel (umbilical hernia) or a defect in the abdominal wall from which loops of the intestine and other abdominal organs may protrude, covered by a thin, membrane-like sac (omphalocele).
Carpenter syndrome diagnosis
Making a diagnosis for a genetic or rare disease can often be challenging. Healthcare professionals typically look at a person’s medical history, symptoms, physical exam, and laboratory test results in order to make a diagnosis.
A diagnosis of Carpenter syndrome may sometimes be suggested before birth (prenatally) based upon certain specialized tests, such as fetoscopy or ultrasound. During fetoscopy, a flexible viewing instrument (endoscope) may be introduced into the uterus through the abdominal wall to directly observe the fetus and, in some cases, to obtain fetal blood or tissue samples. Fetal ultrasonography is a noninvasive diagnostic procedure in which reflected sound waves create an image of the developing fetus. Prenatal molecular genetic testing to confirm a suspected diagnosis of Carpenter syndrome is available using samples derived from fetal tissues, obtained for example, by chorionic villus biopsy or amniocentesis.
In most cases, the diagnosis is made or confirmed at or shortly after birth based upon a thorough clinical examination, identification of characteristic physical findings, and a variety of specialized tests. Such testing may include advanced imaging techniques, such as computerized tomography (CT) scanning or magnetic resonance imaging (MRI), or other diagnostic tests to help detect or characterize certain abnormalities that may be associated with the disorder (e.g., craniosynostosis, polysyndactyly, other skeletal abnormalities, hearing impairment, etc.). During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of internal structures. During MRI, a magnetic field and radio waves create detailed cross-sectional images of certain organs and tissues.
Molecular genetic testing of RAB23 and MEGF8 can confirm a suspected clinical diagnosis of Carpenter syndrome. Molecular genetic testing can detect mutations in the specific genes known to cause the disorder, but is available only on a clinical basis. Array comparative genomic hybridization, to look for chromosome rearrangements including deletion of the GLI3 gene, may be appropriate in some clinical circumstances.
A thorough cardiac evaluation may also be recommended to detect any heart abnormalities that may be associated with the disorder. Such evaluation may include a through clinical examination, during which heart and lung sounds are evaluated through use of a stethoscope, and specialized tests that enable physicians to evaluate the structure and function of the heart (e.g., x-ray studies, electrocardiography [EKG]), echocardiography, cardiac catheterization).
Carpenter syndrome treatment
The treatment of Carpenter syndrome is directed toward the specific symptoms that are apparent in each individual. Such treatment may require the coordinated efforts of a team of medical professionals, such as pediatricians; surgeons; physicians who diagnose and treat disorders of the skeleton, joints, muscles, and related tissues (orthopedists); physicians who specialize in heart disease (cardiologists); physicians who diagnose and treat neurological disorders (neurologists); hearing specialists; and/or other health care professionals.
Specific therapies for individuals with Carpenter syndrome are symptomatic and supportive. Because craniosynostosis may sometimes result in abnormally increased pressure within the skull (intracranial pressure) and on the brain, early surgery may be advised to help prevent or correct premature closure of cranial sutures. Some reports suggest that early surgical intervention may help to prevent intellectual disability in some instances. However, intellectual disability has occurred in some individuals with Carpenter syndrome despite early surgical correction of craniosynostosis. In addition, normal intelligence has been present in some without such surgical intervention.
In some instances, corrective and reconstructive surgery may also be recommended to help correct additional craniofacial malformations, polydactyly and syndactyly, other skeletal defects, or other physical abnormalities potentially associated with the disorder. In addition, for those with congenital heart defects, treatment with certain medications, surgical intervention, and/or other measures may be necessary. The surgical procedures performed will depend upon the severity and location of the anatomical abnormalities, their associated symptoms, and other factors.
For some individuals with hearing impairment, hearing aids may be beneficial. Appropriate use of hearing aids, other supportive techniques, and/or speech therapy may help to prevent or improve speech problems that may occur in some individuals with the disorder.
Early intervention may be important to ensure that children with Carpenter syndrome reach their potential. Special services that may be beneficial to affected children include special education, physical therapy, and/or other medical, social, or vocational services.
Genetic counseling is recommended for affected individuals and their families. Diagnostic evaluations are also important for family members of individuals with the disorder to detect any symptoms and physical characteristics that may be associated with Carpenter syndrome. Psychosocial support for the entire family is essential as well.
Carpenter syndrome life expectancy
The life expectancy for individuals with Carpenter syndrome is shortened but extremely variable 2.
References- Carpenter Syndrome. https://rarediseases.org/rare-diseases/carpenter-syndrome
- Carpenter syndrome. https://ghr.nlm.nih.gov/condition/carpenter-syndrome
- Asadi, Shahin & Amjadi, Hossein & Jamali, Mahsa. (2019). The role of genetics mutations in genes RAB23 & MEGF8 in carpenter syndrome. Journal of Biosciences. 3. 1-8. 10.31248/JBBD2019.108





{kind=link}